
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Komplementierung der Spleißaktivität durch einen Galectin-3 - U1 snRNP Komplex auf Perlen
In diesem Artikel
Zusammenfassung
Dieser Artikel beschreibt die experimentellen Verfahren zur (a) Depletion von U1 snRNP aus Kernextrakten mit gleichzeitigem Verlust der Spleißaktivität; und (b) Rekonstitution der Spleißaktivität im U1-depletierten Extrakt durch Galectin-3 - U1-snRNP-Partikel, die an Perlen gebunden sind, kovalent gekoppelt mit Anti-Galectin-3-Antikörpern.
Zusammenfassung
Klassische Depletions-Rekonstitutionsexperimente deuten darauf hin, dass Galectin-3 ein erforderlicher Spleißfaktor in Kernextrakten ist. Der Mechanismus der Integration von Galectin-3 in den Spleißweg wird in diesem Artikel behandelt. Die Sedimentation von HeLa-Zellkernextrakten auf 12%-32% Glyceringradienten ergibt Fraktionen, die in einem endogenen ~10S-Partikel angereichert sind, das Galectin-3 und U1 snRNP enthält. Wir beschreiben nun ein Protokoll zur Depletion von Kernextrakten von U1 snRNP mit gleichzeitigem Verlust der Spleißaktivität. Die Spleißaktivität im U1-depletierten Extrakt kann durch das Galectin-3 - U1 snRNP-Partikel rekonstituiert werden, das auf Agaroseperlen eingeschlossen ist, kovalent gekoppelt mit Anti-Galectin-3-Antikörpern. Die Ergebnisse deuten darauf hin, dass der galectin-3 - U1 snRNP - pre-mRNA ternäre Komplex ein funktioneller E-Komplex ist, der zu Zwischenprodukten und Produkten der Spleißreaktion führt und dass Galectin-3 durch seine Assoziation mit U1 snRNP in den Spleißweg eintritt. Das Schema der Verwendung von Komplexen, affinitäts- oder immunselektioniert auf Kügelchen zur Rekonstitution der Spleißaktivität in Extrakten, die von einem bestimmten Spleißfaktor erschöpft sind, kann allgemein auf andere Systeme anwendbar sein.
Einleitung
Die Produktion der meisten eukaryotischen Boten-RNAs (mRNAs) beinhaltet die Entfernung von Introns und die Ligatur von Exons in einem Kernprozess, der als Prä-mRNA-Spleißen bezeichnet wird1. Zwei Klassen von RNA-Protein-Komplexen (RNPs) lenken die Verarbeitung von Pre-Messenger-RNA in reife mRNA über spleißosomale Komplexe. Eine Klasse, entstehende Prä-Messenger-RNPs, wird co-transkriptionell durch die Bindung heterogener kerniger RNP-Proteine und anderer RNA-bindender Proteine, einschließlich einiger Mitglieder der SR-Familie, gebildet, was hnRNP-Komplexe ergibt2. Die zweite Klasse, Uracil-reiche kleine Kern-RNPs (U snRNPs mit U1, U2, U4, U5 und U6 snRNAs) ist mit U-spezifischen und Kernproteinen assoziiert3,4. Die U-snRNPs interagieren geordnet mit bestimmten Regionen von Pre-Messenger-RNPs in einem dynamischen Remodeling-Pfad, wenn Introns herausgeschnitten und Exons ligatiert werden, um reife mRNPs zu erzeugen5. Viele weitere Kernproteine sind an diesen Verarbeitungsereignissen beteiligt6.
Galectin-1 (Gal1) und Galectin-3 (Gal3) sind zwei Proteine, die erforderliche Faktoren im Spleißweg sind, wie Ausschöpfungs-Rekonstitutionsstudien zeigen7,8. Die Entfernung beider Galectine aus dem Spleißen kompetenter Kernextrakte (NE) schafft die Spleißosomenmontage und die Spleißaktivität frühzeitig ab. Die Zugabe von Galektin zu einem so doppelt erschöpften NE stellt beide Aktivitäten wieder her. Gal1 und Gal3 sind Komponenten aktiver Spleißosomen, wie durch spezifische Immunpräzipitation von pre-mRNA, spleißenden Zwischenprodukten und reifer mRNA durch Antiserum spezifisch für Gal1 oder Gal39 belegt wird. Wichtig ist, dass Gal3 mit endogener U-snRNA assoziiert ist, die Partikel im NORDOSTEN außerhalb des Spleißweges enthält, wie die Ausfällung von snRNPs durch Anti-Gal3-Antisera10 zeigt. Schließlich verändert das Stummschalten von Gal3 in HeLa-Zellen die Spleißmuster zahlreicher Gene11.
In NE, die vorinkubiert wurden, um vorgeformte Spleißosomen12 zu zerlegen, werden snRNPs in mehreren Komplexen gefunden, die in Glyceringradienten von 7S bis größer als 60S sedimentieren. Obwohl die Glyceringradientenfraktionierung eine gängige Technik zur Isolierung von spleißosomalen Komplexen und Komponenten ist (siehe z.B. Referenzen 13,14,15), haben wir diese Methode erweitert, indem wir spezifische Fraktionen mittels Antikörperimmunpräzipitationen charakterisieren. Eine snRNP-Sedimentation bei 10S enthält nur U1-snRNA zusammen mit Gal3. Die Immunpräzipitation der 10S-Fraktion mit Antiseren, die spezifisch für Gal3 oder U1 snRNP sind, ko-präzipitiert sowohl U1 als auch Gal3, was darauf hindeutet, dass einige der U1-snRNP-Monopartikel an Gal310 gebunden sind. Da U1 snRNP der erste Komplex ist, der in der spleißosomalen Montage an prä-mRNP bindet1,5, stellt dieser Schritt eine potenzielle Eintrittsstelle für Gal3 in den Spleißweg dar. Auf dieser Grundlage zeigten wir, dass 10S Gal3-U1 snRNP-Monopartikel, die an Anti-Gal3-haltige Kügelchen gebunden waren, die Spleißaktivität eines U1-snRNP-erschöpften NE wiederherstellten, was diesen Komplex als einen Mechanismus etablierte, durch den Gal3 in den spleißosomalen Weg rekrutiert wird16. Dies steht im Gegensatz zu Versuchen, Spleißosomen in bestimmten Stadien der Spleißreaktion zu isolieren und die damit verbundenen Faktoren zu katalogisieren17,18. In solchen Studien wird das Vorhandensein bestimmter Faktoren zu einem bestimmten Zeitpunkt festgestellt, nicht jedoch der Mechanismus, durch den sie belastet wurden.
Zuvor hatten wir die Herstellung von NE, das Spleißsubstrat, die Montage des Spleißreaktionsgemisches und die Analyse von Produkten in unserer Dokumentation der Rolle von Galectinen beim Pre-mRNA-Spleißen ausführlich beschrieben19. Wir beschreiben nun die experimentellen Verfahren zur Fraktionierung von Kernextrakten zur Erlangung einer mit Gal3 - U1 snRNP-Komplex angereicherten Fraktion und zur Immunselektion des letzteren Komplexes zur Rekonstitution der Spleißaktivität in einem U1-depletierten Kernextrakt.

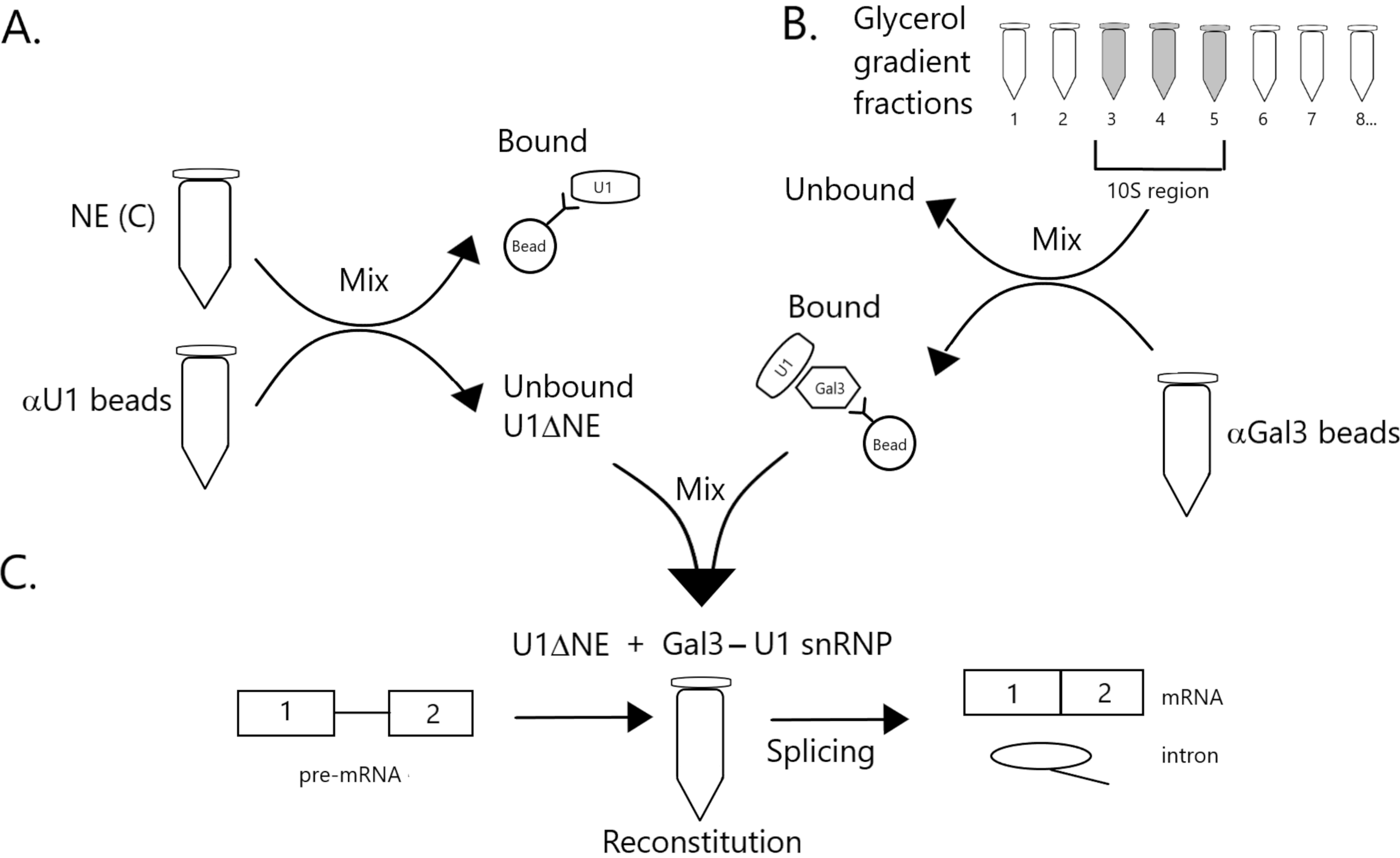
Abbildung 1: Schematische Darstellung der Komplementierung der Spleißaktivität in Kernextrakt, der an U1-snRNP durch einen Gal3-U1-snRNP-Komplex auf Kügelchen abgereichert ist. (A) NE in Puffer C (NE(C)) wird mit Protein A-Sepharose-Perlen kovalent gekoppelt mit Anti-U1-SnRNP (αU1-Perlen) inkubiert. Der ungebundene Anteil ist an U1 snRNP (U1ΔNE) erschöpft. (B) NE in Puffer D (NE(D)) wird über einen 12%-32%igen Glyceringradienten durch Ultrazentrifugation fraktioniert. Fraktionen, die der 10S-Region (Fraktionen 3-5) entsprechen, werden kombiniert und mit Perlen gemischt, die kovalent mit Anti-Gal3-Antikörpern (αGal3-Perlen) gekoppelt sind. Das an die Kügelchen gebundene Material enthält ein Gal3-U1 snRNP-Monopartikel. (C) Der Gal3-U1 snRNP-Komplex aus Teil (B) wird mit U1ΔNE aus Teil (A) in einem Spleißassay unter Verwendung eines 32P-markierten MINX-Prä-mRNA-Substrats gemischt und die Zwischenprodukte und Produkte der Spleißreaktion werden durch Gelelektrophorese und Autoradiographie analysiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll
1. Hinweise zu den allgemeinen Verfahren
- Stellen Sie sicher, dass alle Chemikalien (Pufferkomponenten, Enzyme usw.) frei von Ribonuklease (RNase) gehalten werden. Sequester alle kommerziell gekauften Reagenzflaschen aus dem allgemeinen Laborgebrauch. Tragen Sie Handschuhe für alle Schritte des experimentellen Verfahrens. Verwenden Sie nur Gebackene Gläser und Utensilien (siehe Schritt 1.2 unten) und vorbehandelte Lösungen (siehe Schritt 1.3 unten).
- Alle Gläser (Becher, Kolben, Flaschen, Pipetten usw.) für mindestens 4 h bei 177 °C backen. Wickeln Sie andere Utensilien (Spatel, Rührstangen usw.) vor dem Backen unter den gleichen Bedingungen in Alufolie.
- Eine 0,1%ige (vol/vol) Lösung von Diethylpyrocarbonat (DEPC) in doppelt destilliertem Wasser (ddH2O) herstellen. Mit einem magnetischen Rührstab diese Lösung über Nacht umrühren und dann autoklavieren. Verwenden Sie dieses DEPC-behandelte H2O, um alle Lösungen herzustellen, die Tris enthalten; Filtern Sie dann mit einem Flaschendevakuumfilter. Verwenden Sie normales ddH2O, um alle anderen Lösungen (ohne Tris) vorzubereiten; dann mit DEPC (0,1%, vol/vol) und Autoklav behandeln.
ANMERKUNG: Die in den folgenden experimentellen Verfahren verwendeten Puffer sind in Tabelle 1 alphabetisch aufgeführt.
| Name des Puffers | Zusammensetzung |
| Boratpuffer | 0,2 M Natriumborat, pH 9 |
| Puffer C | 20 mM HEPES, pH 7,9, 25% (vol/vol) Glycerin, 0,42 M NaCl, 1,5 mM MgCl2, 0,2 mM EDTA, 0,5 mM Phenylmethylsulfonylfluorid (PMSF), 0,5 mM Dithiothreitol (DTT) |
| Puffer D | 10 mM HEPES, pH 7,9, 20% (vol/vol) Glycerin, 0,1 M KCl, 0,2 mM EDTA, 0,5 mM PMSF, 0,5 mM DTT |
| 60%D | 60% Puffer D und 40% H2O |
| Ethanolamin | 0,2 M Ethanolamin, pH 8 |
| HEPES-Bindungspuffer | 20 mM HEPES, pH 7,9 |
| HEPES Waschpuffer | 20 mM HEPES, pH 7,9, 0,5 M NaCl |
| RNA-Ladepuffer | 90% Formamid, 20 mM EDTA, pH 8, 0,05% (w/v) Bromphenolblau |
| SDS-PAGE-Puffer | 25 mM Tris, 169 mM Glycin, 0,1% Natriumdodecylsulfat (SDS), pH 8,8 |
| SDS-Probenpuffer | 62,5 mM Tris, pH 6,8, 2% SDS, 10% Glycerin, 5% 2-Mercaptoethanol, 0,1% (w/v) Bromphenolblau |
| FSME-Puffer für RNA-Gele | 89 mM Tris, 89 mM Borsäure, 2,5 mM EDTA, pH 8,3 |
| TE-Puffer | 10 mM Tris, pH 8, 1 mM EDTA |
| Übertragungspuffer | 25 mM Tris, 1,92 M Glycin, 20% Methanol, pH 8,3 |
| T-TBS-Puffer | 10 mM Tris, 0,5 M NaCl, 0,05% Tween 20, pH 7,5 |
| TX Waschpuffer | 0,05% Triton X-100 (TX) in 60%D |
Tabelle 1: Name und Zusammensetzung der Puffer
2. Herstellung von NE erschöpft von U1 snRNP (U1 ΔNE)
- Herstellung von Anti-U1-Kügelchen für die Immunadsorption
- 50 mg Protein A-Sepharose CL-4B Perlen im Überschuss dePC-behandeltes H2O vorschwillieren, um etwa 200 μL geschwollene Perlen zu erzeugen und dann in HEPES-Waschpuffer zu waschen.
- Für diese Wäsche und alle nachfolgenden Wäschen die Kügelchen durch Zentrifugation (1.000 x g in einem schwenkbaren Eimerrotor bei 4 °C für 10-15 s) pelletieren und die ungebundene Wäsche mit einem Mikropipettor entnehmen und entsorgen.
- Mischen Sie 150 μL gewaschene Perlen mit 150 μL humanem Autoimmunserum, das spezifisch für U1 snRNP ist (Volumen des Antikörpers zum Volumen der Perlen im Verhältnis 1: 1).
- Auf der Grundlage des Gesamtvolumens von (~300 μL aus Schritt 2.1.3 oben) wird das Gemisch auf 20 mM HEPES, pH 7,9, entsprechend den Bedingungen des HEPES-Bindungspuffers eingestellt; Inkubieren Sie diese Mischung mit kontinuierlichem Schaukeln bei Raumtemperatur für 60 min.
- Waschen Sie die mit Antikörpern gebundenen Kügelchen mit 1 ml Boratpuffer (0,2 M Natriumborat, pH 9) und resuspendieren Sie in 1 ml desselben Boratpuffers.
- Um den an das Protein A-Sepharose gebundenen Antikörper kovalent zu koppeln, Dimethylpimelimidat in eine Endkonzentration von 20 mM zu geben und bei Raumtemperatur mit Schaukeln für 60 min zu inkubieren.
- Waschen Sie die Perlen mit 1 ml Boratpuffer.
- Um ein nicht umgesetztes Vernetzungsreagenz zu blockieren, fügen Sie 1 ml 0,2 M Ethanolamin (pH 8) hinzu und inkubieren Sie bei Raumtemperatur mit Schaukeln für 60 min.
- Waschen Sie die Antikörper-gekoppelten Kügelchen, im Folgenden als Anti-U1-Kügelchen bezeichnet, zweimal mit 0,5 ml TX-Waschpuffer (0,05% Triton X-100 in 60% D).
- Depletion von U1 snRNP aus NE (siehe Abbildung 1A)
HINWEIS: Das Verfahren zur Herstellung von NE aus HeLa-Zellen wurde ursprünglich von Dignam et al.20 entwickelt. Wir haben die Materialien und detaillierten Methoden zur Herstellung von NE für Spleißassays19 beschrieben (siehe Schritte 2.1 und 3.1 dieser Referenz). NE, wie ursprünglich vorbereitet, befindet sich in Puffer C und wird im Folgenden als NE(C) bezeichnet. NE(C), das gegen Puffer D dialysiert und mit puffer D ausgeglichen ist, wird als NE(D) bezeichnet.- Inkubieren Sie 200 μL NE(C) mit 100 μL Anti-U1-Perlen aus Schritt 2.1.9 oben.
- Fügen Sie 5 μL RNasin zu der Mischung hinzu.
- Drehen Sie das Mikroröhrchen 1 h lang Kopf über Schwanz bei 4 °C.
- Pelletieren Sie die Mischung durch Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s) und sammeln Sie das ungebundene Material (U1ΔNE) mit einer Hamilton-Spritze.
- Dialysieren Sie das gesamte Volumen von U1ΔNE, zusammen mit einem separaten 50 μL Aliquot des ursprünglichen, nicht vollständigen NE(C), in separaten Kompartimenten eines Mikrodialysators unter Rühren für 75 min gegen 60% D unter Verwendung einer Dialysemembran mit 8 K Molekulargewichtsgrenzung.
- Diese Präparate (U1ΔNE und NE in 60% D) werden unmittelbar nach der Dialyse in 20 μL Aliquots aufgeteilt; anschließend in einem Trockeneis/Ethanol-Bad einfrieren und bei -80 °C lagern.
- Analyse des RNA- und Proteingehalts von U1ΔNE und an Anti-U1-Kügelchen gebundenem Material
- Nach dem Entfernen des ungebundenen Materials (U1ΔNE) (Schritt 2.2.4) wird das an den Anti-U1-Kügelchen gebundene Material durch Zugabe von 0,5 ml TX-Waschpuffer gewaschen. Pelletieren Sie die Mischung durch Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s) und entfernen Sie den Überstand mit einem Mikropipettor und entsorgen Sie ihn.
- Wiederholen Sie die Waschschritte 2.3.1 zweimal.
- Entfernen Sie das an die Anti-U1-Kügelchen gebundene Material, indem Sie 100 μL 2x SDS-Probenpuffer zu 100 μL der Kügelchen hinzufügen und für 10 min bei Raumtemperatur inkubieren.
- Pellet die Mischung durch Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s); Entfernen Sie den Überstand mit der Hamilton-Spritze und frieren Sie ihn in einem Trockeneis-/Ethanolbad ein. Bei -80 °C lagern.
- Vergleichen Sie das nicht depleted NE, das erschöpfte NE (U1ΔNE) und das an die Perlen gebundene Material (aus den Kügelchen entfernt durch SDS-Probenpuffer, wie in den Schritten 2.3.3 und 2.3.4 oben beschrieben). Befolgen Sie die Schritte 2.3.6-2.3.8 für die RNA-Analyse oder die Schritte 2.3.9-2.3.10 für die Proteinanalyse.
- Extrahieren Sie für jede Probe die RNA mit 200 μL Phenol-Chloroform (50:50, v/v); anschließend erneut mit 180 μL Chloroform-Isoamylalkohol (25:1, v/v) extrahieren. Nach der Extraktion 300 μL kaltes 200-beständiges Ethanol zugeben, zum Mischen invertieren und die ausgefällte RNA über Nacht bei -20 °C lagern.
- Zentrifugieren Sie die Ethanol-ausgefällte RNA (12.000 x g für 10 min bei 4 °C). Waschen Sie die Pellets mit 150 μL kaltem 70% Ethanol. Zentrifugiere erneut (12.000 x g) bei 4 °C für 15 min. Entfernen Sie den Überstand mit einem Mikropipettor und trocknen Sie die Pellets in einem Geschwindigkeitssauger für 10-15 min ohne Hitze.
- Resuspendieren Sie das getrocknete RNA-Pellet in 10 μL RNA-Ladepuffer, wirbeln Sie vorsichtig, erhitzen Sie es auf 75-85 ° C für 90 s und inkubieren Sie es dann für 2 min auf Eis. Trennen Sie die snRNAs durch Gelelektrophorese (2 h bei 16 mA) durch 13% Polyacrylamid - 8,3 M Harnstoffgele und färben Sie sie dann entweder mit Ethidiumbromid an oder unterliegen Sie einem Northern Blotting10,16.
- Laden Sie die Proteinproben in SDS-Probenpuffer aus Schritt 2.3.5 auf 12,5% Polyacrylamidgele und Elektrophorrese bei 200 V für ca. 45-50 min in SDS-PAGE-Puffer (Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese).
- Übertragen Sie die getrennten Proteine auf die Nitrocellulosemembran bei 400 mA für 2 h im Transferpuffer. Blockieren Sie nach dem Transfer die Membran durch Inkubation über Nacht in T-TBS, das 10% fettfreie Trockenmilch enthält. Dann immunoblot die Membran, um spezifische Proteine aufzudecken8,21.
3. Immunpräzipitation von 10S-Fraktionen von Glyceringradienten durch Anti-Gal3
- Herstellung von Anti-Gal3-Perlen zur Immunadsorption
HINWEIS: Die Ableitung und Charakterisierung von kaninchenpolyklonalen Antiseren gegen Gal3 für Kaninchen #2421 und für Kaninchen #4910 wurden zuvor beschrieben.- Verwenden Sie Präimmunserum von Kaninchen # 49 als Kontrolle.
- Für die Herstellung von Anti-Gal3-Perlen befolgen Sie das zuvor beschriebene Verfahren zur Herstellung von Anti-U1-Perlen (Schritt 2.1), mit der Ausnahme, dass entsprechend Schritt 2.1.3 das Verhältnis von Antiserum (z. B. Anti-Gal3, Nr. 49) zu Perlen 3: 1 beträgt.
- Kurz vor Gebrauch die Antikörper-gekoppelten Kügelchen, im Folgenden als Anti-Gal3-Kügelchen bezeichnet, zweimal mit 0,5 ml TX-Waschpuffer waschen. Entfernen Sie den Überstand, zuerst mit einem Mikropipettor, um den größten Teil der Flüssigkeit herauszuholen, und dann mit einer Hamilton-Spritze, um die Flüssigkeit aus den Perlen zu bekommen; abwerfen.
- Immunpräzipitation von Glyceringradientenfraktionen durch Anti-Gal3 (siehe Abbildung 1B)
- Fraktionieren Sie NE(D) über einen 12%-32%igen Glyceringradienten10. Kombinieren und mischen Sie die Glyceringradientenfraktionen 3, 4 und 5 (nummeriert von der Spitze des Gradienten), die sich in der Nähe des 10S-Bereichs des Gradienten befinden.
- Bereiten Sie zwei Proben mit jeweils 150 μL Aliquot kombinierter Gradientenfraktionen 3-5 (Schritt 3.2.1) vor und geben Sie sie in 50 μL Anti-Gal3-Kügelchen.
- Bereiten Sie parallel zwei Proben mit jeweils 150 μL Fraktion 1 vor (enthält Gal3 nicht im Komplex mit U1 snRNP10; Schritt 3.2.1) und legen Sie sie in 50 μL Anti-Gal3-Kügelchen.
- Als Kontrolle geben Sie 150 μL 60% D in ein anderes Mikroröhrchen mit 50 μL Anti-Gal3-Perlen.
- Mischen Sie vorsichtig, indem Sie auf das Rohr klopfen, und drehen Sie dann das Mikroröhrchen bei 4 ° C für 1 h.
- Pelletieren Sie die Mischung durch schonende Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s).
- Entfernen Sie den Überstand (ungebundenes Material) mit einer Hamilton-Spritze. Waschen Sie die Perlen nicht und verwenden Sie sie sofort für die Zugabe der Spleißreaktionen (Abschnitt 4.2).
- Analyse des RNA- und Proteingehalts im ungebundenen und gebundenen Material aus der Anti-Gal3-Fällung von 10S-Gradientenfraktionen
- Zur Analyse der Komponenten des gebundenen und ungebundenen Materials aus der Anti-Gal3-Ausfällung der 10S-Gradientenfraktionen wird das ungebundene Material (Überstand nach Schritt 3.2.6) aufgefangen, in ein frisches Mikroröhrchen überführt und bei -20 °C eingefroren.
- Die ausgefällten Kügelchen aus Schritt 3.2.6 (die an Anti-Gal3 gebundenes Material enthalten) werden durch Zugabe von 0,5 ml TX-Waschpuffer gewaschen.
- Pellet die Mischung durch schonende Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s); Entfernen Sie den Überstand mit einem Mikropipettor und verwerfen Sie ihn. Wiederholen Sie die Waschschritte noch zweimal.
- 50 μL 2X SDS Probenpuffer zu den gewaschenen und pelletierten Anti-Gal3-Kügelchen geben.
- Mischen Sie die Perlen vorsichtig und inkubieren Sie für 10 min bei Raumtemperatur.
- Pelletieren Sie die Mischung durch schonende Zentrifugation (1.000 x g in einem schwingenden Eimerrotor bei 4 °C für 10-15 s), sammeln Sie den Überstand durch Hamilton Spritze und lagern Sie ihn in einem frischen Mikroröhrchen bei -20 °C.
- Vergleichen Sie das ungebundene Material (Abschnitt 3.3.1) und das gebundene Material (Schritt 3.3.6) der Anti-Gal3-Fällung in Bezug auf RNA- und Proteinkomponenten unter Verwendung der in Schritt 2.3.6 beschriebenen Verfahren. bis 2.3.10.
4. Montage der Spleißreaktion und Analyse der Produkte
- Vorbereitung des Spleißsubstrats
HINWEIS: Das prä-mRNA-Substrat, minX genannt, enthält zwei Exonsequenzen und eine Intronsequenz aus dem Adenovirus22. Die MINX-DNA-Sequenz im Plasmid steht unter der Kontrolle von T3-, T7- oder SP6-RNA-Polymerase-Promotoren. Die Materialien und detaillierten Methoden zur Linearisierung der MINX-Plasmid-DNA mit BamHI-Restriktionsendonuklease, zur Transkription durch SP6-RNA-Polymerase in Gegenwart von α-32P[GTP] und zur Aufreinigung von 32P-markiertem MINX für Spleißassays sind zuvor beschrieben19 (siehe Schritte 2.2 und 3.2 dieser Referenz).- Lagern Sie das radioaktiv markierte MINX als Ethanolausscheidung bei -20 °C; Verwenden Sie das markierte Spleißsubstrat innerhalb von 4-6 Wochen nach der Transkription.
- Kurz vor Gebrauch zentrifugieren Sie das ethanolgeleitete 32P-markierte MINX bei 12.000 x g für 10 min bei 4 °C; Entfernen Sie den Überstand mit einem Mikropipettor und verwerfen Sie ihn.
- 150 μL 70% Ethanol zugeben und bei 12.000 x g für 15 min bei 4 °C zentrifugieren. Entsorgen Sie den Überstand und trocknen Sie das Pellet in Geschwindigkeit vac ohne Hitze für 15 min.
- Rehydrieren Sie das Pellet in 50 μL DEPC-Wasser. Spot 2 μL auf jedem der beiden GF/C-Filter; Tauchen Sie die Filter für 10 min in kalte 5% Trichloressigsäure (TCA). Spülen Sie mit kaltem 5% TCA, gefolgt von 180-beständigem Ethanol auf einer Vakuumflasche. Trocknen Sie die Filter an der Luft und unterziehen Sie die Szintillationszählung in 4 mL Safety-Solve.
- Verdünnen Sie 32P-markiertes MINX in 60% D auf 104 cpm/μL für den Spleißassay.
- Aufbau der Spleißreaktion (siehe Abbildung 1C)
- Auf Eis spleißende Reaktionen in einem Gesamtvolumen von 24 μL (8 μL U1ΔNE (aus Schritt 2.2.6), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM Kreatinphosphat, 0,5 mM DTT, 20 Einheiten RNasin, 4 μL 32P-markiertes MINX-Spleißsubstrat (104 cpm/μL), 60% D) zusammensetzen und in jedes Röhrchen mit Perlen aus Abschnitt 3.2.7 geben. Fügen Sie einen identischen Satz von Spleißreaktionen in einem Gesamtvolumen von 24 μL, aber ohne U1ΔNE zusammen und fügen Sie jedem Kürmchen Perlen aus Schritt 3.2.7 hinzu.
- Vorbereiten einer Kontrollspleißreaktion in einem Gesamtvolumen von 12 μL (4 μL NE(D), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM Kreatinphosphat, 0,5 mM DTT, 20 Einheiten RNasin, 2 μL 32P-markiertes MINX-Spleißsubstrat (104 cpm/μL), 60% D).
- Mischen Sie die Rohre vorsichtig durch Klopfen und drehen Sie es bei 30 ° C für 90 min. Pelletieren Sie die Mischung durch schonende Zentrifugation bei 1.000 x g in einem schwingenden Schaufelrotor bei 4 °C für 10-15 s.
- Stoppen Sie die Reaktion und eluieren Sie die Proteine von den Kügelchen, indem Sie 24 μL 2x SDS-Probenpuffer zu den Röhrchen mit Perlen und 12 μL 2x SDS-Probenpuffer in das Kontrollröhrchen geben, das NE, aber keine Kügelchen enthält. Erhitzen Sie die Rohre bei 100 °C für 7 min.
- Zentrifugieren Sie die Rohre vorsichtig bei 1.000 x g in einem schwenkbaren Schaufelrotor bei 4 °C für 10-15 s.
- Die Überstände (Elutionen) werden in frische Mikroröhrchen überführt: ca. 48 μL aus den Kügelchen und 24 μL aus dem NE-Steuerrohr.
- Proteinase K (20 mg/ml) hinzufügen, um die Proteine zu verdauen und zu lösen: Fügen Sie 5 μL zur 48 μL-Elution aus Perlen hinzu und fügen Sie 2,5 μL zur 24 μL NE-Kontrolle hinzu.
- Die Röhrchen bei 37°C für 40 min inkubieren.
- Vorsichtig zentrifugieren Sie die Rohre bei 1.000 x g in einem schwingenden Schaufelrotor bei 4 °C für 10 s.
- Verdünnen Sie die Perlenelutionen mit 39,5 μL TE und 10 μL 3 M Natriumacetat. Verdünnen Sie die NE-Kontrolle mit 63,5 μL TE und 10 μL 3 M Natriumacetat.
- Extrahieren und analysieren Sie die RNA wie unten beschrieben (Abschnitt 4.3).
- Analyse der Produkte der Spleißreaktion
- Extrahieren Sie die RNAs in jeder Probe mit Phenol-Chloroform, gefolgt von Chloroform-Isoamylalkohol; die RNAs mit Ethanol ausfällen, zentrifugieren, die Pellets waschen, den Überstand entfernen und die Pellets nach dem gleichen Verfahren wie in den Schritten 2.3.6 und 2.3.7 beschrieben trocknen.
- Resuspendieren Sie das getrocknete RNA-Pellet in 10 μL RNA-Ladepuffer, wirbeln Sie vorsichtig, erhitzen Sie es auf 75-85 ° C für 90 s und inkubieren Sie es dann für 2 min auf Eis.
- 20 mL einer Lösung, die 13% Polyacrylamid (Bisacrylamid:Acrylamid, 1,9:50 [gew./gew.]) enthält, in 8,3 M Harnstoff herstellen; gegossene Gele mit einer Länge von 15 cm mit dieser Lösung.
- Sobald das Gel gegossen ist, elektrophorisieren Sie es (ohne geladene Proben) bei 400 V für 20 minuten mit FSME als Laufpuffer. Nach diesem Schritt waschen Sie die Vertiefungen mit FSME-Laufpuffer.
- Laden Sie die RNA-Proben in RNA-Ladepuffer und Elektrophorrese mit FSME-Laufpuffer bei 400 V für 3,5 bis 4 h. Nach der Elektrophorese entfernen Sie den Harnstoff, indem Sie das Gel 10 Minuten lang in destilliertem Wasser eintauchen und drehen.
- Vakuumtrocknen Sie das Gel auf 3 M Filterpapier, zuerst für 2 h 15 min bei 80 °C und dann für 30 min ohne Hitze, um es langsam abzukühlen. Unterziehen Sie das getrocknete Gel einer Autoradiographie auf Film, um die Migrationspositionen der radioaktiven Bestandteile zu erkennen.
Ergebnisse
NE-depletierte U1-snRNP-Komplexe (U1ΔNE aus Abschnitt 2.2.6) und Gal3-U1-snRNP-Komplexe aus der 10S-Region des Glyceringradientenimimpräzipitats durch Anti-Gal3 (Schritt 3.2.7) wurden in einer Spleißreaktion gemischt. Dieses Reaktionsgemisch enthielt U1-snRNA (Abbildung 2A, Bahn 3) sowie das U1-spezifische Protein U1-70K (Abbildung 2B, Bahn 3). Wie erwartet, fiel der Anti-Gal3 Gal3 aus (Abbildung 2B
Diskussion
Dieser Bericht enthält die experimentellen Details, die einen Gal3 - U1 snRNP-Komplex dokumentieren, der auf Anti-Gal3-beschichteten Kügelchen gefangen ist, kann an prä-mRNA-Substrat binden und dieser ternäre Komplex kann die Spleißaktivität zu einem U1 snRNP-depletierten NE wiederherstellen. Gal3 ist ein Mitglied einer Familie von Proteinen, die ursprünglich auf der Grundlage seiner Galactose-spezifischen Kohlenhydratbindungsaktivität isoliert wurden23 . Frühe Immunfluoreszenz- und subze...
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Diese Arbeit wurde durch den National Science Foundation Grant MCB-0092919 und den Michigan State University Intramural Research Grant 09-CDFP-2001 (an RJP) sowie durch den National Institutes of Health Grant GM-38740 und das Michigan AgBioResearch Project MICL02455 (an JLW) unterstützt.
Das minx pre-mRNA-Substrat, das in den Spleißassays verwendet wurde, war ein freundliches Geschenk von Dr. Susan Berget (Baylor College of Medicine, Houston, TX, USA).
Materialien
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
Referenzen
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932 (1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -. F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten