
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Complementación de la actividad de empalme por un complejo de galectina-3 - U1 snRNP en perlas
En este artículo
Resumen
Este artículo describe los procedimientos experimentales para (a) el agotamiento de U1 snRNP de extractos nucleares, con pérdida concomitante de la actividad de empalme; y (b) reconstitución de la actividad de empalme en el extracto agotado de U1 por partículas de galectina-3 - U1 snRNP unidas a perlas covalentemente acopladas con anticuerpos anti-galectina-3.
Resumen
Los experimentos clásicos de agotamiento-reconstitución indican que la galectina-3 es un factor de empalme requerido en los extractos nucleares. El mecanismo de incorporación de galectina-3 en la vía de empalme se aborda en este trabajo. La sedimentación de extractos nucleares de células HeLa en gradientes de glicerol del 12% al 32% produce fracciones enriquecidas en una partícula endógena ~ 10S que contiene galectina-3 y U1 snRNP. Ahora describimos un protocolo para agotar los extractos nucleares de U1 snRNP con pérdida concomitante de la actividad de empalme. La actividad de empalme en el extracto agotado de U1 puede ser reconstituida por la partícula snRNP de galectina-3 - U1 atrapada en las perlas de agarosa acoplada covalentemente con anticuerpos anti-galectina-3. Los resultados indican que el complejo ternario galectina-3 - U1 snRNP - pre-ARNm es un complejo E funcional que conduce a intermediarios y productos de la reacción de empalme y que la galectina-3 entra en la vía de empalme a través de su asociación con U1 snRNP. El esquema de uso de complejos de afinidad o inmunoseleccionados en perlas para reconstituir la actividad de empalme en extractos agotados de un factor de empalme específico puede ser generalmente aplicable a otros sistemas.
Introducción
La producción de la mayoría de los ARN mensajeros eucariotas (ARNm) implica la eliminación de intrones y la ligadura de exones en un proceso nuclear denominado empalme pre-ARNm1. Dos clases de complejos ARN-proteína (RNPs) dirigen el procesamiento del ARN pre-mensajero en ARNm maduro a través de complejos espliceosómicos. Una clase, los RNPs pre-mensajeros nacientes, se forma co-transcripcionalmente por la unión de proteínas RNP nucleares heterogéneas y otras proteínas de unión a ARN, incluyendo algunos miembros de la familia SR, produciendo complejos hnRNP2. Los RNPs nucleares pequeños ricos en uracilo de segunda clase (U snRNPs con SnRNAs U1, U2, U4, U5 y U6) se asocian con proteínas específicas de U y del núcleo3,4. Los snRNPs U interactúan de manera ordenada con regiones específicas de RNPs pre-mensajeros en una vía de remodelación dinámica a medida que los intrones se extirpan y los exones se ligan para producir mRNPs maduros5. Muchas proteínas nucleares adicionales participan en estos eventos de procesamiento6.
La galectina-1 (Gal1) y la galectina-3 (Gal3) son dos proteínas que son factores necesarios en la vía de empalme como lo demuestran los estudios de agotamiento-reconstitución7,8. La eliminación de ambas galectinas del empalme de extractos nucleares competentes (NE) suprime el ensamblaje del espliceosoma y la actividad de empalme en un paso temprano. La adición de cualquiera de las dos galectinas a un NE tan doblemente agotado restaura ambas actividades. Gal1 y Gal3 son componentes de los empaliceosomas activos, como lo demuestra la inmunoprecipitación específica de pre-ARNm, intermedios de empalme y ARNm maduro por antisuero específico para Gal1 o Gal39. Es importante destacar que Gal3 se asocia con partículas endógenas U snRNA que contienen partículas en el NE fuera de la vía de empalme, como lo demuestra la precipitación de snRNPs por anti-Gal3 antisera10. Finalmente, el silenciamiento de Gal3 en células HeLa altera los patrones de empalme de numerosos genes11.
En NE preincubado para desmontar spliceosomas preformados12, los snRNPs se encuentran en múltiples complejos sedimentando en gradientes de glicerol desde 7S hasta mayores de 60S. Aunque el fraccionamiento del gradiente de glicerol es una técnica común para el aislamiento de complejos y componentes espliceosómicos (ver referencias13,14,15 por ejemplo), hemos ampliado este método caracterizando fracciones específicas mediante inmunoprecipitaciones de anticuerpos. Un sedimento snRNP a 10S contiene solo SnRNA U1 junto con Gal3. La inmunoprecipitación de la fracción 10S con antisueros específicos para Gal3 o U1 snRNP co-precipita tanto U1 como Gal3 indicando que algunas de las monopartículas U1 snRNP están unidas a Gal310. Como U1 snRNP es el primer complejo que se une a pre-mRNP en el ensamblaje espliceosómico1,5, este paso representa un sitio de entrada potencial para Gal3 en la vía de empalme. Sobre esta base, demostramos que las monopartículas 10S Gal3-U1 snRNP unidas a perlas que contienen anti-Gal3 restauraron la actividad de empalme a un NE agotado U1 snRNP, estableciendo este complejo como un mecanismo por el cual Gal3 es reclutado en la vía espliceosomal16. Esto contrasta con los intentos de aislar los espliceosomas en etapas específicas de la reacción de empalme y catalogar los factores asociados17,18. En tales estudios, se determina la presencia de ciertos factores en algún momento, pero no el mecanismo por el cual se cargaron.
Anteriormente habíamos descrito en detalle la preparación de NE, el sustrato de empalme, el ensamblaje de la mezcla de reacción de empalme y el análisis de productos en nuestra documentación del papel de las galectinas en el empalme pre-ARNm19. Ahora describimos los procedimientos experimentales para el fraccionamiento de extractos nucleares para obtener una fracción enriquecida en el complejo Gal3 - U1 snRNP y para la inmunoselección de este último complejo para reconstituir la actividad de empalme en un extracto nuclear agotado de U1.

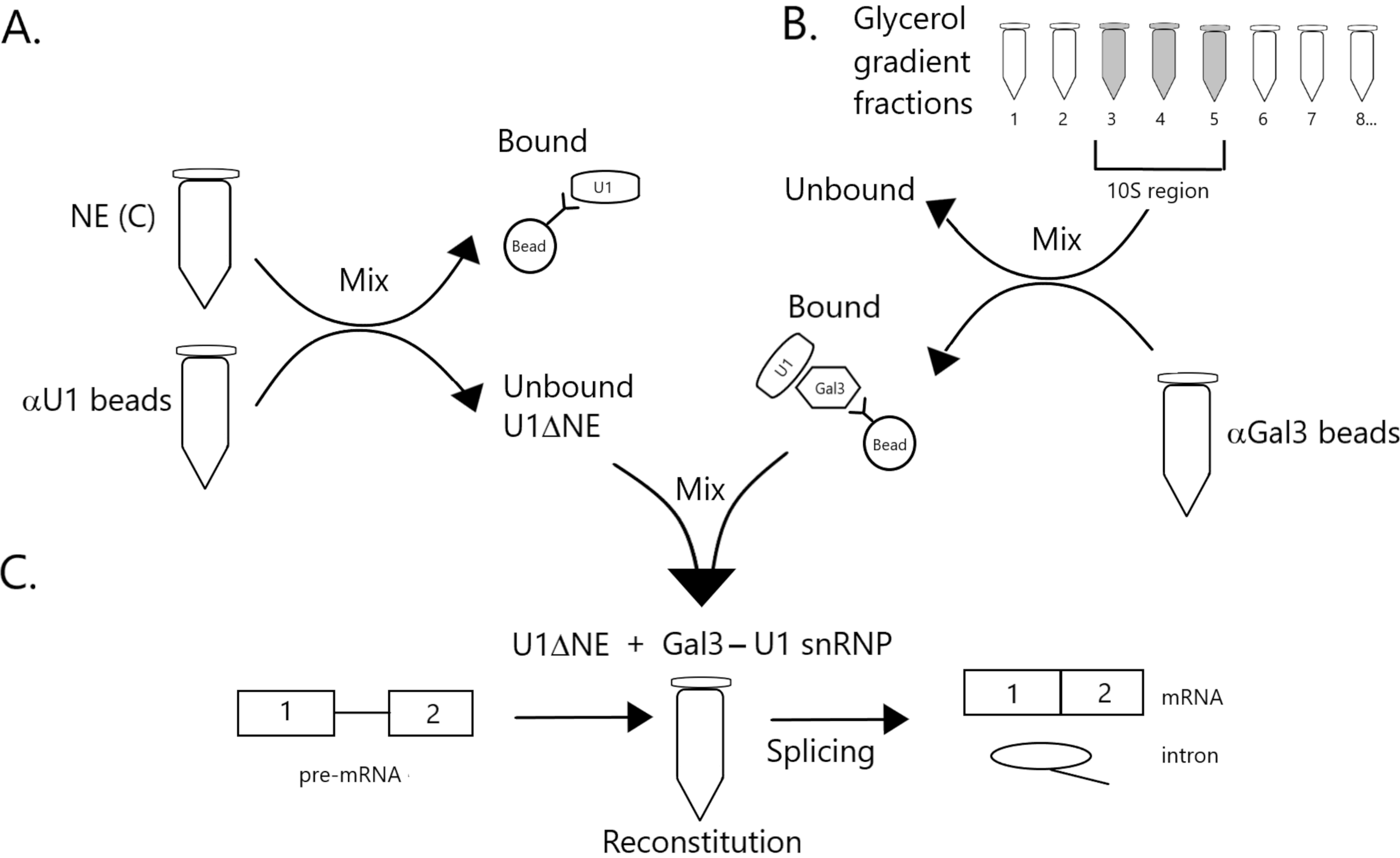
Figura 1: Diagrama esquemático que ilustra la complementación de la actividad de empalme en extracto nuclear agotado de U1 snRNP por un complejo Gal3-U1 snRNP en perlas. (A) NE en Buffer C (NE(C)) se incuba con perlas de proteína A-sefalrosa covalentemente acopladas con snRNP anti-U1 (perlas αU1). La fracción no unida se agota de U1 snRNP (U1ΔNE). (B) NE en el tampón D (NE(D)) se fracciona sobre un gradiente de glicerol de 12%-32% por ultracentrifugación. Las fracciones correspondientes a la región 10S (fracciones 3-5) se combinan y mezclan con perlas covalentemente acopladas con anticuerpos anti-Gal3 (perlas αGal3). El material unido a las perlas contiene una monopartícula Gal3-U1 snRNP. (C) El complejo snRNP Gal3-U1 de la Parte (B) se mezcla con U1ΔNE de la Parte (A) en un ensayo de empalme utilizando sustrato de pre-ARNm MINX marcado con 32P y los intermedios y productos de la reacción de empalme se analizan mediante electroforesis en gel y autorradiografía. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. Notas sobre los procedimientos generales
- Asegúrese de que todos los productos químicos (componentes tampón, enzimas, etc.) se mantengan libres de ribonucleasa (RNasa). Secuestrar todas las botellas de reactivos compradas comercialmente del uso general en el laboratorio. Use guantes para todos los pasos del procedimiento experimental. Utilice solo cristalería y utensilios que hayan sido horneados (consulte el paso 1.2 a continuación) y soluciones que hayan sido tratadas previamente (consulte el paso 1.3 a continuación).
- Hornea toda la cristalería (vasos de precipitados, frascos, botellas, pipetas, etc.) durante un mínimo de 4 h a 177 °C. Envuelva otros utensilios (espátulas, barras de agitación, etc.) en papel de aluminio antes de hornear en las mismas condiciones.
- Preparar una solución al 0,1% (vol/vol) de dietilpirocarbonato (DEPC) en agua de doble destilación (ddH2O). Usando una barra de agitación magnética, revuelva esta solución durante la noche y luego en autoclave. Utilice este H2O tratado con DEPC para hacer todas las soluciones que contengan Tris; luego, esterilice el filtro con un filtro de vacío en la parte superior de la botella. Use ddH2O regular para preparar todas las demás soluciones (sin Tris); luego, tratar con DEPC (0,1%, vol/vol) y autoclave.
NOTA: Los búferes utilizados en el siguiente conjunto de procedimientos experimentales se enumeran alfabéticamente en la Tabla 1.
| Nombre del búfer | Composición |
| Búfer de borato | 0,2 M de borato de sodio, pH 9 |
| Búfer C | 20 mM HEPES, pH 7,9, 25% (vol/vol) glicerol, 0,42 M NaCl, 1,5 mM MgCl2, 0,2 mM EDTA, 0,5 mM fenilmetilsulfonil fluoruro (PMSF), 0,5 mM de ditiotreitol (TDT) |
| Búfer D | 10 mM HEPES, pH 7,9, 20% (vol/vol) glicerol, 0,1 M KCl, 0,2 mM EDTA, 0,5 mM PMSF, 0,5 mM TDT |
| 60%D | 60% Buffer D y 40% H2O |
| Etanolamina | 0.2 M etanolamina, pH 8 |
| Búfer de enlace HEPES | 20 mM HEPES, pH 7.9 |
| Tampón de lavado HEPES | 20 mM HEPES, pH 7.9, 0.5 M NaCl |
| Búfer de carga de ARN | 90% formamida, 20 mM EDTA, pH 8, 0.05% (p/v) azul de bromofenol |
| Búfer SDS-PAGE | 25 mM Tris, 169 mM de glicina, 0,1% de dodecil sulfato de sodio (SDS), pH 8,8 |
| Búfer de muestra SDS | 62.5 mM Tris, pH 6.8, 2% SDS, 10% glicerol, 5% 2-mercaptoetanol, 0.1% (p/v) azul de bromofenol |
| Tampón TBE para geles de ARN | 89 mM Tris, 89 mM de ácido bórico, 2,5 mM de EDTA, pH 8,3 |
| Búfer TE | 10 mM Tris, pH 8, 1 mM EDTA |
| Búfer de transferencia | 25 mM Tris, 1.92 M de glicina, 20% de metanol, pH 8.3 |
| Búfer T-TBS | Tris de 10 mM, 0,5 M de NaCl, 0,05% Tween 20, pH 7,5 |
| Tampón de lavado TX | 0.05% Triton X-100 (TX) en 60%D |
Tabla 1: Nombre y composición de los búferes
2. Preparación del NE agotado de U1 snRNP (U1 ΔNE)
- Preparación de perlas anti-U1 para inmunoadsorción
- Pre-hinchar 50 mg de proteína A-sefalrosa CL-4B perlas en exceso de H2O tratado con DEPC para producir aproximadamente 200 μL de perlas hinchadas y luego lavar en el tampón de lavado HEPES.
- Para este lavado y todos los lavados posteriores, peletizar las perlas por centrifugación (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s) y retirar el lavado sin encuadernar con un micropipettor y desechar.
- Mezcle 150 μL de perlas lavadas con 150 μL de suero autoinmune humano específico para U1 snRNP (volumen de anticuerpos a volumen de perlas en una proporción de 1: 1).
- Ajustar, sobre la base del volumen total de (~300 μL desde el paso 2.1.3 anterior), la mezcla a 20 mM HEPES, pH 7.9, correspondiente a las condiciones del tampón de unión HEPES; incubar esta mezcla con balanceo continuo a temperatura ambiente durante 60 min.
- Lavar las perlas unidas con anticuerpos con 1 ml de tampón de borato (0,2 M de borato de sodio, pH 9) y resuspend en 1 ml del mismo tampón de borato.
- Para acoplar covalentemente el anticuerpo unido a las perlas de proteína A-sefalrosa, agregue dimetilpimelimida a una concentración final de 20 mM e incube a temperatura ambiente con balanceo durante 60 min.
- Lave las perlas con 1 ml de tampón de borato.
- Para bloquear cualquier reactivo reticulado no reaccionado, agregue 1 ml de etanolamina de 0,2 M (pH 8) e incube a temperatura ambiente con balanceo durante 60 min.
- Lave las perlas acopladas a anticuerpos, en lo sucesivo designadas como perlas anti-U1, dos veces con 0,5 ml de tampón de lavado TX (0,05% Tritón X-100 en 60% D).
- Agotamiento de U1 snRNP de NE (ver Figura 1A)
NOTA: El procedimiento para preparar NE a partir de células HeLa fue desarrollado inicialmente por Dignam et al.20. Hemos descrito los materiales y métodos detallados para la preparación de NE para ensayos de empalme19 (véanse los pasos 2.1 y 3.1 de esa referencia). NE, tal como se preparó inicialmente, está en el búfer C y en lo sucesivo se designará como NE(C). NE(C) dializado y equilibrado con buffer D se designará como NE(D).- Incubar 200 μL de NE(C) con 100 μL de perlas anti-U1 del paso 2.1.9 anterior.
- Añadir 5 μL de RNasin a la mezcla.
- Gire la cabeza del microtubo sobre la cola a 4 °C durante 1 h.
- Granular la mezcla por centrifugación (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s) y recoger el material no unido (U1ΔNE) utilizando una jeringa Hamilton.
- Dializar todo el volumen de U1ΔNE, junto con una alícuota separada de 50 μL del NE(C) original no agotado, en compartimentos separados de un microdializador, con agitación, durante 75 min contra 60% D utilizando una membrana de diálisis con un corte de peso molecular de 8 K.
- Inmediatamente después de la diálisis, divida estas preparaciones (U1ΔNE y NE en 60% D) en alícuotas de 20 μL; luego congele en un baño de hielo seco / etanol y guárdelo a -80 ° C.
- Análisis del contenido de ARN y proteína de U1ΔNE y material unido a perlas anti-U1
- Después de la extracción del material no unido (U1ΔNE) (paso 2.2.4), lave el material unido a las perlas anti-U1 agregando 0,5 ml de tampón de lavado TX. Peletizar la mezcla por centrifugación (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s) y retirar el sobrenadante con un micropipettor y desechar.
- Repita los pasos de lavado 2.3.1 dos veces.
- Retire el material unido a las perlas anti-U1 agregando 100 μL de tampón de muestra SDS 2x a 100 μL de las perlas e incubando durante 10 min a temperatura ambiente.
- Pellet la mezcla por centrifugación (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s); retire el sobrenadante con la jeringa Hamilton y congele en un baño de hielo seco / etanol. Conservar a -80 °C.
- Compare el NE no agotado, el NE agotado (U1ΔNE) y el material unido a las perlas (eliminado de las perlas por el tampón de muestra SDS como se describe en los pasos 2.3.3 y 2.3.4 anteriores). Siga los pasos 2.3.6-2.3.8 para el análisis de ARN o los pasos 2.3.9-2.3.10 para el análisis de proteínas.
- Para cada muestra, extraer el ARN con 200 μL de fenol-cloroformo (50:50, v/v); luego extraer de nuevo con 180 μL de cloroformo-isoamilo alcohol (25:1, v/v). Después de la extracción, agregue 300 μL de etanol frío a prueba de 200, invierta para mezclar y almacene el ARN precipitado durante la noche a -20 ° C.
- Centrifugar el ARN precipitado con etanol (12.000 x g durante 10 min a 4 °C). Lavar los pellets con 150 μL de etanol frío al 70%. Centrifugar de nuevo (12.000 x g) a 4 °C durante 15 min. Retire el sobrenadante con un micropipetador y seque los gránulos en una aspiradora de velocidad durante 10-15 minutos sin calor.
- Resuspender el pellet de ARN seco en 10 μL de tampón de carga de ARN, suavemente vórtice, calentar a 75-85 ° C durante 90 s, y luego incubar en hielo durante 2 min. Separe los snRNAs mediante electroforesis en gel (2 h a 16 mA) a través de geles de poliacrilamida al 13% - 8,3 M de urea y luego se tiñe con bromuro de etidio o se someta a northern blotting10,16.
- Cargue las muestras de proteínas, en tampón de muestra SDS desde el paso 2.3.5, en geles de poliacrilamida al 12.5% y electroforesa a 200 V durante aproximadamente 45-50 min en tampón SDS-PAGE (electroforesis en gel de poliacrilamida de dodecil sulfato de sodio).
- Transfiera las proteínas separadas a la membrana de nitrocelulosa a 400 mA durante 2 h en tampón de transferencia. Después de la transferencia, bloquee la membrana incubando durante la noche en T-TBS que contiene 10% de leche seca sin grasa. A continuación, inmunoblote la membrana para revelar proteínas específicas8,21.
3. Inmunoprecipitación de fracciones 10S de gradientes de glicerol por anti-Gal3
- Preparación de perlas anti-Gal3 para inmunoadsorción
NOTA: La derivación y caracterización de antisueros policlonales de conejo frente a Gal3 para conejo #2421 y para conejo #4910 han sido descritos anteriormente.- Use suero preinmune de conejo # 49 como control.
- Para la preparación de perlas anti-Gal3 siga el procedimiento descrito anteriormente para la preparación de perlas anti-U1 (paso 2.1), con la excepción de que correspondiente al paso 2.1.3, la proporción de antisuero (por ejemplo, anti-Gal3, # 49) a cuentas es de 3: 1.
- Justo antes de usar, lave las perlas acopladas a anticuerpos, en lo sucesivo designadas como perlas anti-Gal3, dos veces con 0,5 ml de tampón de lavado TX. Retire el sobrenadante, primero con un micropipetador para sacar la mayor parte del líquido y luego con una jeringa Hamilton para sacar el líquido de las perlas; descartar.
- Inmunoprecipitación de fracciones de gradiente de glicerol por anti-Gal3 (ver Figura 1B)
- Fraccionar NE(D) sobre un gradiente de glicerol del 12%-32%10. Combine y mezcle las fracciones de gradiente de glicerol 3, 4 y 5 (numeradas desde la parte superior del gradiente), que están cerca de la región 10S del gradiente.
- Preparar dos muestras, cada una con alícuota de 150 μL de fracciones de gradiente combinadas 3-5 (paso 3.2.1), y colocar en 50 μL de perlas anti-Gal3.
- Paralelamente, preparar dos muestras cada una con 150 μL de fracción 1 (que contenga Gal3 no en complejo con U1 snRNP10; paso 3.2.1) y colocar en 50 μL de perlas anti-Gal3.
- Como control, coloque 150 μL de 60% D en otro microtubo de cuentas anti-Gal3 de 50 μL.
- Mezcle suavemente golpeando el tubo, luego gire la cabeza sobre la cola del microtubo a 4 ° C durante 1 h.
- Pellet la mezcla por centrifugación suave (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s).
- Retire el sobrenadante (material no unido) con una jeringa Hamilton. No lave las perlas y utilizar inmediatamente para la adición de las reacciones de empalme (sección 4.2).
- Análisis del contenido de ARN y proteína en el material no unido y unido a partir de la precipitación anti-Gal3 de fracciones de gradiente 10S
- Para el análisis de los componentes del material unido y no unido a partir de la precipitación anti-Gal3 de las fracciones de gradiente 10S, recoja el material no unido (sobrenadante después del paso 3.2.6), transfiéralo a un microtubo fresco y congele a -20 °C.
- Lave las perlas precipitadas del paso 3.2.6 (que contiene material unido a anti-Gal3) agregando 0.5 ml de tampón de lavado TX.
- Pellet la mezcla por centrifugación suave (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s); retire el sobrenadante con un micropipetador y deséchelo. Repita los pasos de lavado dos veces más.
- Agregue 50 μL de tampón de muestra 2X SDS a las perlas anti-Gal3 lavadas y granuladas.
- Mezclar las perlas suavemente e incubar durante 10 min a temperatura ambiente.
- Peletizar la mezcla por centrifugación suave (1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s), recoger el sobrenadante mediante jeringa Hamilton y almacenar en un microtubo fresco a -20 °C.
- Comparar el material no unido (sección 3.3.1) y el material unido (paso 3.3.6) de la precipitación anti-Gal3 en términos de COMPONENTES de ARN y proteínas, utilizando los procedimientos descritos en los pasos 2.3.6. a 2.3.10, respectivamente.
4. Montaje de la reacción de empalme y análisis de los productos
- Preparación del sustrato de empalme
NOTA: El sustrato pre-ARNm, designado MINX, contiene dos secuencias de exones y una secuencia de intrón de Adenovirus22. La secuencia de ADN MINX en el plásmido está bajo el control de los promotores de la ARN polimerasa T3, T7 o SP6. Los materiales y métodos detallados para la linealización del ADN plásmido MINX con endonucleasa de restricción BamHI, la transcripción por ARN polimerasa SP6 en presencia de α-32P[GTP] y la purificación de MINX marcado con 32P para ensayos de empalme se describen anteriormente19 (véanse los pasos 2.2 y 3.2 de dicha referencia).- Almacenar el MINX radiomarcado como precipitado de etanol a -20 °C; use el sustrato de empalme etiquetado dentro de las 4-6 semanas posteriores a la transcripción.
- Justo antes de su uso, centrifugue el etanol precipitado con la etiqueta 32P MINX a 12.000 x g durante 10 min a 4 °C; retire el sobrenadante con un micropipetador y deseche.
- Añadir 150 μL de etanol al 70% y centrífuga a 12.000 x g durante 15 min a 4 °C. Deseche el sobrenadante y seque el pellet en speed vac sin calor durante 15 min.
- Rehidratar el pellet en 50 μL de agua DEPC. Punto 2 μL en cada uno de los dos filtros GF/C; sumergir los filtros en ácido tricloroacético (TCA) al 5% en frío durante 10 min. Enjuague con TCA al 5% en frío, seguido de etanol a prueba de 180 en un matraz al vacío. Seque al aire los filtros y se someta a un conteo de centelleo en 4 mL de Safety-Solve.
- Diluir MINX marcado con 32P en 60% D a 104 cpm/μL para el ensayo de empalme.
- Montaje de la reacción de empalme (ver Figura 1C)
- Montar, sobre hielo, las reacciones de empalme en un volumen total de 24 μL (8 μL U1ΔNE (a partir del paso 2.2.6), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de fosfato de creatina, 0,5 mM TDT, 20 unidades de RNasina, sustrato de empalme MINX marcado con 4 μL 32P (104 cpm/μL), 60% D) y añadir a cada tubo de perlas de la sección 3.2.7. Ensamble un conjunto idéntico de reacciones de empalme en un volumen total de 24 μL pero sin U1ΔNE y agréguelo a cada tubo de perlas a partir del paso 3.2.7.
- Preparar una reacción de empalme de control en un volumen total de 12 μL (4 μL NE(D), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de fosfato de creatina, 0,5 mM de TDT, 20 unidades de RNasin, sustrato de empalme MINX marcado con 2 μL 32P (104 cpm/μL), 60% D).
- Mezcle los tubos suavemente golpeando y gire el extremo sobre la cola a 30 ° C durante 90 minutos. Pellet la mezcla por centrifugación suave a 1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s.
- Detenga la reacción y eluya las proteínas de las perlas agregando 24 μL de tampón de muestra SDS 2x a los tubos que contienen perlas, y 12 μL de tampón de muestra SDS 2x al tubo de control que contiene NE pero no perlas. Calentar los tubos a 100 °C durante 7 min.
- Centrifuga los tubos suavemente a 1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10-15 s.
- Transfiera los sobrenadantes (eluciones) a microtubos frescos: aproximadamente 48 μL de los tubos de perlas y 24 μL del tubo de control NE.
- Añadir proteinasa K (20 mg/mL) para digerir y solubilizar las proteínas: añadir 5 μL a la elución de 48 μL de las perlas y añadir 2,5 μL al control ne de 24 μL.
- Incubar los tubos a 37°C durante 40 min.
- Centrifuga suavemente los tubos a 1.000 x g en un rotor de cucharón oscilante a 4 °C durante 10 s.
- Diluir las eluciones de perlas con 39,5 μL de TE y 10 μL de 3 M de acetato de sodio. Diluir el control NE con 63,5 μL TE y 10 μL 3 M de acetato de sodio.
- Extraer y analizar el ARN como se describe a continuación (sección 4.3).
- Análisis de productos de la reacción de empalme
- Extraer los ARN de cada muestra con fenol-cloroformo, seguido de cloroformo-isoamílico alcohol; precipitar los ARN con etanol, centrifugar, lavar los gránulos, retirar el sobrenadante y secar los gránulos siguiendo el mismo procedimiento descrito en los pasos 2.3.6 y 2.3.7.
- Resuspender el pellet de ARN seco en 10 μL de tampón de carga de ARN, suavemente vórtice, calentar a 75-85 ° C durante 90 s, y luego incubar en hielo durante 2 min.
- Preparar 20 ml de una solución que contenga poliacrilamida al 13% (bisacrilamida:acrilamida, 1,9:50 [wt/wt]) en 8,3 M de urea; geles de fundición de 15 cm de longitud utilizando esta solución.
- Una vez que el gel está fundido, electroforesarlo (sin ninguna muestra cargada) a 400 V durante 20 minutos utilizando TBE como amortiguador de funcionamiento. Después de este paso, lave los pozos con el búfer de ejecución TBE.
- Cargue las muestras de ARN, en tampón de carga de ARN, y electroforesa con TBE ejecutando tampón a 400 V durante 3,5 a 4 h. Después de la electroforesis, retire la urea sumergiendo y girando el gel en agua destilada durante 10 min.
- Seque al vacío el gel en papel de filtro de 3 M, primero durante 2 h 15 min a 80 °C y luego durante 30 min sin calor para enfriarlo lentamente. Someter el gel seco a autoradiografía sobre película para detectar las posiciones de migración de los componentes radiactivos.
Access restricted. Please log in or start a trial to view this content.
Resultados
Los complejos NE agotados de U1 snRNP (U1ΔNE de la Sección 2.2.6) y Gal3 - U1 snRNP de la región 10S del gradiente de glicerol inmunoprecipitado por anti-Gal3 (paso 3.2.7) se mezclaron en una reacción de empalme. Esta mezcla de reacción contenía SnRNA U1 (Figura 2A, carril 3), así como la proteína específica U1, U1-70K (Figura 2B, carril 3). Como era de esperar, el anti-Gal3 precipitó Gal3 (Figura ...
Access restricted. Please log in or start a trial to view this content.
Discusión
Este informe proporciona los detalles experimentales que documentan que un complejo snRNP Gal3 - U1 atrapado en perlas recubiertas anti-Gal3 puede unirse al sustrato pre-ARNm y este complejo ternario puede restaurar la actividad de empalme a un NE agotado de U1 snRNP. Gal3 es un miembro de una familia de proteínas originalmente aisladas sobre la base de su actividad de unión a carbohidratos específicos de galactosa23 . Los estudios tempranos de inmunofluorescencia y fraccionamiento subcelular p...
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo ha sido apoyado por national Science Foundation Grant MCB-0092919 y Michigan State University Intramural Research Grant 09-CDFP-2001 (a RJP) y por National Institutes of Health Grant GM-38740 y Michigan AgBioResearch Project MICL02455 (a JLW).
El sustrato minx de pre-ARNm utilizado en los ensayos de empalme fue un amable regalo de la Dra. Susan Berget (Baylor College of Medicine, Houston, TX, EE. UU.).
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
Referencias
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932(1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados