
Method Article
Sequenziamento di lettura ultra-lungo per analisi di DNA Genomic intero
In questo articolo
Riepilogo
Lungo-ha letto sequenze notevolmente facilitano l'assemblaggio di complessi genomi e caratterizzazione di variazione strutturale. Descriviamo un metodo per generare sequenze ultra-lunghi di piattaforme nanopore di sequenziamento. L'approccio adotta un'estrazione di DNA ottimizzata seguita da preparazioni di libreria modificata per generare centinaia di kilobase letture con moderata copertura da cellule umane.
Abstract
Terza generazione di tecnologie di sequenziamento del DNA di singola molecola offrono significativamente più leggere lunghezza che possa facilitare l'assemblaggio di complessi genomi e analisi delle varianti strutturali complessi. NanoPore piattaforme eseguire sequenziamento di singola molecola misurando direttamente le modifiche correnti mediate dal passaggio di DNA attraverso i pori e possono generare centinaia di letture kilobasi (kb) con il minimo costo di capitale. Questa piattaforma è stata adottata da molti ricercatori per una varietà di applicazioni. Raggiungimento di lunghezze per saperne di sequenziamento è il fattore più critico di sfruttare il valore di piattaforme di sequenziamento nanopore. Per generare letture ultra-lunghe, particolare attenzione è necessaria per evitare rotture del DNA e guadagnare in efficienza per generare modelli produttivi sequenziamento. Qui, forniamo il protocollo dettagliato di sequenziamento del DNA ultra-lungo compresa l'estrazione di DNA ad alto peso molecolare (HMW) dalle cellule fresche o surgelate, costruzione della libreria di tosatura meccanica o transposase frammentazione e la sequenziazione su un dispositivo nanopore. Da 20-25 µ g di DNA HMW, può raggiungere il metodo N50 leggere lunghezza di 50-70 kb con taglio meccanico e N50 di 90-100 kb leggere lunghezza con transposase mediata frammentazione. Il protocollo può essere applicato a DNA Estratto da cellule di mammifero per eseguire il sequenziamento dell'intero genoma per il rilevamento di varianti strutturali e assemblaggio del genoma. Ulteriori miglioramenti sull'estrazione del DNA e reazioni enzimatiche ulteriormente aumentare la lunghezza di lettura ed espandere la sua utilità.
Introduzione
Nell'ultimo decennio, massicciamente parallelo e tecnologie di seconda generazione ad alta velocità altamente accurato sequenziamento hanno guidato un'esplosione di biomedica scoperta e innovazione tecnologica1,2,3. Nonostante i progressi tecnici, i breve-lettura dati generati dalle piattaforme di seconda generazione sono inefficaci nel risolvere complesse regioni genomiche e sono limitati nella rilevazione delle varianti strutturali genomiche (SVs), che svolgono i ruoli importanti nell'uomo evoluzione e malattie4,5. Inoltre, breve-lettura dati sono in grado di risolvere la ripetizione variazione e non sono adatti per esigenti aplotipo scaglionamento delle varianti genetiche6.
Recenti progressi nel sequenziamento di singola molecola offre significativamente più lungo leggere lunghezza, che può facilitare l'individuazione dello spettro completo di SVs7,8,9e offerte accurate e complete di assemblaggio del complesso genomi microbici e mammiferi6,10. La piattaforma nanopore esegue sequenziamento di singola molecola misurando direttamente le modifiche correnti mediate da passaggio di DNA attraverso i pori11,12,13. A differenza di qualsiasi esistente chimica di sequenziamento del DNA, sequenziamento nanopore può generare lunga (decine di migliaia di kilobasi) letture in tempo reale senza fare affidamento sulla cinetica della polimerasi o artificiale amplificazione del campione del DNA. Di conseguenza, nanopore long-lettura sequenziamento (NLR-seq) detiene grande promessa per la generazione di ultra-lunghe lunghezze lettura ben oltre 100 kb, che avrebbe notevolmente progredire analisi genomiche e biomedica14, in particolare nella bassa complessità o ricchi di ripetizione regioni del genoma15.
La caratteristica unica di sequenziamento nanopore è suo potenziale per generare lunga legge senza una limitazione di lunghezza teorica. Pertanto, la lettura lunghezza dipende la lunghezza fisica del DNA che risente direttamente la qualità di modello di integrità e sequenziamento del DNA. Inoltre, a seconda dell'entità della manipolazione e il numero di passaggi, come forze di pipettaggio e condizioni di estrazione, la qualità del DNA è altamente variabile. Di conseguenza, è difficile per uno a cedere lunghe letture applicando solo i protocolli di estrazione del DNA standard e metodi di costruzione di libreria in dotazione dal produttore. A questo scopo abbiamo sviluppato un robusto metodo per generare lungo ultra leggere (centinaia di kilobasi) dati di sequenziamento a partire da palline delle cellule raccolte. Abbiamo adottato diversi miglioramenti nelle procedure di preparazione di DNA estrazione e biblioteca. Abbiamo semplificato il protocollo per escludere le procedure inutili che causano danni e degradazione del DNA. Questo protocollo è composto di alto peso molecolare (HMW) estrazione del DNA, ultra-lunga costruzione della libreria del DNA e l'ordinamento su una piattaforma nanopore. Per un biologo molecolare ben addestrato, che in genere dura 6 ore dalla raccolta al completamento dell'estrazione del DNA HMW, 90 min o 8 h per la costruzione della libreria a seconda del metodo di taglio e fino a un ulteriore 48 h per il sequenziamento del DNA delle cellule. L'uso del protocollo consentirà alla comunità di genomica di migliorare la nostra comprensione della complessità del genoma e una visione nuova variazione del genoma in malattie umane.
Protocollo
Nota: Il protocollo NLR-seq è costituito da tre passaggi consecutivi: 1) estrazione di grande molecolarità peso DNA genomico (HMW); 2) si conclude la costruzione della libreria del DNA ultra-lunga, che comprende la frammentazione del DNA HMW nella misura desiderata e legatura degli adattatori di sequenziamento del DNA; e 3) caricamento del DNA adattatore-legati sulle matrici di nanopori (Figura 1).
1. estrazione del DNA HMW
- Installazione di reagente. Fai 1 x tampone fosfato (PBS) soluzione salina tamponata (1.000 mL) aggiungendo 100 mL di PBS (10x) a 900 mL di acqua e mescolare bene. Rendere il buffer di lisi (50 mL) aggiungendo 43,5 mL di acqua per un tubo da 50 mL. Aggiungere 500 μL di Tris (1 M, pH 8.0), 1 mL di sodio cloruro (NaCl) (5 M), 2,5 mL dell'acido etilendiamminotetraacetico (EDTA) (0.5 M, pH 8.0) e 2,5 mL di sodio dodecil solfato (SDS) (10%, wt/vol) nella provetta e mescolare bene.
Nota: Questo tampone PBS può essere conservato a 4 ° C fino a 6 mesi. Il buffer di lisi premade può essere conservato a RT fino a 2 mesi. - Controllare la mortalità delle cellule e contare le celle. Assicurarsi che il rapporto dal vivo è > 85% e il numero totale delle cellule è 30 x 106.
Nota: Le celle utilizzate nel presente protocollo sono dalla linea cellulare HG00733, una varietà di cellula linfoblastoidi umane di origine portoricana, ampiamente utilizzati nel Consorzio genoma 1000 per variazione strutturale analisi (Vedi tabella materiali per informazioni sugli ordini), che appartiene risorsa di campione internazionale genoma. - Raccogliere le cellule mediante centrifugazione a 200 x g per 5 min a RT. scartare il mezzo e risospendere il pellet cellulare (30 x 106 cellule) con 5 mL di tampone PBS 1X. Centrifugare nuovamente a 200 x g per 5 min a RT e scartare il surnatante.
Nota: 25-35 x 106 delle cellule sono accettabili per questo approccio. Ulteriore variazione nella quantità di celle utilizzate sarà necessario ulteriore ottimizzazione. Il pellet cellulare possa essere conservato a − 80 ° C per fino a 6 mesi. - Risospendere il pellet cellulare in 200 μL di tampone PBS 1X. Se si utilizza un pellet cellulare congelato, lavare con 5 mL di tampone PBS 1X. Centrifugare la soluzione a 200 x g per 5 min a RT, scartare il surnatante e risospendere le cellule in 200 μL di tampone PBS 1X.
- Preparare 10 mL di tampone di lisi in un tubo da 50 mL. Aggiungere la sospensione cellulare 200 μL per il buffer di lisi e vortexare alla massima velocità per 3 s. Incubare la soluzione a 37 ° C per 1 h.
- Aggiungere 2 μL della RNasi A (100 mg/mL) MU. Ruotare delicatamente il tubo da 50 mL per mescolare il campione. Incubare la soluzione a 37 ° C per 1 h.
- Aggiungere proteinasi di 50 μL K (20 mg/mL) MU. Ruotare delicatamente il tubo da 50 mL per mescolare il campione. Incubare la soluzione a 50 ° C per 2 h. Durante l'incubazione, mescolare delicatamente il campione ogni 30 min.
- Rimuovere il tubo da 50 mL da 50 ° C e lasciate riposare a temperatura ambiente per 5 min.
- Aggiungere 10 mL di livello di fenolo del fenolo: cloroformio: alcool isoamilico (25:24:1, vol/vol/vol) MU e ruotare il tubo su un mixer di rotatore (Vedi Tabella materiali) avvolge il cappuccio del tubo con parafilm per evitare perdite di RT in una cappa a 20 rpm per 10 min. durante la rotazione.
- Preparare due provette di gel da 50 mL (Vedi Tabella materiali) mediante centrifugazione a 1.500 x g per 2 minuti a TA.
Nota: Il gel forma una barriera stabile tra la fase acquosa contenente dell'acido nucleico e il solvente organico. - Versare la soluzione di campione/fenolo in uno dei preparati 50 mL gel tubi da 1.10 passaggio. Centrifughi la soluzione a 3.000 x g per 10 minuti a TA.
- Versare il surnatante in una nuova provetta da 50 mL. Aggiungere 10 mL di livello di fenolo del fenolo: cloroformio: alcool isoamilico (25:24:1, vol/vol/vol) e ruotare il tubo su un mixer di rotatore a RT in una cappa a 20 giri per 10 min.
- Ripetere il tubo del gel passo 1.11 una volta con il secondo preparato.
- Versare il surnatante in una nuova provetta da 50 mL. Aggiungere 25 mL di etanolo al 100% ghiacciata e ruotare delicatamente il tubo a mano fino a quando non precipita il DNA (Figura 2).
Nota: L'approccio di precipitazione aiuta a stabilizzare il DNA HMW. - Piegare un suggerimento 20 μL per fare un gancio. Con attenzione estrarre il DNA HMW con il gancio e sbollire la goccia di liquido.
- Posto il DNA HMW in una provetta da 50 mL contenente 40 mL di etanolo al 70%. Lavare il DNA capovolgendo delicatamente la provetta 3 volte.
- Ripetere il passaggio 1,15 una volta a raccogliere il DNA dal tubo di etanolo al 70%.
- Posto il DNA HMW in una provetta da 2 mL contenente 1,8 mL di etanolo al 70%.
- Centrifugare il DNA HMW lavato a 10.000 x g per 3 s a RT. Remove tanto dell'etanolo residuo possibile pipettando.
Nota: Non disturbare il pellet di DNA quando l'etanolo residuo di pipettaggio. - Incubare la provetta da 2 mL a 37 ° C per 10 min con il coperchio aperto ad asciugare il campione.
- Se continuando con passaggio 2.1 (con taglio meccanico e 1 D legatura Sequencing Kit), aggiungere 1 mL di TE (10 mM Tris e 1 mM EDTA, pH 8.0) nella provetta da 2 mL.
- Se continuando con Step 2.2 (con frammentazione transposase-basato e rapida Sequencing Kit), aggiungere 200 μL di 10 mM Tris (pH 8.0) con 0,02% Triton X-100.
Nota: Non disturbare il pellet di DNA. Lasciando il tubo stand a 4 ° C nel buio per 48 h aiuterà completamente risospendere il campione. Il DNA di HMW può essere conservato a 4 ° C fino a 2 settimane. Più lungo tempo di immagazzinamento o altre condizioni di conservazione possono introdurre più brevi frammenti.
2. ultra-lunga costruzione della libreria del DNA
Nota: Esistono due modi per costruire le librerie di DNA ultra-lunghe basate su due diversi metodi di taglio accoppiati con nanopore Kit di sequenziamento. Una libreria basata su tosatura meccanica produce dati con un N50 del 50-70 kb, prendendo circa 8 h per la costruzione della libreria. Una libreria di base di frammentazione di transposase produce un N50 di 90-100 kb di dati, prendendo solo 90 min per la costruzione della libreria. Il protocollo di taglio meccanico dà maggiore resa dal DNA stesso input utilizzando versioni identiche di adattatore di sequenziamento e la qualità delle celle di flusso nanopore.
- Costruzione della libreria basata su taglio meccanico
- Disgelo e mescolare i reagenti da legatura kit (Vedi Tabella materiali). Disgelo FFPE DNA riparazione buffer e buffer di riparazione/dA-tailing fine sul ghiaccio, poi vortice e spin giù per mix. È possibile scongelare adattatore mix (AMX) e adattatore perlina associazione buffer (ABB) sul ghiaccio, poi pipetta e spin giù per mescolare. Disgelo in esecuzione buffer con mix di combustibili (RBF) e buffer di eluizione (ELB) RT, poi vortice e spin per mescolare. Scongelare biblioteca caricamento Perline (LLB) a RT e pipetta miscelare prima dell'uso.
- Una volta scongelato, mantenere tutti i componenti del kit sul ghiaccio. Estrarre gli enzimi solo quando necessario. Portare i branelli magnetici a RT per l'uso.
Nota: Per consigli sui branelli magnetici da utilizzare, vedere la tabella dei materiali.
- Una volta scongelato, mantenere tutti i componenti del kit sul ghiaccio. Estrarre gli enzimi solo quando necessario. Portare i branelli magnetici a RT per l'uso.
- Controllare la qualità e la quantità del DNA HMW dal passaggio 1.21.1. Pipetta fuori 20 μL di DNA in nuovi tubi 1,5 mL da tre posizioni diverse nel tubo del DNA HMW utilizzando P200 ampio foro suggerimenti. Prendere 1 μL dalle tre aliquote per rilevare la concentrazione utilizzando un fluorimetro e la qualità di un UV lettura. Controllare più volte per confermare i risultati.
Nota: I risultati attesi sono mostrati in Figura 3A. Il valore di260/280 OD è circa 1.9 e il valore di260/230 OD è circa 2.3. - Trasferimento il restante 940 μL di DNA HMW in un tappo tubo 50 mL con un P1000 ampio foro punta.
- Aspirare tutto il DNA in una siringa da 1 mL senza ago.
- Mettere l'ago 27 G sulla siringa ed espellere tutto il DNA nella PAC delicatamente e lentamente (~ 10 s). Togliere l'ago 27 G dalla siringa.
- Ripetere i passaggi 2.1.4 e 2.1.5 per 29 volte per un totale di 30 pass attraverso l'ago.
Nota: Il DNA di HMW tranciate può essere conservato a 4 ° C al buio per fino a 24 h. controllo di qualità (QC) è altamente raccomandato tramite l'elettroforesi del gel del pulsare-campo, ma è costoso e richiede tempo. Se esegue la QC un impulso automatico campo gel elettroforesi computer utilizzano un protocollo di 5-150 kb per un 20 h eseguire. I risultati attesi sono mostrati in Figura 4. - Preparare la reazione di riparazione del DNA in una provetta da 0,2 mL aggiungendo 100 µ l di DNA HMW tranciate (20 μg), 15 μL di tampone di riparazione del DNA FFPE, 12 μL di miscela di riparazione del DNA FFPE e 16 μL di acqua esente da nucleasi. Mescolare la reazione scorrendo delicatamente 6 volte e spin giù per rimuovere le bolle.
- Incubi la reazione a 20 ° C per 60 min. trasferimento del campione in una nuova provetta da 1,5 mL con una punta di ampio foro P200.
- Risospendere i branelli magnetici pipettando o Vortex. Aggiungere 143 perline μL (1x) per la reazione di riparazione del DNA e mescolare delicatamente, spostando il tubo 6 volte. Ruotare il tubo su un mixer di rotatore a RT a 20 giri/min per 30 min.
- Rotazione verso il basso il campione a 1.000 x g per 2 s a RT. posto il tubo su un rack magnetico per 10 min. tenere il tubo sul rack magnetico e scartare il surnatante.
- Tenendo il tubo sul rack magnetico, aggiungere 400 μL di etanolo al 70% preparata al momento senza disturbare il pellet. Rimuovere l'etanolo al 70% dopo 30 s.
- Ripetere il passaggio 2.1.11 una volta.
- Rotazione verso il basso il campione a 1.000 x g per 2 s al posto di RT. il tubo indietro sulla griglia magnetica. Rimuovere qualsiasi residuo etanolo e asciugare per 30 s. Non asciugare oltre il pellet.
- Togliere il tubo dal rack magnetico e aggiungere 103 μL di TE (10 mM Tris e 1 mM EDTA, pH 8.0). Delicatamente flick il tubo per garantire che le perline sono coperti nel buffer e incubare delicatamente su un mixer di rotatore a RT per 30 min. Scorri il tubo ogni 5 min per facilitare la risospensione del pellet.
- Pallina, le perline sul rack magnetico per almeno 10 min trasferire 100 µ l di eluato con un P200 ampio foro di punta in una provetta da 0,2 mL.
- Preparare la fine riparazione e dA-tailing reazione in un tubo di 0,2 mL aggiungendo 100 µ l di DNA HMW riparato, 14 μL di tampone di fine riparazione/dA-tailing e 7 μL di miscela di riparazione/dA-tailing fine. Mescolare la reazione scorrendo delicatamente 6 volte e spin giù per rimuovere le bolle.
- Incubare la reazione a 20 ° C per 60 min seguita da 65 ° C per 20 min e quindi tenere a 22° C. Trasferimento il campione in una nuova provetta da 1,5 mL utilizzando un P200 ampio foro suggerimento.
- Risospendere i branelli magnetici pipettando o Vortex. Aggiungere 48 μL di perline (0.4 x) per la reazione di riparazione/dA-tailing fine e mescolare delicatamente, spostando il tubo 6 volte. Ruotare il tubo su un mixer di rotatore a RT a 20 giri/min per 30 min.
- Ripetere i passaggi 2.1.10-2.1.13 una volta.
- Togliere il tubo dal rack magnetico e aggiungere 33 μL di TE (10 mM Tris e 1 mM EDTA, pH 8.0). Delicatamente flick il tubo per garantire che le perline sono coperti nel buffer e incubare delicatamente su un mixer di rotatore a RT per 30 min. Scorri il tubo ogni 5 min per facilitare la risospensione del pellet.
- Pallina, le perline sul rack magnetico per almeno 10 min trasferire 30 μL di eluato con un P200 ampio foro di punta in una nuova provetta da 1,5 mL. Prendere il μL di extra 1-2 per rilevare la concentrazione utilizzando un fluorimetro.
Nota: Il recupero di 5-6 μg in questa fase è previsto. - Preparare la reazione di legatura nella provetta del campione di 1,5 mL di aggiunta 30 μL di fine-riparato DNA HMW, 20 μL di miscela di adattatore (AMX 1D) e 50 μL di mix master di legatura di blunt/TA. Mescolare la reazione scorrendo delicatamente 6 volte tra ogni aggiunta sequenziale e spin giù per rimuovere le bolle.
- Incubare la reazione a RT per 60 min.
- Risospendere i branelli magnetici pipettando o Vortex. Aggiungere 40 perle μL (0.4 x) per la reazione di legatura e mescolare delicatamente, spostando il tubo 6 volte. Ruotare il tubo su un mixer di rotatore a RT a 20 giri/min per 30 min.
- Ripetere il punto 2.1.10 una volta.
- Aggiungere 400 μL di tampone di associazione (ABB) perlina adattatore nel tubo. Scorri il tubo dolcemente 6 volte per risospendere le perline. Riposizionare il tubo sul rack magnetico per separare le perle dal buffer ed eliminare il surnatante.
- Ripetere il passaggio 2.1.26 una volta.
- Rotazione verso il basso il campione a 1.000 x g per 2 s al posto di RT. il tubo indietro sulla griglia magnetica. Rimuovere qualsiasi residuo buffer e asciugare per 30 s. Non asciugare oltre il pellet.
- Rimuovere il tubo dal rack magnetico e risospendere il pellet in 43 μL di tampone di eluizione. Delicatamente flick il tubo per garantire che le perline sono coperti nel buffer e incubare delicatamente su un mixer di rotatore a RT per 30 min. Scorri il tubo ogni 5 min per facilitare la risospensione del pellet.
- Pallina, le perline sul rack magnetico per almeno 10 min trasferimento 40 μL di eluato con un P200 ampio foro di punta in una nuova provetta da 1,5 mL. Prendere il μL di extra 1-2 per rilevare la concentrazione utilizzando un fluorimetro.
Nota: Il recupero di 1-2 μg in questa fase è previsto. La libreria di tosatura-base meccanica è pronta per il caricamento. La libreria possa essere conservata nel ghiaccio per fino a 2 h fino a carico per la sequenza se necessario.
- Disgelo e mescolare i reagenti da legatura kit (Vedi Tabella materiali). Disgelo FFPE DNA riparazione buffer e buffer di riparazione/dA-tailing fine sul ghiaccio, poi vortice e spin giù per mix. È possibile scongelare adattatore mix (AMX) e adattatore perlina associazione buffer (ABB) sul ghiaccio, poi pipetta e spin giù per mescolare. Disgelo in esecuzione buffer con mix di combustibili (RBF) e buffer di eluizione (ELB) RT, poi vortice e spin per mescolare. Scongelare biblioteca caricamento Perline (LLB) a RT e pipetta miscelare prima dell'uso.
- Costruzione della libreria di base di frammentazione transposase
- Disgelo i reagenti dalla transposase kit (Vedi Tabella materiali). Scongelare il mix di frammentazione (FRA) e rapido adattatore (RAP) su ghiaccio e pipetta per mescolare. Scongelare i buffer di sequenziamento (SQB), perline (LB) di carico, svuotare il buffer (FLB) e sciacquare tether (FLT) a RT e pipettare per mescolare. Scongelare il caricamento beads (LB) a RT e pipetta miscelare prima dell'uso. Una volta scongelato, mantenere tutti i componenti del kit sul ghiaccio. Estrarre gli enzimi solo quando necessario.
- Controllare la qualità e la quantità del DNA HMW dal passaggio 1.21.2. Pipetta fuori 20 μL di DNA in nuovi tubi 1,5 mL da tre posizioni diverse nel tubo del DNA HMW utilizzando P200 ampio foro suggerimenti. Prendere 1 μL dalle tre aliquote per rilevare la concentrazione utilizzando un fluorimetro e la qualità di un UV lettura. Controllare più volte per confermare i risultati.
Nota: I risultati attesi sono mostrati in Figura 3B. Il valore di260/280 OD è circa 1.9 e il valore di260/230 OD è circa 2.3. - Preparare la reazione di tagmentation di DNA in un tubo di 0,2 mL aggiungendo 22 μL di DNA HMW, 1 μL di 10 mM Tris (pH 8.0) con 0,02% Triton X-100 e 1 μL di frammentazione mescolare (FRA). Mix di pipettaggio con un P200 ampio foro punta più lentamente possibile 6 volte, facendo attenzione a non introdurre bolle.
- Incubare la reazione a 30 ° C per 1 min seguita da 80 ° C per 1 min e quindi tenere a 4 ° C. Il mix di trasferimento in una nuova provetta da 1,5 mL con un P200 ampio foro punta e vai al passaggio successivo immediatamente.
- Aggiungere 1 μL di rapida adattatore (RAP) nella provetta da 1,5 mL. Mix di pipettaggio con un P200 ampio foro punta più lentamente possibile 6 volte, facendo attenzione a non introdurre bolle.
- Incubare la reazione a RT per 60 min.
Nota: La libreria di base di frammentazione di transposase è pronta per il caricamento. La libreria possa essere conservata nel ghiaccio per fino a 2 h fino a carico per la sequenza se necessario.
3. sequenziamento sul dispositivo nanopore
- Controllare che l'apparecchio di sequenziamento nanopore (Vedi Tabella materiali). Assicurarsi che sia l'hardware e il software stanno lavorando e c'è abbastanza spazio di archiviazione.
- Controllare la cella di flusso. Aprire una nuova cella di flusso e inserire la cella di flusso nel dispositivo nanopore. Controllare che la casella della posizione della cella di flusso è stata inserita (X1-X5). Selezionare il tipo di cella di flusso corretto. Fare clic su Verifica celle di flusso di flusso di lavoro. Fare clic sul pulsante Avvia Test per avviare la cella di flusso, analisi QC.
Nota: Se il numero segnalato poro attivo totale è inferiore a 800, è possibile utilizzare una nuova cella di flusso differenti per la sequenza. - Preparare il tampone di adescamento. Per una libreria basata su taglio meccanica, aggiungere 576 μL di tampone con mix di combustibili (RBF) e 624 μL di acqua esente da nucleasi in una provetta da 1,5 mL. Vortex e centrifugare fino a mix il buffer di adescamento. Per una libreria basata su frammentazione transposase, aggiungere 30 μL di filo tether (FLT) al tubo di svuotamento buffer (FLB). Vortex e centrifugare fino a mix il buffer di adescamento.
- Nella cella di flusso, spostare il coperchio della porta di adescamento in senso orario per esporre la porta di adescamento.
- Impostare una pipetta P1000 a 100 μL e inserire la punta nella porta di adescamento. Tirarsi indietro una piccola quantità di tampone (meno di 30 μL) per rimuovere eventuali bolle da cella di flusso. Smettere di pipettaggio una volta che una piccola quantità di liquido giallo entra la punta.
- Utilizzare una pipetta P1000 per caricare 800 μL del mix adescamento in cella di flusso tramite la porta di adescamento. Per evitare di introdurre bolle, aggiungere 30 μL del mix per coprire la parte superiore della porta adescamento in primo luogo, quindi inserire la punta alla porta di adescamento e aggiungere lentamente il resto del mix adescamento adescamento. Prendere la punta quando c'è circa 50 μL di sinistra. Aggiungere il resto del mix di adescamento sulla parte superiore del porto di adescamento. Il liquido andrà all'interno di sé.
- Lasciare il programma di installazione per incubare per 5 min. Nel frattempo, preparare la miscela di biblioteca nel tubo da 1,5 mL contenente la libreria.
Nota: Per una libreria basata su tosatura meccanica aggiungere 35 µ l di tampone di corsa con mix di combustibili (RBF) a 40 µ l della libreria del DNA. Per una libreria di base di frammentazione di transposase aggiungere 34 µ l di tampone di sequenziamento (SQB) e 16 µ l di acqua priva di nucleasi a 25 µ l della libreria del DNA. - Aprire il flusso delle cellule campione porta coperchio delicatamente per esporre nel pozzetto del campione. Utilizzare una pipetta P1000 per aggiungere 200 μL del mix di adescamento attraverso la porta di adescamento nella cella di flusso, come descritto al punto 3.5. Assicurarsi che il mix di adescamento non è caricato nella cella di flusso attraverso la porta campione.
- Impostare una pipetta P200 80 μL. Mescolare delicatamente la libreria con un ampio foro punta pipettando su e giù per 6 volte solo prima del caricamento.
- Caricare il mix di libreria goccia a goccia attraverso l'orificio di campione nella cella di flusso. Aggiungere ogni goccia solo dopo che il precedente calo è completamente caricato nella porta.
- Rimettere il coperchio porta campione delicatamente e assicurarsi che la porta campione è completamente coperto. Spostare il coperchio della porta di adescamento in senso antiorario per coprire la porta di adescamento. Chiudere il coperchio del dispositivo.
- Fare clic su Nuovo esperimento flusso di lavoro. Digitare il nome della libreria, selezionare il kit corretto secondo le procedure utilizzate e verifica che le impostazioni siano corrette (48 h esegue, in tempo reale base-calling ON).
- Fare clic su Start Esegui. Dopo 10 min, registrare l'ID di cella di flusso e i numeri di nanopore attivo (numero totale e numeri ogni quattro gruppi) dalle informazioni di esecuzione.
- Analisi dei dati. Copiare i dati in un computer locale o in un cluster in qualsiasi momento il sequenziamento o quando viene completata l'esecuzione. Utilizzare Minimap216 (https://github.com/lh3/minimap2) per allineare i dati di sequenza del genoma di riferimento. Riassumere le prestazioni di sequenziamento dai dati grezzi sequenza e gli allineamenti di NanoPlot17 (https://github.com/wdecoster/NanoPlot).
Risultati
Il protocollo di sequenziamento DNA ultra-lungo si applica HMW DNA per la costruzione della libreria. Pertanto, è fondamentale scegliere celle ben coltivate con il rapporto dal vivo > 85% dalla cella in fase di raccolta. La quantità di celle utilizzate per l'estrazione di DNA influenzerà la qualità e la quantità del DNA HMW. La lisi delle cellule non funziona bene se a partire con troppe cellule. Usando troppo poche cellule non genera abbastanza DNA per la costruzione della libreria perché la precipitazione del DNA HMW viene eseguita utilizzando delicata rotazione a mano invece di centrifugazione ad alta velocità. Un esempio del DNA HMW dopo l'aggiunta di etanolo 100% ghiacciata e rotazione è indicato come il precipitato bianco di cotone-come nella Figura 2.
È importante controllare la qualità dell'input del DNA prima di iniziare la costruzione della libreria. Degradazione, non corretta quantificazione, contaminazione (ad es., proteine, RNA, detergenti, tensioattivo e residua fenolo o etanolo) e basso peso molecolare di DNA possono avere un effetto significativo sulle procedure successive e sul finale leggere lunghezza. Si consiglia di eseguire l'analisi QC usando il DNA da tre diverse posizioni nella provetta contenente il DNA HMW. Da UV lettura dei risultati del DNA HMW, il valore di280 OD260/OD è circa 1.9 e il valore di230 di OD260/OD è circa 2.3 (Figura 3AB). Questi valori di rapporto sono coerenti tra le tre prove per un buon campione di DNA HMW. Diversi metodi di taglio richiede diversi volumi di ingresso del DNA. La concentrazione di DNA HMW deve essere > 200 ng / µ l per la tosatura meccanica, mentre deve essere > 1 µ g / µ l per transposase frammentazione. La concentrazione rilevata da un fluorimetro è di poco inferiore a UV lettura. Tuttavia, il coefficiente di variazione della concentrazione dello stesso campione di DNA HMW deve essere inferiore al 15% con il fluorimetro e UV lettura di saggi. Cesoia meccanica, si applica una siringa con un ago per rompere il DNA HMW, in modo che il numero di passaggi attraverso l'ago avrà un impatto la dimensione del DNA tranciata e finale leggere lunghezza. È consigliabile eseguire la dimensione QC dopo ago tosatura per garantire la maggior parte del DNA HMW è maggiore di 50 kb come illustrato nella Figura 4. Nel metodo di taglio meccanico, 30 pass generato i migliori risultati di sequenziamento, considerando la lunghezza e l'uscita.
La N50 di una libreria basata su taglio meccanica è 50-70 kb mentre è una libreria di base di frammentazione di transposase 90-100 kb. I risultati di quattro esecuzioni utilizzando la linea cellulare HG00733 sono riportati nella tabella 1. Tutti i quattro funzionamenti hanno oltre 2.300 letture con lunghezza più di 100 kb. La lunghezza massima è più lunga nelle transposase basati su frammentazione librerie (455 kb e 489 kb) confrontate con le librerie basate su taglio meccaniche (348 kb e 387 kb) mentre il secondo prodotto più totale letture, che indica un più alto rendimento. La costruzione della libreria base di frammentazione transposase ha un minor numero di passaggi e tempi di preparazione più brevi in modo che introdurrà meno brevi frammenti. Le due esecuzioni utilizzando transposase hanno una lunghezza media più lunga (> 30 kb) e la lunghezza mediana (> 10 kb). Inoltre, i dati mostrano alta qualità costante in tutte le esecuzioni (Punteggio di qualità media è circa 10.0, ~ 90% di precisione base). Oltre il 97% delle basi totali sono stato allineato al genoma umano riferimento (hg19) utilizzando Minimap216 con le impostazioni predefinite. Le distribuzioni di dimensione prevista del crudi si legge sono illustrate nella Figura 5. Tutte le esecuzioni hanno una grande percentuale di dati superiore a 50 kb mentre librerie basate su frammentazione transposase hanno un più alto rapporto di ultra-lunghe letture (ad es. > 100 kb). Questo protocollo è stato applicato con successo nelle linee cellulari umane multiple (complementare tabella 1).

Figura 1: Panoramica schematica del flusso di lavoro lungo-lettura sequenziamento (NLR-seq) nanopore. Orange, la transposase complesso. Giallo-verde, l'adattatore nanopore. Clicca qui per visualizzare una versione più grande di questa figura.
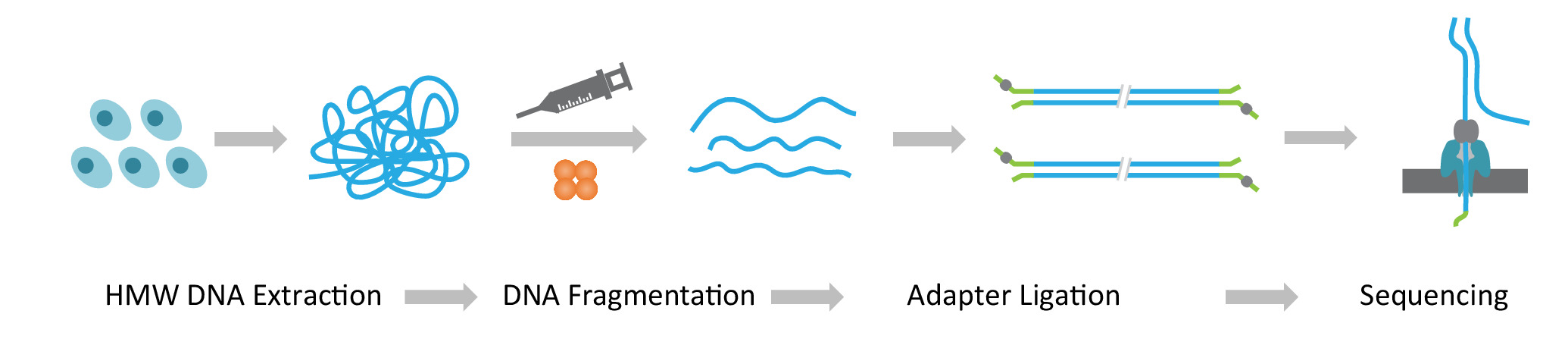{kind=link}

Figura 2: precipitazione del DNA rappresentativi dal metodo di estrazione del fenolo-cloroformio. La freccia bianca indica il DNA HMW. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: risultati di esempio QC del DNA HMW da rilevazione UV. (A) DNA HMW dal passaggio 1.21.1 pronto per la costruzione della libreria di tosatura-base meccanica. (B) DNA HMW 1.21.2 nel passaggio per la costruzione della libreria di base di frammentazione transposase. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: risultati QC dell'ago Tosato HMW DNA tramite l'elettroforesi del gel del pulsare-campo. L1: Quick-carico 1 kb DNA ladder; L2: Quick-carico estendere 1 kb DNA ladder. 1-8: DNA con passaggio diverse volte attraverso la tosatura dell'ago. 1-3, nessun taglio; 4, 10 volte; 5, 20 volte; 6, 30 volte; 7, 40 volte; 8, 50 volte. Questo passaggio QC è facoltativo. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: previsto distribuzioni di dimensione delle librerie del DNA ultra-lunghe nanopore. MS, librerie basate su taglio meccaniche. TF, librerie basate su frammentazione transposase. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Shearing_rep1 meccanica | Shearing_rep2 meccanica | Transposase fragmentation_rep1 | Transposase fragmentation_rep2 | |
| Linea cellulare | HG00733 | HG00733 | HG00733 | HG00733 |
| N50 del legge | 55.180 | 63.007 | 98.237 | 95.629 |
| Numero di letture più di 100 Kb | 2.500 | 3.082 | 2.386 | 2.355 |
| Numero di letture totale | 97.859 | 80.465 | 24.166 | 21.032 |
| Lunghezza massima (bp) | 348.482 | 387.113 | 454.660 | 489.426 |
| Lunghezza media (bp) | 17.861 | 20.395 | 33.528 | 38.175 |
| Lunghezza mediana (bp) | 5.335 | 5.894 | 10.249 | 15.656 |
| Sinonimo di qualità della legge | 10.0 | 10.1 | 9.9 | 10.0 |
| Basi totali di crude letture | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| Basi totali di allineati letture | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| Rapporto mappata di basi totali (hg19, Minimap2) | 96,9% | 98,0% | 97,7% | 97,0% |
| Numero di pori di attivi | 1225: 480, 402, 254, 89 | 1058: 480, 356, 176, 46 | 958: 452, 328, 148, 30 | 1092: 487, 367, 195, 43 |
Tabella 1: Riepilogo dalle metriche delle prestazioni viene eseguito con diversi protocolli di tosatura.
| Libreria 1 | Libreria 2 | |
| Linea cellulare | K562 | GM19240 |
| Cella di dati per l'ordine | ATCC, cat. No. CCL-243 | Coriell Institute, cat. No. GM19240 |
| Protocollo | taglio meccanico | taglio meccanico |
| N50 del legge | 60.063 | 55.295 |
| Numero di letture totale | 193.783 | 120.807 |
| Lunghezza mediana (bp) | 1.843 | 4.688 |
| Lunghezza media (bp) | 9.825 | 17.408 |
| Lunghezza massima (bp) | 548.780 | 212.338 |
| Basi totali di crude letture | 1,903,989,686 | 2,103,015,331 |
| Basi totali di allineati letture | 1,837,350,047 | 1,997,419,761 |
| Rapporto mappata di basi totali (hg19, Minimap2) | 96,6% | 95,0% |
| Numero di pori di attivi | 1111: 482, 371, 203, 55 | 1032: 447, 333, 196, 56 |
Complementare tabella 1: Sintesi delle due esecuzioni di NLR-seq utilizzando altre linee cellulari con il protocollo di taglio meccanico.
Discussione
In linea di principio, è in grado di generare 100 kb a megabase letture in lunghezza11,12,13nanopore sequenziamento. Quattro principali fattori influirà sulle prestazioni della qualità di esecuzione e i dati di sequenziamento: 1) poro attivo numeri e l'attività dei pori; 2) motore proteina, che controlla la velocità del DNA passando attraverso il nanoporo; 3) modello DNA (lunghezza, purezza, qualità, massa); 4) sequenziamento adattatore legatura efficienza, che determina il DNA utilizzabile dall'esempio di input. I primi due fattori dipendono dalla versione di cella di flusso e il kit di sequenziamento fornito dal produttore. I secondi due fattori sono punti critici in questo protocollo (estrazione del DNA HMW, tosatura e legatura).
Questo protocollo richiede pratica e pazienza. La qualità del DNA HMW è importante per ultra-lungo di librerie di DNA6. Il protocollo inizia con le cellule raccolte con elevata redditività (> 85% di cellule vitali preferita), limitando il DNA degradato dalle cellule morte. Qualsiasi processo difficili che può introdurre danni al DNA (ad es., forte inquietante, agitazione, vortice, più pipettaggio, ripetuto congelamento e scongelamento) dovrebbe essere evitato. Nella progettazione del protocollo, omettiamo di pipettaggio in tutto il processo di estrazione del DNA. Ampio foro suggerimenti devono essere utilizzati quando il pipettaggio è necessario dopo la tosatura meccanica durante la costruzione della libreria e il sequenziamento. Come i nanopori sono sensibili le composizioni chimiche nella camera tampone12, ci dovrebbe essere come pochi residui contaminanti (ad esempio, i detergenti, tensioattivi, fenolo, etanolo, proteine RNA, ecc.) come possibile nel DNA. Considerando la lunghezza e la resa, il metodo di estrazione del fenolo Mostra i risultati migliori e più riproducibili rispetto ai molteplici metodi di estrazione differenti provati finora.
Nonostante la capacità di questo protocollo di produrre sequenze di lungo-ha letto, restano ancora diverse limitazioni. In primo luogo, questo protocollo è stato ottimizzato basato sul dispositivo di sequenziamento nanopore disponibile al momento della pubblicazione; quindi, esso è limitato alla chimica selettiva di sequenziamento nanopore e poteva essere non ottimale quando eseguita in altri tipi di dispositivi di lettura lunga sequenza. In secondo luogo, il risultato è altamente dipendente dalla qualità del DNA estratto dal materiale di partenza (tessuti o cellule). Lunghezza di lettura sarà compromessa se il DNA di partenza è già degradato o danneggiato. In terzo luogo, anche se più passaggi di QC sono incorporati nel protocollo per controllare la qualità del DNA, la resa finale e la lunghezza della legge può essere influenzate da cella di flusso e attività, che potrebbe essere variabile in questa fase iniziale della piattaforma di sequenziamento nanopore del poro sviluppo.
Il protocollo descritto qui utilizza campioni di linea cellulare umana sospensione per estrazione del DNA. Abbiamo ottimizzato i tempi di passaggio nell'ago di tosatura, il rapporto di DNA HMW al transposase ed il tempo di legatura per produrre i risultati descritti. Il protocollo può essere espansa in quattro modi. In primo luogo, gli utenti possono avviare con altre cellule di mammiferi coltivate e con diverse quantità di cellule, tessuti, campioni clinici o altri organismi. Ulteriore ottimizzazione per tempo di incubazione di Lisi, il volume di reazione e centrifugazione sarà necessari. In secondo luogo, è difficile prevedere le dimensioni di destinazione per la sequenza di lettura ultra-lungo. Se le lunghezze di lettura sono più breve del previsto, gli utenti possono regolare i tempi di passaggio nel metodo basato su taglio meccanico o modificare il rapporto del DNA HMW transposase nel metodo basato su frammentazione transposase. Tempo maggiore di associazione e di eluizione durante la procedura di pulizia sono utili perché il DNA HMW è altamente viscoso. In terzo luogo, con dispositivi di sequenziamento nanopore diversi, si può regolare la quantità e il volume del DNA per soddisfare i criteri del sequencer. In quarto luogo, solo quelli DNA legato agli adattatori di sequenziamento sarà sequenziati. Per migliorare ulteriormente l'efficienza di legatura, si può tentare di titolare le concentrazioni di adattatore e ligasi. Tempo di legatura modificate e agenti molecolari affollamento come PEG18 possono essere applicati in futuro. Il protocollo di sequenziamento DNA ultra-lungo combinato con CRISPR19,20 può offrire uno strumento efficace per il sequenziamento di arricchimento di destinazione.
Divulgazioni
Gli autori dichiarano di non avere nessun concorrenti interessi finanziari.
Riconoscimenti
Gli autori ringraziano Y. Zhu per i suoi commenti sul manoscritto. Ricerca riportata in questa pubblicazione è stata parzialmente sostenuta dal National Cancer Institute del National Institutes of Health, sotto Premio numero P30CA034196. Il contenuto è di esclusiva responsabilità degli autori e non rappresentano necessariamente il punto di vista ufficiale del National Institutes of Health.
Materiali
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
Riferimenti
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326 (2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239 (2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287 (2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615 (2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati