
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Complementação da Atividade de Emenda por um Complexo Galectin-3 - U1 snRNP on Beads
Neste Artigo
Resumo
Este artigo descreve os procedimentos experimentais para (a) esgotamento do U1 snRNP a partir de extratos nucleares, com perda concomitante de atividade de emenda; e (b) reconstituição da atividade de emenda no extrato empobrecido U1 por partículas de galectina-3 - U1 snRNP ligadas a contas covalentemente acopladas a anticorpos anti-galectina-3.
Resumo
Experimentos clássicos de esgotamento-reconstituição indicam que a galectina-3 é um fator de emenda necessário em extratos nucleares. O mecanismo de incorporação da galectina-3 na via de emenda é abordado neste artigo. A sedimentação de extratos nucleares de células HeLa em 12%-32% gradientes de glicerol produz frações enriquecidas em uma partícula endógena ~10S que contém galectina-3 e U1 snRNP. Descrevemos agora um protocolo para esgotar extratos nucleares do U1 snRNP com perda concomitante de atividade de emenda. A atividade de emenda no extrato empobrecido U1 pode ser reconstituída pela partícula de galectin-3 - U1 snRNP presa em contas de agarose covalentemente acopladas com anticorpos anti-galectina-3. Os resultados indicam que o complexo ternário pré-mRNA é um complexo E funcional que leva a intermediários e produtos da reação de emenda e que a galectina-3 entra no caminho de emenda através de sua associação com o U1 snRNP. O esquema de utilização de complexos de afinidade ou imuno-selecionados em contas para reconstituir a atividade de emenda em extratos esgotados de um fator de emenda específico pode ser geralmente aplicável a outros sistemas.
Introdução
A produção da maioria dos RNAs (mRNAs) do mensageiro eucariótico envolve a remoção de introns e a ligadura de exons em um processo nuclear chamado de emenda pré-mRNA1. Duas classes de complexos de proteína RNA (RNPs) direcionam o processamento do RNA pré-mensageiro em mRNA maduro através de complexos spliceossômicos. Uma classe, rnps pré-mensageiro nascente, é formada co-transcrição pela vinculação de proteínas heterogêneas nucleares RNP e outras proteínas de ligação de RNA, incluindo alguns membros da família SR, produzindo complexos hnRNP2. A segunda classe, rica em uracil, pequenas RNPs nucleares (U snRNPs com U1, U2, U4, U5 e U6 snRNAs) está associada com proteínas específicas e core u3,4. Os SNRNPs da U interagem de forma ordenada com regiões específicas de RNPs pré-mensageiros em um caminho de remodelação dinâmica à medida que os introns são extirpados e exons são ligados para produzir mRNPs5 maduros. Muitas proteínas nucleares adicionais participam desses eventos de processamento6.
Galectin-1 (Gal1) e galectin-3 (Gal3) são duas proteínas que são fatores necessários na via de emenda, como mostrado pelos estudos de esgotamento-reconstituição7,8. A remoção de ambas as galectinas de emendas extratos nucleares competentes (NE) abolia a montagem de emendas e a atividade de emenda em um passo inicial. A adição de uma galectina a um NE duplamente esgotado restaura ambas as atividades. Gal1 e Gal3 são componentes de emendas ativas, evidenciadas por imunoprecipitação específica de pré-mRNA, intermediários de emenda e mRNA maduro por antiserum específico para Gal1 ou Gal39. É importante ressaltar que a Gal3 associa-se ao snRNA u endógeno contendo partículas no NE fora da via de emenda, como mostrado pela precipitação de snRNPs por anti-Gal3 antisera10. Finalmente, o silenciamento de Gal3 em células HeLa altera padrões de emenda de numerosos genes11.
No NE pré-incubado para desmontar os spliceosomes pré-formados12, snRNPs são encontrados em múltiplos complexos sedimentando em gradientes de glicerol de 7S para maiores de 60S. Embora o fracionamento do gradiente de glicerol seja uma técnica comum para o isolamento de complexos e componentes spliceossômicos (ver referências13,14,15 por exemplo), ampliamos esse método caracterizando frações específicas usando imunoprecipitações de anticorpos. Um sedimento snRNP em 10S contém apenas u1 snRNA juntamente com Gal3. A imunoprecipitação da fração 10S com antisera específica para Gal3 ou U1 snRNP co-precipita tanto u1 quanto Gal3 indicando que algumas das mon partículas do U1 snRNP estão ligadas a Gal310. Como o U1 snRNP é o primeiro complexo que se liga ao pré-mRNP em conjuntos emendasomais1,5, esta etapa representa um potencial local de entrada para Gal3 na via de emenda. Com base nisso, mostramos que as monpartículas 10S Gal3-U1 snRNP ligadas à anti-Gal3 contendo contas restauradas para um NE esgotado U1 snRNP, estabelecendo este complexo como um mecanismo pelo qual Gal3 é recrutado para o caminho emendalomal16. Isso contrasta com as tentativas de isolar os emendas em estágios específicos na reação de emenda e catalogação dos fatores associados17,18. Em tais estudos, a presença de certos fatores em algum momento é apurada, mas não o mecanismo pelo qual foram carregados.
Havia descrito anteriormente em detalhes a preparação do NE, o substrato de emenda, a montagem da mistura de reação de emenda, e a análise de produtos em nossa documentação do papel das galectinas no splicing pré-mRNA19. Descrevemos agora os procedimentos experimentais para o fracionamento de extratos nucleares para obter uma fração enriquecida no complexo Gal3 - U1 snRNP e para a imuno-seleção deste último complexo para reconstituir a atividade de emenda em um extrato nuclear esgotado pelo U1.

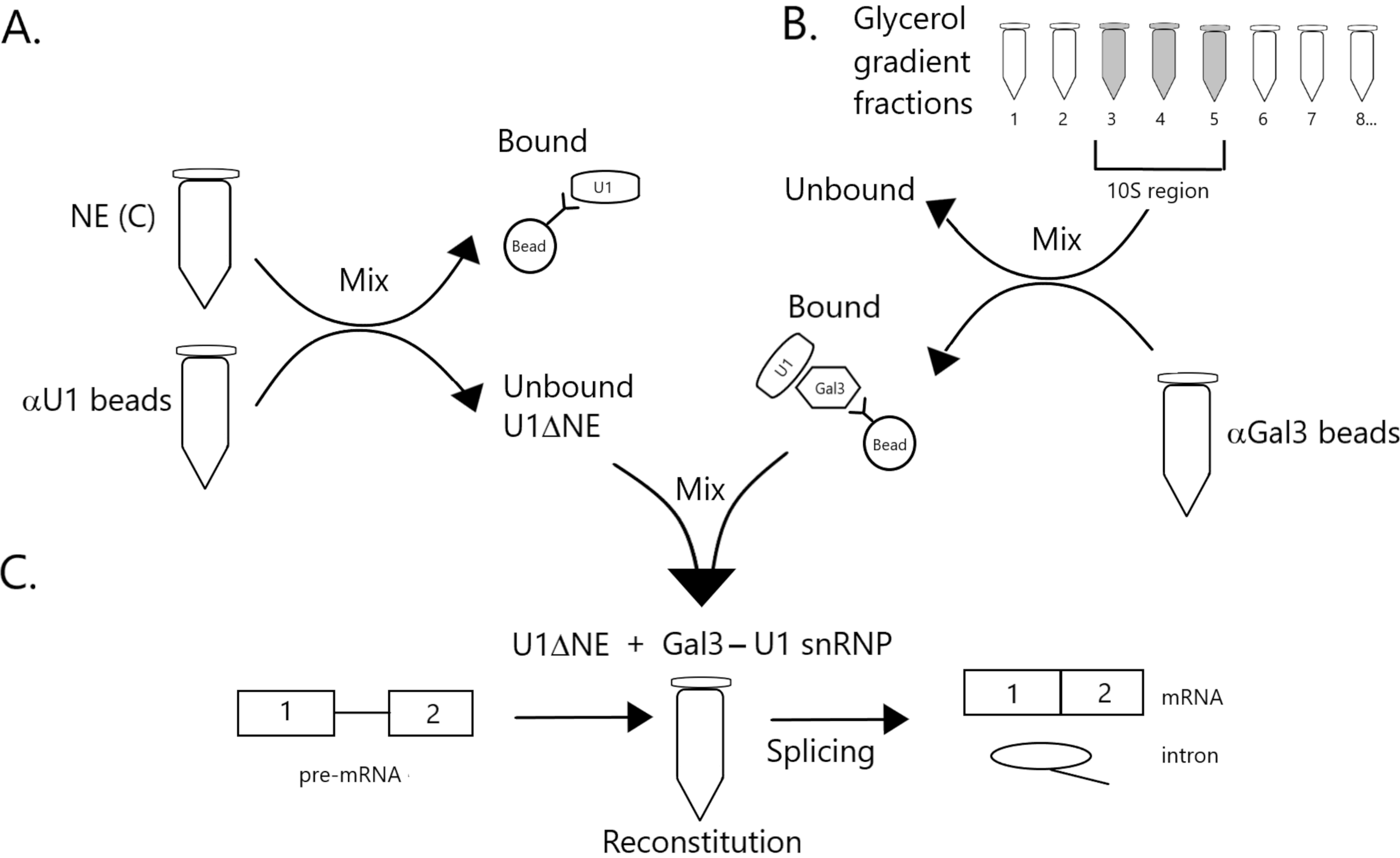
Figura 1: Diagrama esquemático ilustrando a complementação da atividade de emenda em extrato nuclear esgotado de U1 snRNP por um complexo de snRNP Gal3-U1 em contas. (A) NE em Buffer C (NE(C)) é incubado com contas de proteína A-Sepharose covalentemente acopladas com anti-U1 snRNP (contas αU1). A fração desvinculada está esgotada de U1 snRNP (U1ΔNE). (B) NE em Buffer D (NE(D)) é fracionado sobre um gradiente de glicerol de 12%-32% por ultracentrifugação. Frações correspondentes à região 10S (frações 3-5) são combinadas e misturadas com contas covalentemente acopladas com anticorpos anti-Gal3 (contas αGal3). O material ligado às contas contém uma monopartícula de SnRNP Gal3-U1. (C) O complexo de snRNP Gal3-U1 da Parte (B) é misturado com U1ΔNE da Parte (A) em um ensaio de emenda usando substrato minx pré-mRNA com 32P e os intermediários e produtos da reação de emenda são analisados por eletroforese gel e autoradiografia. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Protocolo
1. Notas sobre procedimentos gerais
- Certifique-se de que todos os produtos químicos (componentes tampão, enzimas, etc.) sejam mantidos livres de ribonuclease (RNase). Sequester todas as garrafas de reagente compradas comercialmente do uso geral do laboratório. Use luvas para todas as etapas do procedimento experimental. Utilize apenas vidros e utensílios assados (veja o passo 1.2 abaixo) e soluções que foram pré-tratadas (veja o passo 1.3 abaixo).
- Asse todos os vidros (béquers, frascos, garrafas, pipetas, etc.) por um mínimo de 4h a 177 °C. Enrole outros utensílios (espátulas, barras de mexida, etc.) em papel alumínio antes de assar sob as mesmas condições.
- Prepare uma solução de 0,1% (vol/vol) de dietilpirato (DEPC) em água duplamente destilada (ddH2O). Usando uma barra de agitação magnética, mexa esta solução durante a noite e, em seguida, autoclave. Use este H2O tratado com DEPC para fazer todas as soluções que contenham Tris; em seguida, esterilize o filtro usando um filtro de vácuo de tampa de garrafa. Use ddH2O regular para preparar todas as outras soluções (sem Tris); em seguida, trate com DEPC (0,1%, vol/vol) e autoclave.
NOTA: Os buffers utilizados no seguinte conjunto de procedimentos experimentais estão listados alfabeticamente na Tabela 1.
| Nome do buffer | Composição |
| Tampão de borate | 0,2 M borato de sódio, pH 9 |
| Tampão C | 20 mM HEPES, pH 7.9, 25% (vol/vol) glicerol, 0,42 M NaCl, 1,5 mM MgCl2, 0,2 mM EDTA, 0,5 mM fenilmetiletilasulfonil fluoreto (PMSF), 0,5 mM dithiothreitol (DTT) |
| Buffer D | 10 mM HEPES, pH 7.9, 20% (vol/vol) glicerol, 0,1 M KCl, 0,2 mM EDTA, 0,5 mM PMSF, 0,5 mM DTT |
| 60%D | 60% Buffer D e 40% H2O |
| Etanolamina | 0,2 M etanolamina, pH 8 |
| Tampão de ligação HEPES | 20 mM HEPES, pH 7.9 |
| Tampão de lavagem HEPES | 20 mM HEPES, pH 7.9, 0,5 M NaCl |
| Tampão de carregamento de RNA | 90% de formamida, 20 mM EDTA, pH 8, 0,05% (w/v) azul bromofenol |
| Buffer SDS-PAGE | 25 mM Tris, 169 mM de gliccina, 0,1% sulfato de dodecyl de sódio (SDS), pH 8,8 |
| Tampão de amostra SDS | 62,5 mM Tris, pH 6.8, 2% SDS, 10% glicerol, 5% 2-mercaptoetanol, 0,1% (w/v) azul bromofenol |
| Tampão TBE para géis de RNA | 89 mM Tris, ácido bórico de 89 mM, 2,5 mM EDTA, pH 8.3 |
| Tampão de TE | 10 mM Tris, pH 8, 1 mM EDTA |
| Buffer de transferência | 25 mM Tris, 1,92 M de gliccina, 20% metanol, pH 8.3 |
| Tampão T-TBS | 10 mM Tris, 0,5 M NaCl, 0,05% Tween 20, pH 7.5 |
| Tampão de lavagem TX | 0,05% Triton X-100 (TX) em 60%D |
Tabela 1: Nome e Composição de Buffers
2. Preparação do NE esgotado de U1 snRNP (U1 ΔNE)
- Preparação de contas anti-U1 para imunoadsorção
- Pré-inchar 50 mg Proteína A-Sepharose CL-4B contas em excesso DEPC-tratado H2O para produzir aproximadamente 200 μL de contas inchadas e, em seguida, lavar em tampão de lavagem HEPES.
- Para esta lavagem e todas as lavagens subsequentes, pelota as contas por centrifugação (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s) e remova a lavagem sem saída usando um micropipettor e descarte.
- Misture 150 μL de contas lavadas com 150 μL de soro autoimune humano específico para U1 snRNP (volume de anticorpos ao volume de contas em uma razão de 1:1).
- Ajuste, com base no volume total de (~300 μL da etapa 2.1.3 acima), a mistura para 20 mM HEPES, pH 7.9, correspondente às condições do tampão de ligação HEPES; incubar esta mistura com balanço contínuo à temperatura ambiente por 60 minutos.
- Lave as contas amarradas com anticorpos com 1 mL de tampão de bólato (0,2 M de borato de sódio, pH 9) e resuspend em 1 mL do mesmo tampão de borate.
- Para acoplar o anticorpo ligado às contas de proteína A-Sepharose, adicione dimetilpimelimidato a uma concentração final de 20 mM e incubar à temperatura ambiente com balanço por 60 minutos.
- Lave as contas com 1 mL de tampão de borate.
- Para bloquear qualquer reagente transversal não redigido, adicione 1 mL de etanolamina de 0,2 M (pH 8) e incubar em temperatura ambiente com balanço por 60 min.
- Lave as contas acopladas a anticorpos, a partir de agora designadas como contas anti-U1, duas vezes com 0,5 mL de tampão de lavagem TX (0,05% Triton X-100 em 60% D).
- Esgotamento do U1 snRNP do NE (ver Figura 1A)
NOTA: O procedimento de preparação do NE a partir de células HeLa foi inicialmente desenvolvido por Dignam et al.20. Descrevemos os materiais e métodos detalhados para a elaboração do NE para os ensaios de emenda19 (ver as etapas 2.1 e 3.1 dessa referência). NE, como inicialmente preparado está em Buffer C e será designado para o futuro como NE(C). NE(C) dialisado contra e equilibrado com Buffer D será designado como NE(D).- Incubar 200 μL de NE(C) com 100 μL de contas anti-U1 da etapa 2.1.9 acima.
- Adicione 5 μL de RNasin à mistura.
- Gire a cabeça sobre a cauda do microtube a 4 °C por 1 h.
- Pellule a mistura por centrifugação (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s) e colete o material sem entrada (U1ΔNE) usando uma seringa Hamilton.
- Dique todo o volume de U1ΔNE, juntamente com uma alíquota separada de 50 μL do NE (C) original, em compartimentos separados de um microdilipador, com agitação, por 75 min contra 60% D usando uma membrana de diálise com corte de peso molecular de 8 K.
- Imediatamente após a diálise, divida essas preparações (U1ΔNE e NE em 60% D) em alíquotas de 20 μL; em seguida, encaixe o congelamento em um banho seco de gelo/etanol e armazene a -80 °C.
- Análise do RNA e teor de proteína de U1ΔNE e material vinculado a contas anti-U1
- Após a remoção do material desvinculado (U1ΔNE) (etapa 2.2.4), lave o material vinculado às contas anti-U1 adicionando 0,5 mL de tampão de lavagem TX. Pelotar a mistura por centrifugação (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s) e remova o sobrenante usando um micropipettor e descarte.
- Repita as etapas de lavagem 2.3.1 duas vezes.
- Remova o material vinculado às contas anti-U1 adicionando 100 μL de tampão de amostra SDS de 2x a 100 μL das contas e incubando por 10 minutos à temperatura ambiente.
- Pelotar a mistura por centrifugação (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s); remova o supernatante pela seringa Hamilton, e escorra o congelamento em um banho seco de gelo/etanol. Armazém a -80 °C.
- Compare o NE não-diluído, o NE empobrecido (U1ΔNE) e o material vinculado às contas (removido das contas pelo buffer de amostra SDS descrito nas etapas 2.3.3 e 2.3.4 acima). Siga os passos 2.3.6-2.3.8 para análise de RNA ou etapas 2.3.9-2.3.10 para análise de proteínas.
- Para cada amostra, extrair o RNA com 200 μL de fenol-clorofórmio (50:50, v/v); em seguida, extrair novamente com 180 μL de clorofórmio-isoamyl álcool (25:1, v/v). Após a extração, adicione 300 μL de etanol frio à prova de 200, inverta para misturar e armazene o RNA precipitado durante a noite a -20 °C.
- Centrifugar o RNA precipitado pelo etanol (12.000 x g por 10 min a 4 °C). Lave as pelotas com 150 μL de etanol frio de 70%. Centrifugar novamente (12.000 x g) a 4 °C por 15 min. Remova o supernatante usando um micropipetador e seque as pelotas em uma velocidade vac por 10-15 min sem calor.
- Resuspenque a pelota RNA seca em 10 μL de tampão de carregamento de RNA, suavemente vórtice, aqueça a 75-85 °C por 90 s e, em seguida, incubar no gelo por 2 min. Separe os snRNAs por eletroforese de gel (2 h a 16 mA) através de 13% de poliacrilamida - 8,3 M de géis de ureia e, em seguida, colorir com brometo de etídio ou sujeito a manchas do norte10,16.
- Carregue as amostras de proteína, em tampão amostral de SDS a partir da etapa 2.3.5, em 12,5% de géis de poliacrilamida e eletroforese a 200 V por aproximadamente 45-50 min no buffer de gel de sulfato de sódio (sulfato de sódio poliacrilamida de gel eletroforese).
- Transfira as proteínas separadas para a membrana nitrocelulose a 400 mA por 2 h no buffer de transferência. Após a transferência, bloqueie a membrana incubando durante a noite em T-TBS contendo 10% de leite seco sem gordura. Em seguida, imunoblote a membrana para revelar proteínas específicas8,21.
3. Imunoprecipitação de frações 10S de gradientes de glicerol por anti-Gal3
- Preparação de contas anti-Gal3 para imunoadsorção
NOTA: A derivação e caracterização da antisera policlonal do coelho contra Gal3 para coelho #2421 e para coelho #4910 foram descritas anteriormente.- Use soro pré-imune do coelho #49 como controle.
- Para a preparação de contas anti-Gal3 siga o procedimento previamente descrito para a preparação de contas anti-U1 (etapa 2.1), com exceção de que correspondente ao passo 2.1.3, a razão de antiserum (por exemplo, anti-Gal3, #49) para contas é de 3:1.
- Pouco antes do uso, lave as contas acopladas a anticorpos, a seguir designadas como contas anti-Gal3, duas vezes com 0,5 mL de tampão de lavagem TX. Remova o supernatante, primeiro com um micropipettor para tirar a maior parte do líquido e, em seguida, com uma seringa Hamilton para tirar o líquido das contas; descartar.
- Imunoprecipitaton de frações de gradiente de glicerol por anti-Gal3 (ver Figura 1B)
- Fracionar NE(D) sobre um gradiente de glicerol de 12%-32%. Misture e misture frações de gradiente de glicerol 3, 4 e 5 (numeradas a partir da parte superior do gradiente), que estão perto da região 10S do gradiente.
- Prepare duas amostras, cada uma com 150 μL aliquot de frações de gradiente combinadas 3-5 (etapa 3.2.1), e coloque em 50 μL de contas anti-Gal3.
- Em paralelo, prepare duas amostras cada uma com 150 μL de fração 1 (contendo Gal3 não em complexo com U1 snRNP10; passo 3.2.1) e coloque em 50 μL de contas anti-Gal3.
- Como controle, coloque 150 μL de 60% D em outro microtubo de 50 μL anti-Gal3 contas.
- Misture suavemente tocando o tubo e gire a cabeça sobre a cauda do microtube a 4°C por 1h.
- Pelle a mistura por centrifugação suave (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s).
- Remova o supernatante (material sem entrada) usando uma seringa Hamilton. Não lave as contas e use imediatamente para a adição das reações de emenda (seção 4.2).
- Análise do teor de RNA e proteína no material desvinculado e vinculado da precipitação anti-Gal3 de frações de gradiente de 10S
- Para a análise de componentes do material vinculado e desvinculado da precipitação anti-Gal3 das frações de gradiente de 10S, colete o material desvinculado (sobrenatante após a etapa 3.2.6), transfira em um microtubo fresco e congele a -20 °C.
- Lave as contas precipitadas da etapa 3.2.6 (contendo material vinculado ao anti-Gal3) adicionando 0,5 mL de tampão de lavagem TX.
- Pelotar a mistura por centrifugação suave (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s); remover o supernante usando um micropipetador e descartar. Repita os passos de lavagem mais duas vezes.
- Adicione 50 μL de tampão de amostra SDS 2X às contas anti-Gal3 lavadas e pelladas.
- Misture as contas suavemente e incubar por 10 minutos à temperatura ambiente.
- Pelotar a mistura por centrifugação suave (1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s), coletar o sobrenante por seringa Hamilton e armazenar em um microtube fresco a -20 °C.
- Compare o material desvinculado (seção 3.3.1) e o material vinculado (etapa 3.3.6) da precipitação anti-Gal3 em termos de RNA e componentes proteicos, utilizando procedimentos conforme descrito nas etapas 2.3.6. para 2.3.10, respectivamente.
4. Montagem de emendas de reação e análise de produtos
- Preparação do substrato de emenda
NOTA: O substrato pré-mRNA, designado MINX, contém duas sequências de exon e uma sequência intron de Adenovirus22. A sequência de DNA MINX no plasmídeo está sob o controle de promotores de polimerase T3, T7 ou SP6. Os materiais e métodos detalhados para a linearização do DNA plasmídeo MINX com endonuclease de restrição BamHI, transcrição por sp6 RNA polymerase na presença de α-32P[GTP] e a purificação de MINX com 32P rotulados para ensaios de emenda são descritos anteriormente19 (ver etapas 2.2 e 3.2 dessa referência).- Armazene o MINX radiolabeled como um etanol precipitado a -20 °C; use o substrato de emenda rotulado dentro de 4-6 semanas após a transcrição.
- Pouco antes do uso, centrifugar o etanol precipitado MINX 32P a 12.000 x g para 10 min a 4 °C; remover o supernatante com um micropipettor e descartar.
- Adicione 150 μL de 70% de etanol e centrífuga a 12.000 x g por 15 min a 4 °C. Descarte o supernatante e seque a pelota em velocidade vac sem calor por 15 minutos.
- Reidratar a pelota em 50 μL de água DEPC. Ponto 2 μL em cada um dos dois filtros GF/C; imerge os filtros em ácido tricloroacético frio de 5% (TCA) por 10 minutos. Enxágüe com TCA frio de 5%, seguido de etanol à prova de 180 em um frasco de vácuo. Aersse os filtros e sujeito à contagem de cintilação em 4 mL de Safety-Solve.
- Diluir MINX com 32P em 60% D a 104 cpm/μL para o ensaio de emenda.
- Montagem da reação de emenda (ver Figura 1C)
- Montar, no gelo, emendando reações em um volume total de 24 μL (8 μL U1ΔNE (a partir do passo 2.2.6), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de fosfato de creatina, 0,5 mM DTT, 20 unidades RNasin, 4 μL 32P rotulado substrato minx (104 cpm/μL), 60% D) e adicionar a cada tubo de contas da seção 3.2.7. Monte um conjunto idêntico de reações de emenda em um volume total de 24 μL, mas sem U1ΔNE e adicione a cada tubo de contas a partir da etapa 3.2.7.
- Prepare uma reação de emenda de controle em um volume total de 12 μL (4 μL NE(D), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de fosfato de creatina, 0,5 mM DTT, 20 unidades RNasin, 2 μL 32P-labeld MINX splicing substrato (104 cpm/μL), 60% D).
- Misture os tubos suavemente tocando e gire a 30°C por 90 min. Pelle a mistura por centrifugação suave a 1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s.
- Pare a reação e elute as proteínas das contas adicionando 24 μL de tampão de amostra de SDS 2x aos tubos contendo contas, e 12 μL de tampão de amostra SDS de 2x ao tubo de controle contendo NE, mas sem contas. Aqueça os tubos a 100 °C por 7 min.
- Centrifugar os tubos suavemente a 1.000 x g em um rotor de balde balançando a 4 °C para 10-15 s.
- Transfira os supernantes (eluções) para microtubos frescos: aproximadamente 48 μL dos tubos de contas e 24 μL do tubo de controle NE.
- Adicione Proteinase K (20 mg/mL) para digerir e solubilizar as proteínas: adicione 5 μL à elução de 48 μL de contas e adicione 2,5 μL ao controle ne de 24 μL.
- Incubar os tubos a 37°C por 40 min.
- Centrifufique suavemente os tubos a 1.000 x g em um rotor de balde balançando a 4 °C por 10 s.
- Diluir as eluções de contas com 39,5 μL TE e acetato de sódio de 10 μL 3 M. Diluir o controle NE com 63,5 μL TE e 10 μL 3 M de acetato de sódio.
- Extrair e analisar o RNA conforme descrito abaixo (seção 4.3).
- Análise dos produtos da reação de emenda
- Extrair as RNAs em cada amostra com fenol-clorofórmio, seguido de clorofórmio-isoamyl; precipitar as RNAs com etanol, centrífuga, lavar as pelotas, remover o sobrenante e secar as pelotas seguindo o mesmo procedimento descrito nas etapas 2.3.6 e 2.3.7.
- Resuspenque a pelota RNA seca em 10 μL de tampão de carregamento de RNA, suavemente vórtice, aqueça a 75-85 °C por 90 s e, em seguida, incubar no gelo por 2 min.
- Prepare 20 mL de uma solução contendo 13% de poliacrilamida (bisacrilamida:acrilamida, 1.9:50 [wt/wt]) em 8,3 M de ureia; géis fundidos de 15 cm de comprimento usando esta solução.
- Uma vez que o gel é lançado, eletroforese-o (sem qualquer amostra carregada) a 400 V por 20 minutos usando TBE como tampão em execução. Após esta etapa, lave os poços com tampão de execução TBE.
- Carregue as amostras de RNA, no tampão de carregamento de RNA, e eletroforese com tampão de execução TBE a 400 V por 3,5 a 4 h. Após a eletroforese, remova a ureia imergindo e girando o gel em água destilada por 10 minutos.
- Seque o gel em papel filtro de 3 M, primeiro por 2 h 15 min a 80 °C e depois por 30 minutos sem calor para esfriá-lo lentamente. Submeter o gel seco à autoradiografia em filme para detectar as posições de migração dos componentes radioativos.
Resultados
NE esgotado de U1 snRNP (U1ΔNE da Seção 2.2.6) e Gal3 - U1 snRNP complexos da região 10S do gradiente de glicerol imunoprecipitado por anti-Gal3 (passo 3.2.7) foram misturados em uma reação de emenda. Esta mistura de reação continha U1 snRNA (Figura 2A, pista 3), bem como a proteína específica U1, U1-70K (Figura 2B, pista 3). Como esperado, o anti-Gal3 precipitou Gal3 (Figura 2B, pista 3)....
Discussão
Este relatório fornece os detalhes experimentais que documentam um complexo Gal3 - U1 snRNP preso em contas revestidas anti-Gal3 pode se ligar ao substrato pré-mRNA e este complexo ternário pode restaurar a atividade de emenda a um NE er. . Os estudos de imunofluorescência precoce e fracionamento subcelular forneceram o indício inicial de uma associação de Gal3 com componentes da máquina de emenda: colocalização em manchas nucleares com polipeptídeos de núcleo Sm de snRNPs e serina e a...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho foi apoiado pela National Science Foundation Grant MCB-0092919 e Michigan State University Intramural Research Grant 09-CDFP-2001 (para RJP) e pelos Institutos Nacionais de Saúde Grant GM-38740 e Michigan AgBioResearch Project MICL02455 (para JLW).
O substrato minx pré-mRNA usado nos ensaios de emenda foi um presente gentil da Dra.
Materiais
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
Referências
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932 (1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -. F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados