
Method Article
Sequenciamento de leitura ultralonga para análise de DNA genômico completo
Neste Artigo
Resumo
Long-leitura sequências facilitam grandemente a montagem de genomas complexas e caracterização da variação estrutural. Nós descrevemos um método para gerar as sequências ultra-longa pelas plataformas de sequenciamento baseado em nanopore. A abordagem adota uma extração de DNA otimizada seguida de preparações de biblioteca modificada para gerar centenas de mil leituras com cobertura moderada de células humanas.
Resumo
Terceira geração de tecnologias de sequenciamento de DNA único-molécula oferecem significativamente mais ler comprimento que pode facilitar a montagem de genomas complexas e análise das variantes estruturais complexas. Nanopore plataformas executam sequenciamento de único-molécula medindo diretamente as alterações atuais mediadas pela passagem de DNA através dos poros e podem gerar centenas de leituras de quilobase (kb) com custo de capital mínimo. Esta plataforma tem sido adotada por muitos pesquisadores para uma variedade de aplicações. Alcançar longas distâncias de leitura de sequenciamento é o fator mais crítico para alavancar o valor das plataformas de sequenciamento de nanopore. Para gerar leituras ultra-longa, consideração especial é necessário para evitar quebras de DNA e ganhar eficiência para gerar modelos de sequenciamento produtivo. Aqui, nós fornecemos o protocolo detalhado de ultra-longa sequenciamento de DNA, incluindo a extração de DNA de alto peso molecular (HMW) de células frescas ou congeladas, construção de biblioteca por mecânica de corte ou fragmentação de transposase e sequenciamento em um dispositivo de nanopore. De 20 a 25 µ g de DNA HMW, o método pode atingir N50 ler comprimento de 50-70 kb com corte mecânico e N50 de 90-100 kb ler comprimento com transposase mediada por fragmentação. O protocolo pode ser aplicado a DNA extraído de células de mamíferos, para realizar o sequenciamento do genoma inteiro para a detecção de variantes estruturais e montagem do genoma. As melhorias adicionais na extração de DNA e reações enzimáticas mais aumentará o comprimento Leia e expandir sua utilidade.
Introdução
Na última década, maciçamente paralelo e tecnologias de segunda geração altamente precisos da elevado-produção sequenciamento conduziram a uma explosão da inovação tecnológica1,2,3e descoberta de biomédica. Apesar dos progressos técnicos, os curto-ler dados gerados pelas plataformas da segunda geração são ineficazes para resolver complexas regiões genômicas e são limitados na detecção de variantes estruturais genômicas (SVs), que desempenham papéis importantes em humanos evolução e doenças4,5. Além disso, curto-ler dados são incapazes de resolver a variação de repetição e são impróprios para discernir haplótipo eliminação de variantes genéticas6.
Recentes progressos em único-molécula sequenciamento oferece significativamente mais ler o comprimento, o que pode facilitar a detecção de todo o espectro da SVs7,8,9e ofertas exactas e completas de montagem do complexo microbiana e mamíferos genomas6,10. A plataforma nanopore executa single-molécula sequenciamento medindo diretamente as alterações atuais mediadas pela passagem de DNA através dos poros11,12,13. Ao contrário de qualquer existente química de sequenciamento de DNA, sequenciamento de nanopore pode gerar long (dezenas de milhares de kilobases) leituras em tempo real, sem depender de cinética de polimerase ou artificial amplificação da amostra do DNA. Portanto, nanopore longa leitura sequenciamento (NLR-seq) detém grande promessa para gerar ultra-longa comprimentos Leia bem além de 100kb, que avançou grandemente análises genômicas e biomédica14, particularmente na baixa complexidade ou repetição-rico regiões dos genomas15.
A característica única de sequenciamento de nanopore é seu potencial para gerar longo lê sem uma limitação do comprimento teórico. Portanto, o comprimento de leitura é dependente do comprimento físico do DNA que é diretamente afetado pela qualidade de modelo de integridade e sequenciamento do DNA. Além disso, dependendo da extensão da manipulação e o número de etapas envolvidas, tais como forças de pipetagem e condições de extração, a qualidade do DNA é altamente variável. Portanto, é desafiador para um ceder tempo leituras aplicando apenas os protocolos padrão de extração de DNA e métodos de construção de biblioteca fornecido do fabricante. Para este fim, nós desenvolvemos um robusto método para gerar ultra-longa ler (centenas de kilobases) dados de sequenciamento a partir de células colhidos Pelotas. Adotamos várias melhorias nos procedimentos de preparação de extração e biblioteca de DNA. Racionalizámos o protocolo para excluir procedimentos desnecessários que causam danos e degradação do DNA. Este protocolo é composto de alto peso molecular (HMW) extração de DNA, construção de biblioteca de DNA ultra-longa e sequenciamento em uma plataforma de nanopore. Para um biólogo molecular bem treinado, que normalmente demora 6h da célula da colheita para a realização de extração de DNA de HMW, 90 min ou 8 h para construção de biblioteca, dependendo do método de corte e até um mais 48 h para sequenciamento de DNA. O uso do protocolo irá capacitar a Comunidade genômica para melhorar a nossa compreensão da complexidade do genoma e ganhar a introspecção nova variação do genoma em doenças humanas.
Protocolo
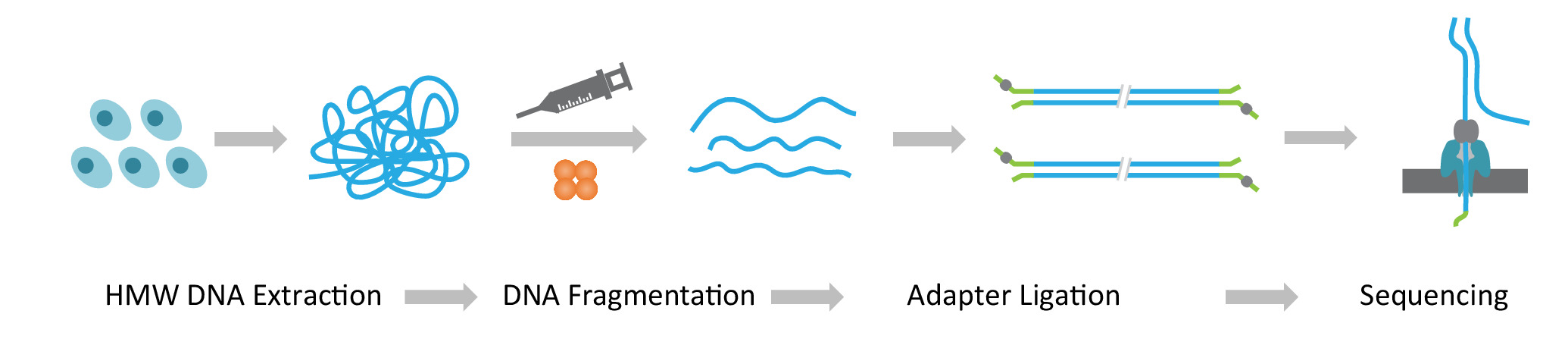
Nota: O protocolo NLR-seq consiste de três etapas consecutivas: 1) extração de alta-molecular peso DNA genômico (HMW); 2) ultralonga construção de biblioteca de DNA, que inclui a fragmentação do ADN para o tamanho desejado HMW e ligadura dos adaptadores de sequenciamento do DNA termina; e 3) carregamento do DNA adaptador-ligados para as matrizes de nanoporos (Figura 1).
1. extração de DNA HMW
- Instalação de reagente. Fazer 1x tampão de fosfato salina tamponada (PBS) (1.000 mL) adicionando-se 100 mL de PBS (10x) a 900 mL de água e misture bem. Fazer o tampão de lise (50 mL) adicionando 43,5 mL de água para um tubo de 50 mL. Adicionar 500 μL de Tris (1 M, pH 8,0), 1 mL de cloreto de sódio (NaCl) (5m), 2,5 mL de ácido etilenodiaminotetracético (EDTA) (0,5 M, pH 8.0) e 2,5 mL de sódio Dodecil sulfato (SDS) (10%, wt/vol) ao tubo e misture bem.
Nota: Este tampão PBS pode ser armazenado a 4 ° C por até 6 meses. A lise do premade pode ser armazenado em RT por até 2 meses. - Verificar a mortalidade de células e contagem das células. Certifique-se que o rácio ao vivo > 85% e o número total é de 30 x 106.
Nota: As células utilizadas no presente protocolo são da linha celular de HG00733, uma linha de celular lymphoblastoid humano de origem porto-riquenha, amplamente utilizada no consórcio de genoma de 1000 para a análise da variação estrutural (ver tabela de materiais para solicitar informações), que pertence a recurso de amostra internacional do genoma. - Coletar as células por centrifugação a 200 x g durante 5 min à RT. Discard a médio e ressuspender as células (30 x 106 células) com 5 mL de tampão de PBS x 1. Centrifugar novamente a 200 x g durante 5 min à RT e descartar o sobrenadante.
Nota: 25-35 x 106 células são aceitáveis para esta abordagem. Mais variação na quantidade de células usadas vai precisar ainda mais de otimização. O centrifugado pode ser armazenado em −80 ° C por até 6 meses. - Ressuspender as células em 200 μL de tampão de PBS x 1. Se usando um pellet de células congeladas, lave com 5 mL de 1X PBS tampão. A solução a 200 x g durante 5 min à RT de centrifugação, descartar o sobrenadante e ressuspender as células em 200 µ l de tampão PBS 1x.
- Prepare-se 10 mL de tampão de Lise em um tubo de 50 mL. Adicione a suspensão de células de 200 μL a Lise e vórtice na velocidade máxima por 3 s. Incubar a solução a 37 ° C por 1h.
- Adicione 2 µ l de RNase A (100 mg/mL) para o lisado. Gire suavemente o tubo de 50 mL para misturar a amostra. Incube a solução a 37 ° C por 1h.
- Adicione 50 proteinase μL K (20 mg/mL) para o lisado. Gire suavemente o tubo de 50 mL para misturar a amostra. Incube a solução a 50 ° C por 2 h. Durante a incubação, misture suavemente a amostra cada 30 min.
- Retire o tubo de 50 mL do 50 ° C e deixe descansar na RT por 5 min.
- Adicionar 10 mL de álcool isoamílico: fenol: clorofórmio (25:24:1, vol/vol/vol) a camada de fenol para o lisado e gire o tubo em um misturador de rotor (veja a Tabela de materiais) em RT em uma coifa a 20 rpm durante 10 min. envolva a tampa do tubo com parafilm para evitar fugas durante a rotação.
- Prepare-se dois tubos de gel 50 mL (ver Tabela de materiais) por centrifugação a 1.500 x g por 2 min em RT
Nota: O gel forma uma barreira estável entre a fase aquosa contendo ácido nucleico e o solvente orgânico. - Despeje a solução de amostra/fenol dentre os preparados 50 mL gel de tubos de passo 1.10. Centrifugue a solução a 3.000 x g durante 10 minutos a RT
- Despeje o líquido sobrenadante para um novo tubo de 50 mL. Adicionar 10 mL de camada de fenol de álcool isoamílico: fenol: clorofórmio (25:24:1, vol/vol/vol) e gire o tubo em um misturador de rotador no RT em uma coifa a 20 rpm durante 10 min.
- Repita o tubo de gel de passo 1.11 uma vez com o segundo preparado.
- Despeje o líquido sobrenadante para um novo tubo de 50 mL. Adicionar 25 mL de etanol a 100% gelada e Gire suavemente o tubo com a mão até o DNA precipita (Figura 2).
Nota: A abordagem de precipitação ajuda a estabilizar o DNA HMW. - Dobre uma ponta de 20 μL para fazer um gancho. Cuidadosamente Retire o DNA HMW com o gancho e deixe a líquido gota.
- Coloque o DNA HMW num tubo de 50 mL contendo 40 mL de etanol a 70%. Lave o DNA delicadamente vezes por inversão do tubo 3.
- Repita a etapa 1.15 uma vez para coletar DNA do tubo de etanol a 70%.
- Coloque o DNA HMW em um tubo de 2 mL contendo 1,8 mL de etanol a 70%.
- Centrifugue o DNA HMW lavado a 10.000 x g durante 3 s em RT. Remove tanto do etanol residual possível por pipetagem.
Nota: Não perturbe a pelota do ADN quando pipetar o etanol residual. - Incube o tubo 2 mL a 37 ° C durante 10 minutos com a tampa aberta para secar a amostra.
- Se continuar com um passo 2.1 (com corte mecânico e 1 Kit de sequenciamento de ligadura D), adicione 1 mL do TE (10 mM Tris e 1 mM EDTA, pH 8.0) para o tubo de 2 mL.
- Se continuar com passo 2.2 (com fragmentação transposase-baseado e rápida Sequencing Kit), adicionar 200 μL de 10mm Tris (pH 8.0) com 0,02% Triton X-100.
Nota: Não perturbe a pelota do ADN. Deixar o tubo fique a 4 ° C, no escuro por 48 h ajudará a amostra totalmente ressuspender. O DNA HMW podem ser armazenado a 4 ° C por até 2 semanas. Tempo de armazenamento ou outras condições de armazenamento podem introduzir mais fragmentos curtos.
2. ultralonga construção de biblioteca de DNA
Nota: Existem duas maneiras para construir as bibliotecas de DNA ultra-longa com base em dois diferentes métodos de corte juntamente com kits de sequenciamento de nanopore. Uma biblioteca baseada em cisalhamento mecânica produz dados com um N50 de 50-70 kb, levando cerca de 8 h para a construção da biblioteca. Uma biblioteca de fragmentação-based transposase produz um N50 de 90-100 kb de dados, tendo apenas 90 min para a construção da biblioteca. O protocolo de corte mecânico dá maior rendimento de DNA mesmo usando versões idênticas de adaptador de sequenciamento e qualidade de nanopore células de fluxo de entrada.
- Construção mecânica biblioteca baseada em cisalhamento
- Degelo e mistura os reagentes da ligadura kit (veja a Tabela de materiais). Degelo FFPE DNA reparo buffers e reparação/dA-tailing final no gelo, então vórtice e girar para baixo a mistura. Descongele o adaptador mix (AMX) e buffer de vinculação de grânulo adaptador (ABB) no gelo, em seguida, pipeta e spin para baixo para misturar. Degelo correndo buffer com mistura de combustível (RBF) e tampão de eluição (ELB) em RT, em seguida, vórtice e rotação para misturar. Descongele o carregamento grânulos (LLB) da biblioteca em RT e pipeta para misturar antes de usar.
- Uma vez descongelado, mantenha todos os componentes do kit no gelo. Tire as enzimas somente quando necessário. Trazer os grânulos magnéticos para RT para uso.
Nota: Para recomendações sobre os grânulos magnéticos para usar, consulte a tabela de materiais.
- Uma vez descongelado, mantenha todos os componentes do kit no gelo. Tire as enzimas somente quando necessário. Trazer os grânulos magnéticos para RT para uso.
- Verifique a qualidade e quantidade do DNA da etapa 1.21.1 HMW. Pipeta de 20 μL de DNA em novos tubos de 1,5 mL de três locais diferentes no tubo de DNA HMW usando P200 grande furo dicas. Tome 1 μL das três alíquotas para detectar a concentração usando um dados e a qualidade usando um UV lendo. Verifique várias vezes para confirmar os resultados.
Nota: Os resultados esperados são mostrados na Figura 3A. O valor de260/280 OD é aproximadamente 1.9 e o valor de260/230 OD é aproximadamente 2,3. - Transferência a 940 restantes μL de DNA HMW, em uma tampa de tubo de 50 mL com uma largura de P1000 furo ponta.
- Aspire todo o ADN em uma seringa de 1 mL sem a agulha.
- Coloque a seringa com a agulha 27G e ejetar todo o ADN na PAC, devagar e com cuidado (~ 10 s). Tire a agulha 27G da seringa.
- Repita as etapas 2.1.4 e 2.1.5 para 29 vezes para um total de 30 passes através da agulha.
Nota: O DNA HMW cortado pode ser armazenado a 4 ° C, no escuro por até 24 h. controle de qualidade (QC) é altamente recomendado por eletroforese em gel de campo pulsado, mas é caro e demorado. Se executar QC em uma automatizado pulso campo gel de electroforese máquina use um protocolo de 5-150 kb para um 20 h executar. Os resultados esperados são mostrados na Figura 4. - Prepare a reação de reparo de DNA em um tubo de 0,2 mL, adicionar 100 μL de DNA de HMW cortado (20 μg), 15 μL de tampão de reparação do ADN FFPE, 12 μL de mistura de reparação do ADN FFPE e 16 μL de água livre de nuclease. Misture a reação por sacudir suavemente 6 vezes e girar para baixo para remover as bolhas.
- Incube a reação a 20 ° C por 60 min. transferência da amostra em um tubo de 1,5 mL novo com uma ponta de furo largo P200.
- Resuspenda as esferas magnéticas pipetagem ou num Vortex. Adicione 143 grânulos μL (1x) para a reação de reparação do ADN e misture suavemente por agredir o tubo 6 vezes. Gire o tubo em um misturador de rotor em RT a 20 rpm por 30 min.
- Girar a amostra a 1.000 x g durante 2 s no lugar do RT. o tubo em um rack magnético para 10 min. Mantenha o tubo sobre o rack magnético e descartar o sobrenadante.
- Mantendo o tubo sobre o rack magnético, adicione 400 μL de etanol a 70% acabadas sem perturbar o sedimento. Remover o etanol a 70% após 30 s.
- Repita a etapa 2.1.11 uma vez.
- Girar a amostra a 1.000 x g durante 2 s no lugar do RT. o tubo de volta na prateleira magnética. Remover qualquer etanol residual e ar seco por 30 s. Não seque a pelota.
- Retire o tubo do rack magnético e adicionar 103 μL de et (10 mM Tris e 1 mM EDTA, pH 8.0). Suavemente o tubo para garantir que os grânulos são cobertos no buffer e incubam delicadamente em um misturador de rotor em RT por 30 min. de filme flick o tubo a cada 5 min para auxiliar a ressuspensão da pelota.
- Granule os grânulos no rack magnético para ponta de diâmetro de pelo menos 10 min. Transfira 100 μL de eluído com um P200 ampla em um tubo de 0,2 mL.
- Prepare a reparação final e dA-tailing reação em um tubo de 0,2 mL, adicionando 100 μL de DNA de HMW reparado, 14 μL de tampão, reparação/dA-tailing final e 7 μL de mistura de reparação/dA-tailing final. Misture a reação por sacudir suavemente 6 vezes e girar para baixo para remover as bolhas.
- Incubar a reação a 20 ° C por 60 min, seguido de 65 ° C por 20 min e em seguida, mantenha a 22° C. Dica de transferência a amostra para um tubo de 1,5 mL novo usando um P200 largura do furo.
- Resuspenda as esferas magnéticas pipetagem ou num Vortex. Adicionar 48 μL de grânulos (0.4 x) para a reação de reparação/dA-tailing final e misture suavemente por agredir o tubo 6 vezes. Gire o tubo em um misturador de rotor em RT a 20 rpm por 30 min.
- Repita as etapas 2.1.10-2.1.13 uma vez.
- Retire o tubo do rack magnético e adicionar 33 μL de et (10 mM Tris e 1 mM EDTA, pH 8.0). Suavemente o tubo para garantir que os grânulos são cobertos no buffer e incubam delicadamente em um misturador de rotor em RT por 30 min. de filme flick o tubo a cada 5 min para auxiliar a ressuspensão da pelota.
- Granule os grânulos no rack magnético para ponta de diâmetro de pelo menos 10 min. Transfira 30 μL de eluído com um P200 ampla para um novo tubo de 1,5 mL. Leve o μL de 1-2 extra para detectar a concentração usando um dados.
Nota: A recuperação de 5-6 μg nesta etapa é esperada. - Prepare a reação da ligadura no tubo de amostra de 1,5 mL por adição 30 μL de DNA de HMW final-reparado, 20 μL da mistura de adaptador (AMX 1D) e 50 μL de mistura de mestre sem corte/TA da ligadura. Misture a reação por sacudir suavemente 6 vezes entre cada adição sequencial e spin para baixo para remover as bolhas.
- Incube a reação na RT por 60 min.
- Resuspenda as esferas magnéticas pipetagem ou num Vortex. Adicionar 40 grânulos μL (0.4 x) para a reação da ligadura e misture suavemente por agredir o tubo 6 vezes. Gire o tubo em um misturador de rotor em RT a 20 rpm por 30 min.
- Repita a etapa 2.1.10 uma vez.
- Adicione 400 μL de tampão de ligação (ABB) de grânulo de adaptador para o tubo. Agite o tubo delicadamente 6 vezes para Ressuspender os grânulos. Coloque o tubo de volta na prateleira magnética para separar os grânulos do buffer e descartar o sobrenadante.
- Repita a etapa 2.1.26 uma vez.
- Girar a amostra a 1.000 x g durante 2 s no lugar do RT. o tubo de volta na prateleira magnética. Remover qualquer reserva residual e ar seco por 30 s. Não seque a pelota.
- Retire o tubo do rack magnético e resuspenda o pellet em 43 μL de tampão de eluição. Suavemente o tubo para garantir que os grânulos são cobertos no buffer e incubam delicadamente em um misturador de rotor em RT por 30 min. de filme flick o tubo a cada 5 min para auxiliar a ressuspensão da pelota.
- Granule os grânulos no rack magnético para ponta de diâmetro de pelo menos 10 min. Transfira 40 μL de eluído com um P200 ampla para um novo tubo de 1,5 mL. Leve o μL de 1-2 extra para detectar a concentração usando um dados.
Nota: A recuperação de 1-2 μg nesta etapa é esperada. A biblioteca baseada em cisalhamento mecânica está pronta para o carregamento. A biblioteca pode ser armazenada no gelo para até 2 h até carregamento para sequenciamento se necessário.
- Degelo e mistura os reagentes da ligadura kit (veja a Tabela de materiais). Degelo FFPE DNA reparo buffers e reparação/dA-tailing final no gelo, então vórtice e girar para baixo a mistura. Descongele o adaptador mix (AMX) e buffer de vinculação de grânulo adaptador (ABB) no gelo, em seguida, pipeta e spin para baixo para misturar. Degelo correndo buffer com mistura de combustível (RBF) e tampão de eluição (ELB) em RT, em seguida, vórtice e rotação para misturar. Descongele o carregamento grânulos (LLB) da biblioteca em RT e pipeta para misturar antes de usar.
- Construção de biblioteca baseada em fragmentação transposase
- Descongele os reagentes do transposase kit (veja a Tabela de materiais). Descongele o mix de fragmentação (FRA) e adaptador rápida (RAP) em gelo e pipeta para misturar. Descongelar o buffer de sequenciamento (SQB), carregamento de grânulos (LB), liberar o buffer (FLB) e liberar o baraço (FLT) no RT e pipetar para misturar. Descongele o carregamento grânulos (LB) em RT e pipeta para misturar antes de usar. Uma vez descongelado, mantenha todos os componentes do kit no gelo. Tire as enzimas somente quando necessário.
- Verifique a qualidade e quantidade do DNA da etapa 1.21.2 HMW. Pipeta de 20 μL de DNA em novos tubos de 1,5 mL de três locais diferentes no tubo de DNA HMW usando P200 grande furo dicas. Tome 1 μL das três alíquotas para detectar a concentração usando um dados e a qualidade usando um UV lendo. Verifique várias vezes para confirmar os resultados.
Nota: Os resultados esperados são mostrados na Figura 3B. O valor de260/280 OD é aproximadamente 1.9 e o valor de260/230 OD é aproximadamente 2,3. - Prepare a reação de tagmentation de DNA em um tubo de 0,2 mL, adicionando 22 μL de DNA HMW, 1 μL de 10mm Tris (pH 8.0) com 0,02% Triton X-100 e 1 μL de fragmentação misturar (FRA). Mix por pipetagem com um P200 grande furo ponta mais lentamente possível 6 vezes, tomando cuidado para não introduzir bolhas.
- Incubar a reação a 30 ° C por 1 min, seguido de 80 ° C por 1 min e mantenha a 4 ° C. Transferência a mistura em um novo tubo de 1,5 mL com um P200 ampla furo ponta e vá para o próximo passo imediatamente.
- Adicione 1 μL do adaptador rápida (RAP) ao tubo de 1,5 mL de amostra. Mix por pipetagem com um P200 grande furo ponta mais lentamente possível 6 vezes, tomando cuidado para não introduzir bolhas.
- Incube a reação na RT por 60 min.
Nota: Biblioteca baseada em fragmentação transposase está pronta para o carregamento. A biblioteca pode ser armazenada no gelo para até 2 h até carregamento para sequenciamento se necessário.
3. sequenciamento do dispositivo nanopore
- Verifique o dispositivo de sequenciamento de nanopore (ver Tabela de materiais). Certifique-se que tanto o software e o hardware estão funcionando e não há espaço de armazenamento suficiente.
- Verifique se a célula de fluxo. Abra uma nova célula de fluxo e inserir o dispositivo de nanopore a célula de fluxo. Verifique se que a caixa da localização da célula de fluxo foi inserida (X1-X5). Selecione o tipo de célula de fluxo correto. Clique em Verificar células de fluxo de fluxo de trabalho. Clique no botão Iniciar teste para começar a célula de fluxo análise QC.
Nota: Se o número de poros ativo total relatado é menos de 800, use uma nova célula de fluxo diferentes para sequenciamento. - Prepare o buffer de escorva. Para uma biblioteca de tosquia-based mecânica, adicionar 576 μL de tampão com mistura de combustível (RBF) e 624 μL de água livre de nuclease num tubo de 1,5 mL. Vórtice e spin para baixo a mistura o buffer de escorva. Para uma biblioteca de fragmentação-based transposase, adicione 30 μL de baraço nivelado (FLT) para o tubo de descarga buffer (FLB). Vórtice e spin para baixo a mistura o buffer de escorva.
- Na célula de fluxo, mova a tampa de porta de escorva no sentido horário para expor a porta de escorva.
- Defina uma pipeta P1000 para 100 μL e insira a ponta na porta de escorva. Puxe para trás um pequeno volume de buffer (menos de 30 μL) para remover quaisquer bolhas da célula de fluxo. Pare de pipetagem, uma vez que uma pequena quantidade de líquido amarelo entra a ponta.
- Use uma pipeta P1000 para carregar 800 μL da mistura escorva para a célula de fluxo através da porta de escorva. Para evitar a introdução de bolhas, adicione 30 μL da mistura escorva para cobrir o topo da porta de escorva primeiro, em seguida, insira a ponta na porta de escorva e lentamente adicione o restante da mistura escorva. Tire a ponta quando há cerca de 50 μL de esquerda. Adicione o restante da mistura de escorva na parte superior da porta de escorva. O fluido vai dentro, por si só.
- Deixe a instalação para incubar durante 5 min. Entretanto, prepare a mistura de biblioteca no tubo de 1,5 mL contendo a biblioteca.
Nota: Para uma biblioteca baseada em cisalhamento mecânica adicione 35 µ l de tampão em execução com mistura de combustível (RBF) a 40 µ l da biblioteca de DNA. Para uma biblioteca de fragmentação-based transposase adicionar 34 µ l de tampão de sequenciamento (SQB) e 16 µ l de água livre de nuclease a 25 µ l da biblioteca de DNA. - Abra a tampa do porta do fluxo celular amostra suavemente para expor a porta amostra. Use uma pipeta P1000 adicionar 200 μL da mistura através da porta de escorva escorva para a célula de fluxo, conforme descrito na etapa 3.5. Certifique-se que a mistura de escorva não é carregada para a célula de fluxo através da porta de amostra.
- Defina uma pipeta P200 para 80 μL. Misture a biblioteca suavemente com uma ponta de furo largo pipetando subindo e descendo 6 vezes só previamente ao seu embarque.
- Carrega o mix de biblioteca gota a gota através da porta de amostra para a célula de fluxo. Adicione cada gota somente após a queda anterior é completamente carregada no porto.
- Volte a colocar a tampa do porta amostra suavemente e certifique-se de que o porto de amostra é totalmente coberto. Mova a tampa de porta de escorva no sentido anti-horário para cobrir o porto de escorva. Feche a tampa do dispositivo.
- Clique sobre o fluxo de trabalho da Nova experiência . Digite o nome da biblioteca, selecione o kit correto de acordo com os procedimentos utilizados e verifique se as configurações estão correta (48 h executar, em tempo real ON base-chamada).
- Clique em Iniciar Executar. Depois de 10 min, grave o ID da célula de fluxo e os números nanopore ativo (número total e números dos cada quatro grupos), a partir das informações de execução.
- Análise de dados. Copie os dados para um computador local ou em um cluster, a qualquer momento, do sequenciamento, ou quando a execução é concluída. Use Minimap216 (https://github.com/lh3/minimap2) para alinhar os dados de sequência para o genoma de referência. Resuma o desempenho de sequenciamento de dados sequência primas e os alinhamentos por NanoPlot17 (https://github.com/wdecoster/NanoPlot).
Resultados
O protocolo de sequenciamento de DNA ultra-longa aplica HMW DNA para construção de biblioteca. Portanto, é fundamental escolher pilhas bem cultivadas com relação ao vivo > 85% na célula colheita passo. A quantidade de células usadas para extração de DNA afetará a qualidade e a quantidade de DNA HMW. A lise celular não funciona bem se iniciando com muitas células. Usar poucas células não gera DNA suficiente para construção de biblioteca porque a precipitação do DNA HMW é executada usando a rotação suave à mão em vez de centrifugação de alta velocidade. Um exemplo do DNA HMW depois adicionando gelada 100% de etanol e de giro é mostrado como o precipitado branco de algodão-como na Figura 2.
É importante verificar a qualidade da entrada DNA antes de iniciar a construção da biblioteca. Degradação, quantificação incorreta, contaminação (por exemplo, proteínas, RNAs, detergentes, surfactante e fenol residual ou etanol) e baixo peso molecular de ADN podem ter um efeito significativo sobre os procedimentos subsequentes e leia o comprimento final. Recomendamos realizar a análise QC usando o DNA de três locais diferentes no tubo contendo DNA HMW. De UV, lendo os resultados para o DNA HMW, o valor de280 OD260/OD é aproximadamente 1.9 e o valor de230 OD260/OD é aproximadamente 2.3 (Figura 3AB). Estes valores de proporção são consistentes entre os três ensaios para uma boa amostra de DNA HMW. Diferentes métodos de corte exige diferentes volumes de entrada DNA. A concentração de DNA HMW precisa ser > 200 ng / µ l para a tosquia mecânica enquanto ele precisa ser > 1 µ g / µ l para fragmentação transposase. A concentração detectada por um dados é um pouco menor do que a UV lendo. No entanto, o coeficiente de variação da concentração da amostra de DNA HMW do mesma é necessário para ser menos de 15% com ambos os dados e o UV ensaios de leitura. Cisalhamento mecânico aplica-se uma seringa com uma agulha para quebrar o HMW DNA para que o número de passes através da agulha terá impacto sobre o tamanho do DNA cortado e o final leia o comprimento. É aconselhável executar tamanho QC depois agulha de corte para garantir a maioria do DNA HMW é maior do que 50 kb, conforme ilustrado na Figura 4. No método de corte mecânico, 30 passes gerou os melhores resultados de sequenciamento, considerando o comprimento e a saída.
O N50 de uma biblioteca baseada no corte mecânica é 50-70 kb, enquanto uma biblioteca baseada na fragmentação de transposase é 90-100 kb. Os resultados de quatro corridas, usando a linha de celular HG00733 são mostrados na tabela 1. Todas as quatro corridas tem mais de 2.300 leituras com comprimento maior que 100 kb. O comprimento máximo é mais longo nas transposase baseado na fragmentação bibliotecas (455 kb e 489 kb) em comparação com as bibliotecas baseadas em corte mecânicas (348 kb e 387 kb) enquanto o último produzido mais leituras totais, indicando um rendimento mais elevado. A construção da biblioteca baseada em fragmentação transposase tem menos etapas e menor tempo de preparação para que introduzirá menos fragmentos curtos. As execuções de dois usando transposase têm um comprimento mais longo (> 30 kb) e comprimento médio (> 10 kb). Além disso, os dados mostram consistente alta qualidade em todas as execuções (índice de qualidade média é aproximadamente 10.0, ~ 90% base de precisão). Mais de 97% das bases totais foram alinhados para o genoma humano de referência (hg19) usando Minimap216 com as configurações padrão. As distribuições de tamanho esperado do cru diz são mostradas na Figura 5. Todas as execuções têm uma grande proporção de dados acima de 50 kb, enquanto transposase baseado na fragmentação bibliotecas têm uma maior taxa de leituras ultra longas (por exemplo, > 100 kb). Este protocolo tem sido aplicado com sucesso em várias linhas de células humanas (complementar a tabela 1).

Figura 1: visão geral esquemática de fluxo de trabalho longa leitura sequenciamento (NLR-seq) nanopore. Laranja, o transposase complexo. Amarelo-verde, o adaptador nanopore. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: precipitação do DNA representativa do método de extração do fenol-clorofórmio. A seta branca indica o DNA HMW. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: resultados de exemplo QC do DNA da leitura UV HMW. (A) HMW DNA da etapa 1.21.1 pronto para construção mecânica biblioteca baseada em corte. (B) HMW DNA da etapa 1.21.2 para construção de biblioteca baseada em fragmentação transposase. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: resultados QC da agulha cisalhadas HMW DNA por eletroforese em gel de campo pulsado. L1: Quick-load kb 1 escada de DNA; L2: Quick-carga 1KB estender a escada de DNA. 1-8: DNA com tempos diferentes de passagem através da agulha de corte. 1-3, sem distorção; 4, 10 vezes; 5, 20 vezes; 6, 30 vezes; 7, 40 vezes; 8, 50 vezes. Esta etapa QC é opcional. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: espera-se distribuições de tamanho das bibliotecas de DNA ultra-longa nanopore. MS, bibliotecas baseadas em corte mecânicas. TF, transposase baseado na fragmentação bibliotecas. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Mecânica shearing_rep1 | Mecânica shearing_rep2 | Transposase fragmentation_rep1 | Transposase fragmentation_rep2 | |
| Linha celular | HG00733 | HG00733 | HG00733 | HG00733 |
| N50 das leituras | 55.180 | 63.007 | 98.237 | 95.629 |
| Número de leituras mais do que 100 Kb | 2.500 | 3.082 | 2.386 | 2.355 |
| Número de leituras totais | 97.859 | 80.465 | 24.166 | 21.032 |
| Comprimento máximo (bp) | 348.482 | 387.113 | 454.660 | 489.426 |
| Comprimento (bp) | 17.861 | 20.395 | 33.528 | 38.175 |
| Comprimento médio (bp) | 5.335 | 5.894 | 10.249 | 15.656 |
| Significa qualidade das leituras | 10.0 | 10.1 | 9.9 | 10.0 |
| Bases totais de leituras crus | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| Bases totais de leituras alinhadas | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| Mapeada relação das bases totais (hg19, Minimap2) | 96,9% | 98,0% | 97,7% | 97.0% |
| Número de poros ativos | 1225: 480, 402, 254, 89 | 1058: 480, 356, 176, 46 | 958: 452, 328, 148, 30 | 1092: 487, 367, 195, 43 |
Tabela 1: Resumo das métricas de desempenho é executado com protocolos diferentes de corte.
| Biblioteca 1 | Biblioteca 2 | |
| Linha celular | K562 | GM19240 |
| Célula de informação requisitando | ATCC, gato. Não. CCL-243 | Instituto Coriell, gato. Não. GM19240 |
| Protocolo | corte mecânico | corte mecânico |
| N50 das leituras | 60.063 | 55.295 |
| Número de leituras totais | 193.783 | 120.807 |
| Comprimento médio (bp) | 1.843 | 4.688 |
| Comprimento (bp) | 9.825 | 17.408 |
| Comprimento máximo (bp) | 548.780 | 212.338 |
| Bases totais de leituras crus | 1,903,989,686 | 2,103,015,331 |
| Bases totais de leituras alinhadas | 1,837,350,047 | 1,997,419,761 |
| Mapeada relação das bases totais (hg19, Minimap2) | 96,6% | 95.0% |
| Número de poros ativos | 1111: 482, 371, 203, 55 | 1032: 447, 333, 196, 56 |
Complementar tabela 1: Resumo das duas corridas NLR-seq usando outras linhas celulares com o protocolo de corte mecânico.
Discussão
Em princípio, sequenciamento de nanopore é capaz de gerar 100 kb para megabase leituras no comprimento11,12,13. Quatro principais fatores afetarão o desempenho de qualidade de dados e executar o sequenciamento: 1) números ativo dos poros e a atividade dos poros; 2) proteína motor, que controla a velocidade do DNA de passagem da nanopore; 3) modelo DNA (comprimento, pureza, qualidade, massa); 4) sequenciamento adaptador ligadura eficiência, que determina o DNA utilizável do exemplo de entrada. Os dois primeiros fatores dependem da versão da célula de fluxo e o kit de sequenciamento, fornecido pelo fabricante. Os segundo dois fatores são passos críticos no presente protocolo (extração de DNA HMW, cisalhamento e ligadura).
Este protocolo requer paciência e prática. A qualidade do DNA HMW é importante para ultra-longa de bibliotecas de DNA6. O protocolo começa com células coletadas com alta viabilidade (> 85% de células viáveis preferida), limitando o DNA degradado de células mortas. Qualquer processo áspero que pode apresentar danos ao DNA (por exemplo, forte perturbando, tremendo, vórtice, múltiplos pipetagem, repetidos congelamento e descongelamento) deve ser evitado. Na concepção do protocolo, omitimos pipetagem em todo o processo de extração de DNA. Dicas de furo grande precisam ser usado quando pipetagem é necessário após o corte mecânico durante a construção da biblioteca e sequenciamento. Como os nanoporos são sensíveis os produtos químicos na câmara reserva12, deveria haver tão poucos contaminantes residuais (por exemplo, os detergentes, surfactantes, fenol, etanol, proteínas, RNAs, etc) quanto possível no DNA. Considerando o comprimento e o rendimento, o método de extração do fenol mostra os resultados melhores e mais reprodutíveis em comparação com vários métodos de extração diferentes testados até agora.
Apesar da capacidade do presente protocolo para produzir sequências de leitura longa, várias limitações ainda permanecem. Em primeiro lugar, este protocolo foi otimizado com base no dispositivo de sequenciamento de nanopore disponível no momento da publicação; assim, é limitado para a química seletiva de sequenciamento baseado em nanopore e pode ser de qualidade inferior quando executadas em outros tipos de dispositivos de sequenciamento de long-leitura. Em segundo lugar, o resultado é altamente dependente da qualidade do DNA extraído de matérias-primas (tecidos ou células). Comprimento de leitura pode ser comprometido se o DNA de partida já está degradado ou danificado. Em terceiro lugar, apesar de várias etapas QC são incorporadas no protocolo para verificar a qualidade do DNA, o rendimento final e comprimento das leituras podem ser afetadas pela célula de fluxo e pore a atividade, que pode ser variável nesta fase inicial da plataforma de sequenciamento de nanopore desenvolvimento.
O protocolo descrito aqui usa amostras de linha de células humanas de suspensão para extração de DNA. Otimizamos os tempos de passagem na agulha de corte, a proporção de DNA HMW transposase e o tempo de ligadura para produzir os resultados descritos. O protocolo pode ser expandido de quatro maneiras. Primeiro, os usuários podem começar com outras culturas de células de mamíferos e com quantidade diferente de células, tecidos, amostras clínicas ou outros organismos. Serão necessários mais otimização no tempo de incubação de Lise, volume de reação e centrifugação. Em segundo lugar, é difícil prever o tamanho de destino para o sequenciamento de leitura ultra-longa. Se os comprimentos de leitura são mais curtos do que o esperado, os usuários podem ajustar os tempos de passagem no método baseado em corte mecânico ou alterar a proporção do DNA HMW para transposase no método baseado na fragmentação transposase. Tempo de ligação e eluição durante as etapas de limpeza são úteis porque o DNA HMW é altamente viscoso. Em terceiro lugar, com nanopore diferentes dispositivos de sequenciamento, um pode ajustar a quantidade e o volume do DNA para satisfazer os critérios de sequenciador. Em quarto lugar, apenas aqueles DNA ligado a adaptadores de sequenciamento vai ser sequenciado. Para melhorar ainda mais a eficiência da ligadura, um pode tentar dosear as concentrações de adaptador e ligase. Tempo de ligadura modificados e moleculares crowding agentes como PEG18 podem ser aplicados no futuro. O protocolo de sequenciamento de DNA ultra-longa combinado com CRISPR19,20 pode oferecer uma ferramenta eficaz para o sequenciamento de enriquecimento do alvo.
Divulgações
Os autores declaram que eles têm não tem interesses financeiro concorrente.
Agradecimentos
Os autores Y. Zhu agradecer seus comentários sobre o manuscrito. Pesquisa reportada nesta publicação foi parcialmente suportada pelo Instituto Nacional de câncer do institutos nacionais da saúde sob prêmio número P30CA034196. O conteúdo é exclusivamente da responsabilidade dos autores e não representa necessariamente a opinião oficial do institutos nacionais da saúde.
Materiais
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
Referências
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326(2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239(2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287(2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615(2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados