
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Комплементация сплайсинговой активности комплексом Галектин-3 - U1 snRNP на бусинах
В этой статье
Резюме
В данной статье описаны экспериментальные процедуры для (а) истощения U1 snRNP из ядерных экстрактов с сопутствующей потерей сплайсинговой активности; и (b) восстановление сплайсинговой активности в U1-обедненном экстракте частицами snRNP галектина-3-U1, связанными с ковалентно связанными с анти-галектин-3 антителами.
Аннотация
Классические эксперименты по истощению-восстановлению показывают, что галектин-3 является обязательным фактором сплайсинга в ядерных экстрактах. Механизм включения галектина-3 в сплайсинговый путь рассматривается в данной работе. Осаждение клеточных ядерных экстрактов HeLa на 12%-32% градиентах глицерина дает фракции, обогащенные эндогенной частицей ~10S, содержащей галектин-3 и U1 snRNP. Теперь мы описываем протокол для истощения ядерных экстрактов U1 snRNP с сопутствующей потерей сплайсинговой активности. Сплайсинговая активность в U1-истощенном экстракте может быть восстановлена частицей snRNP галектина-3 - U1, захваченной на бусинах агарозы, ковалентно связанной с антителами против галектина-3. Результаты показывают, что троичный комплекс галектин-3 - U1 snRNP - пре-мРНК является функциональным комплексом Е, приводящим к промежуточным продуктам и продуктам реакции сплайсинга, и что галектин-3 попадает в путь сплайсинга через его ассоциацию с U1 snRNP. Схема использования комплексов аффинно- или иммуно-, выбранных на шариках, для восстановления сплайсинговой активности в экстрактах, обедненных специфическим фактором сплайсинга, может быть в целом применима к другим системам.
Введение
Производство большинства эукариотических мессенджерных РНК (мРНК) включает удаление интронов и лигирование экзонов в ядерном процессе, называемом сплайсингом премрнК1. Два класса РНК-белковых комплексов (RNP) направляют обработку пре-мессенджерной РНК в зрелую мРНК через сплайсеосомальные комплексы. Один класс, зарождающиеся предмаршированные RNP, образуется котранскрипционно путем связывания гетерогенных ядерных белков RNP и других РНК-связывающих белков, включая некоторых членов семейства SR, давая комплексы hnRNP2. Второй класс, богатый урацилом малые ядерные RNP (U snRNP с U1, U2, U4, U5 и U6 snRNAs) связан с U-специфическими и основными белками3,4. U snRNP взаимодействуют упорядоченным образом с определенными областями предмарш-мессенджерных RNP в динамическом пути ремоделирования, поскольку интроны иссекаются, а экзоны лигируются для получения зрелых mRNPs5. Многие дополнительные ядерные белки участвуют в этих событиях обработки6.
Галектин-1 (Gal1) и галектин-3 (Gal3) являются двумя белками, которые являются необходимыми факторами в пути сплайсинга, как показано исследованиями истощения-восстановления7,8. Удаление обоих галектинов из сращивания компетентных ядерных экстрактов (NE) отменяет сплайсеосомную сборку и сплайсинговую деятельность на ранней стадии. Добавление любого галектина к такому вдвойне истощенному NE восстанавливает обе активности. Gal1 и Gal3 являются компонентами активных сплайсеосом, о чем свидетельствует специфическая иммунопреципитация пре-мРНК, сплайсинговых промежуточных продуктов и зрелой мРНК антисывороткой, специфичной для Gal1 или Gal39. Важно отметить, что Gal3 ассоциируется с эндогенной U snRNA, содержащей частицы в NE вне пути сплайсинга, как показано осаждением snRNP анти-Gal3 antisera10. Наконец, глушение Gal3 в клетках HeLa изменяет паттерны сплайсинга многочисленных генов11.
В NE, предварительно инкубированном для разборки предварительно сформированных сплайсеосом12, snRNP обнаруживаются в нескольких комплексах, осажденных в градиентах глицерина от 7S до более 60S. Хотя фракционирование градиента глицерина является распространенным методом выделения сплайсеосомальных комплексов и компонентов (см., например, ссылки13,14,15), мы расширили этот метод, охарактеризовав специфические фракции с использованием иммунопреципитаций антител. SnRNP осадок при 10S содержит только U1 snRNA вместе с Gal3. Иммунопреципитация фракции 10S с антисыворотки, специфичной для Gal3 или U1 snRNP, осаждает как U1, так и Gal3, указывая на то, что некоторые из моночастиц U1 snRNP связаны с Gal310. Поскольку U1 snRNP является первым комплексом, который связывается с pre-mRNP в сплайсеосомальной сборке1,5, этот шаг представляет собой потенциальный входной участок для Gal3 в путь сплайсинга. На этой основе мы показали, что моночастицы 10S Gal3-U1 snRNP, связанные с бусинами, содержащими анти-Gal3, восстанавливают активность сплайсинга к U1 snRNP истощенной NE, устанавливая этот комплекс как один механизм, с помощью которого Gal3 набирается в сплайсеосомальный путь16. Это контрастирует с попытками выделения сплайсеосом на определенных этапах реакции сплайсинга и каталогизации связанных с ними факторов17,18. В таких исследованиях констатируется наличие определенных факторов в какой-то момент времени, но не механизм, с помощью которого они были загружены.
Ранее мы подробно описали подготовку NE, сплайсинговую подложку, сборку реакционной смеси сплайсинга и анализ продуктов в нашей документации о роли галектинов в сплайсинге премрнК19. Теперь описаны экспериментальные процедуры фракционирования ядерных экстрактов для получения фракции, обогащенной комплексом SnRNP Gal3 - U1, и иммуноотбора последнего комплекса для восстановления сплайсинговой активности в ядерном экстракте, обедненном U1.

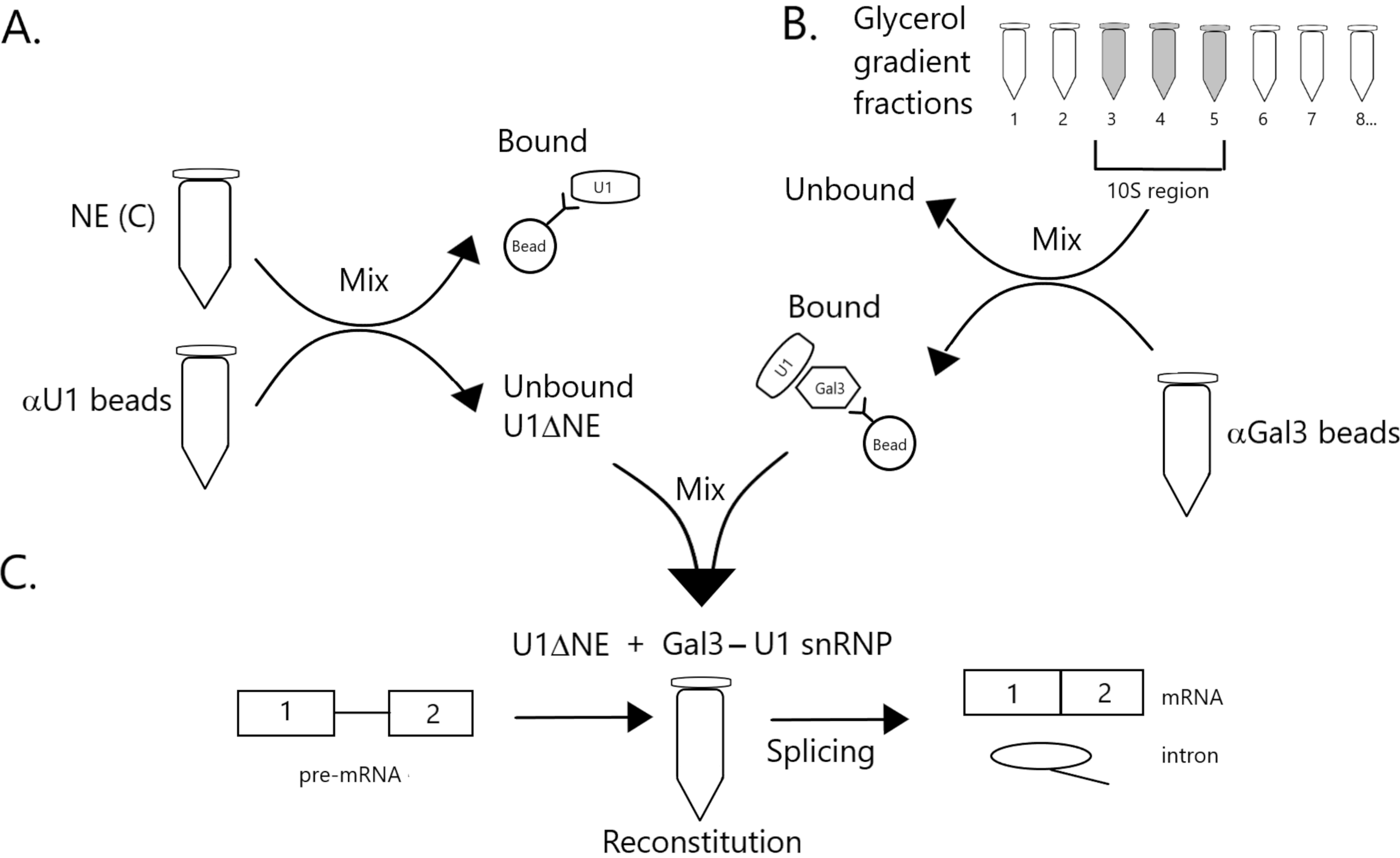
Рисунок 1: Принципиальная диаграмма, иллюстрирующая дополнение сплайсинговой активности в ядерном экстракте, обедненном U1 snRNP комплексом Gal3-U1 snRNP на шариках. (A) NE в буфере C (NE(C)) инкубируют с белком A-Sepharose бусинами, ковалентно связанными с анти-U1 snRNP (бусины αU1). Несвязанная фракция обедняется U1 snRNP (U1ΔNE). (B) NE в буфере D (NE(D)) фракционируют по 12%-32% градиенту глицерина путем ультрацентрифугирования. Фракции, соответствующие области 10S (фракции 3-5), объединяют и смешивают с бусинами, ковалентно связанными с антителами против Gal3 (шарики αGal3). Материал, связанный с шариками, содержит моночастицу Gal3-U1 snRNP. (C) Комплекс SnRNP Gal3-U1 из части (B) смешивают с U1ΔNE из части (A) в сплайсинговом анализе с использованием 32P-меченого субстрата MINX pre-mRNA, а промежуточные продукты и продукты реакции сплайсинга анализируют с помощью гелевого электрофореза и ауторадиографии. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
протокол
1. Примечания по общим процедурам
- Убедитесь, что все химические вещества (буферные компоненты, ферменты и т. д.) не содержат рибонуклеазы (РНКазы). Изолируйте все коммерчески приобретенные бутылки с реагентами от общего лабораторного использования. Надевайте перчатки на все этапы экспериментальной процедуры. Используйте только стеклянную посуду и посуду, которые были запечены (см. шаг 1.2 ниже) и растворы, которые были предварительно обработаны (см. шаг 1.3 ниже).
- Выпекайте всю стеклянную посуду (стаканы, колбы, бутылки, пипетки и т.д.) не менее 4 ч при 177 °C. Заверните другую посуду (шпатели, перемешиватели и т.д.) в алюминиевую фольгу перед выпечкой в тех же условиях.
- Готовят 0,1% (об./об.) раствор диэтилпирокарбоната (DEPC) в двухдистиллированной воде (ddH2O). Используя магнитный перемешиватель, перемешайте этот раствор в течение ночи, а затем автоклав. Используйте этот обработанный DEPC H2O для приготовления всех растворов, содержащих Tris; затем фильтр стерилизуют с помощью вакуумного фильтра для бутылок. Используйте обычный ddH2O для приготовления всех остальных растворов (без Tris); затем обрабатывают DEPC (0,1%, об/об) и автоклавом.
ПРИМЕЧАНИЕ: Буферы, используемые в следующем наборе экспериментальных процедур, перечислены в алфавитном порядке в таблице 1.
| Имя буфера | Состав |
| Боратный буфер | 0,2 М бората натрия, рН 9 |
| Буфер C | 20 мМ HEPES, рН 7,9, 25% (об./об.) глицерин, 0,42 М NaCl, 1,5 мМ MgCl2, 0,2 мМ ЭДТА, 0,5 мМ фенилметилсульфонилфторид (PMSF), 0,5 мМ дитиотрейтол (DTT) |
| Буфер D | 10 мМ HEPES, рН 7,9, 20% (об./об.) глицерин, 0,1 М KCl, 0,2 мМ ЭДТА, 0,5 мМ PMSF, 0,5 мМ DTT |
| 60%Д | 60% буфер D и 40% H2O |
| Этаноламин | 0,2 М этаноламина, рН 8 |
| Буфер связывания HEPES | 20 мМ HEPES, рН 7,9 |
| Буфер для промывки HEPES | 20 мМ HEPES, рН 7,9, 0,5 М NaCl |
| Буфер загрузки РНК | 90% формамид, 20 мМ ЭДТА, рН 8, 0,05% (мас./об.) бромфенола синего |
| Буфер SDS-PAGE | 25 мМ Tris, глицин 169 мМ, 0,1% додецилсульфата натрия (SDS), рН 8,8 |
| Буфер образцов SDS | 62,5 мМ Tris, pH 6,8, 2% SDS, 10% глицерин, 5% 2-меркаптоэтанол, 0,1% (мас./об.) бромфенол-синий |
| Буфер TBE для РНК-гелей | 89 мМ Tris, борная кислота 89 мМ, ЭДТА 2,5 мМ, рН 8,3 |
| Буфер TE | 10 мМ Трис, рН 8, 1 мМ ЭДТА |
| Буфер передачи | 25 мМ Tris, 1,92 М глицина, 20% метанола, рН 8,3 |
| Буфер T-TBS | 10 мМ Tris, 0,5 М NaCl, 0,05% анимации 20, рН 7,5 |
| TX буфер промывки | 0,05% Тритон X-100 (TX) в 60% D |
Таблица 1: Название и состав буферов
2. Препарат NE истощен U1 snRNP (U1 ΔNE)
- Препарат шариков против U1 для иммуносансорбции
- Предварительно набухайте 50 мг бусин белка A-Sepharose CL-4B в избытке H2O, обработанном DEPC, чтобы произвести приблизительно 200 мкл набухших шариков, а затем промыть в буфере для промывки HEPES.
- Для этой промывки и всех последующих промывок гранулируют шарики центрифугированием (1000 х г в качающемся роторе ведра при 4 °C в течение 10-15 с) и удаляют несвязанную промывку с помощью микропипеттера и выбрасывают.
- Смешать 150 мкл промытых шариков со 150 мкл аутоиммунной сыворотки человека, специфичной для U1 snRNP (объем антитела к объему шариков в соотношении 1:1).
- Отрегулируйте, исходя из общего объема (~300 мкл из шага 2.1.3 выше), смесь до 20 мМ HEPES, рН 7,9, соответствующего условиям буфера связывания HEPES; инкубируют эту смесь с непрерывным раскачиванием при комнатной температуре в течение 60 мин.
- Промыть шарики, связанные антителами, 1 мл боратного буфера (0,2 М бората натрия, рН 9) и повторно суспендировать в 1 мл того же боратного буфера.
- Чтобы ковалентно соединить антитело, связанное с белком А-сефарозные шарики, добавляют диметилпимелимидат до конечной концентрации 20 мМ и инкубируют при комнатной температуре с раскачиванием в течение 60 мин.
- Вымойте шарики 1 мл боратного буфера.
- Чтобы заблокировать любой непрореагировавший сшивающий реагент, добавляют 1 мл 0,2 М этаноламина (рН 8) и инкубируют при комнатной температуре с раскачиванием в течение 60 мин.
- Промывайте бусины, связанные с антителами, в дальнейшем обозначаемые как бусины против U1, дважды с 0,5 мл буфера промывки TX (0,05% Triton X-100 в 60% D).
- Истощение U1 snRNP из NE (см. Рисунок 1A)
ПРИМЕЧАНИЕ: Процедура получения NE из клеток HeLa была первоначально разработана Dignam et al.20. Мы описали материалы и подробные методы подготовки NE к анализу сращивания19 (см. шаги 2.1 и 3.1 этой ссылки). NE, как первоначально подготовлено, находится в буфере C и в дальнейшем будет обозначен как NE(C). NE(C), диализованный по отношению к буферу D и уравновешенный буфером D, будет обозначен как NE(D).- Инкубировать 200 мкл NE(C) со 100 мкл шариков против U1 со стадии 2.1.9 выше.
- Добавьте в смесь 5 мкл РНАзина.
- Поверните микротрубку головой над хвостом при 4 °C в течение 1 ч.
- Гранулируйте смесь центрифугированием (1000 х г в качающемся роторе ковша при 4 °C в течение 10-15 с) и собирайте несвязанный материал (U1ΔNE) с помощью шприца Гамильтона.
- Диализуйте весь объем U1ΔNE вместе с отдельной 50-мкл аликвотой исходного неображенного NE(C) в отдельных отсеках микродиализатора, с перемешиванием, в течение 75 мин против 60% D с использованием диализной мембраны с отсечкой молекулярной массы 8 К.
- Сразу после диализа разделить эти препараты (U1ΔNE и NE в 60% D) на 20 мкл аликвот; затем заморозить в ванне с сухим льдом/этанолом и хранить при -80 °C.
- Анализ содержания РНК и белка в U1ΔNE и материала, связанного на бусинах против U1
- После удаления несвязанного материала (U1ΔNE) (этап 2.2.4) промыть материал, связанный с шариками против U1, добавив 0,5 мл буфера промывки TX. Гранулируйте смесь центрифугированием (1000 х г в качающемся роторе ковша при 4 °C в течение 10-15 с) и удаляйте супернатант с помощью микропипеттера и выбрасывайте.
- Повторите шаги стирки 2.3.1 дважды.
- Удалите материал, связанный с шариками против U1, добавив 100 мкл 2x буфера образцов SDS к 100 мкл шариков и инкубируя в течение 10 минут при комнатной температуре.
- Гранулирование смеси центрифугированием (1 000 х г в качающемся роторе ковша при 4 °С в течение 10-15 с); удалить супернатант шприцем Гамильтона и заморозить в ванне с сухим льдом / этанолом. Хранить при температуре -80 °C.
- Сравните неразрушенный NE, истощенный NE (U1ΔNE) и материал, связанный с шариками (удаленный из бусин буфером образцов SDS, как описано в шагах 2.3.3 и 2.3.4 выше). Выполните шаги 2.3.6-2.3.8 для анализа РНК или шаги 2.3.9-2.3.10 для анализа белка.
- Для каждого образца экстрагируют РНК с 200 мкл фенол-хлороформа (50:50, v/v); затем снова экстрагировать 180 мкл хлороформ-изоамилового спирта (25:1, v/v). После экстракции добавляют 300 мкл холодного 200-стойкого этанола, инвертируют для смешивания и хранят осажденную РНК в течение ночи при -20 °C.
- Центрифугируют осажденную этанолом РНК (12 000 х г в течение 10 мин при 4 °C). Промыть гранулы 150 мкл холодного 70% этанола. Центрифуга снова (12 000 х г) при 4 °C в течение 15 мин. Удалите супернатант с помощью микропипеттера и высушите гранулы в вакууме со скоростью в течение 10-15 мин без нагрева.
- Повторно суспендируют высушенную РНК-гранулу в 10 мкл РНК-нагрузочного буфера, осторожно вихрь, нагревают до 75-85 °С в течение 90 с, а затем инкубируют на льду в течение 2 мин. Разделяют snRNAs гелевым электрофорезом (2 ч при 16 мА) через 13% полиакриламид - 8,3 М гелей мочевины и затем либо окрашивают бромидом этидия, либо подвергают северному блоттингу10,16.
- Загрузите образцы белка в буфере образцов SDS со стадии 2.3.5 на 12,5% полиакриламидные гели и электрофорез при 200 В в течение приблизительно 45-50 мин в буфере SDS-PAGE (электрофорез полиакриламидного геля додецилсульфата натрия).
- Перенос отделенных белков на нитроцеллюлозную мембрану при 400 мА в течение 2 ч в трансферном буфере. После переноса блокируют мембрану путем инкубации на ночь в T-TBS, содержащем 10% обезжиренного сухого молока. Затем иммуноблотируют мембрану, чтобы выявить специфические белки8,21.
3. Иммунопреципитация 10S фракций градиентов глицерина анти-Gal3
- Препарат шариков анти-Gal3 для иммуносансорбции
ПРИМЕЧАНИЕ: Происхождение и характеристика поликлональной антисыворотки кролика против Gal3 для кролика #2421 и для кролика #4910 были описаны ранее.- Используйте преиммунную сыворотку от кролика No49 в качестве контроля.
- Для приготовления бусин анти-Gal3 следуйте процедуре, ранее описанной для приготовления бусин anti-U1 (этап 2.1), за исключением того, что, соответствующее шагу 2.1.3, отношение антисыворотки (например, анти-Gal3, #49) к бусинам составляет 3:1.
- Непосредственно перед использованием промывайте бусины, связанные с антителами, в дальнейшем обозначаемые как бусины против Gal3, дважды с 0,5 мл буфера промывки TX. Удалите супернатант, сначала с помощью микропипеттера, чтобы получить большую часть жидкости, а затем с помощью шприца Гамильтона, чтобы получить жидкость из шариков; отбрасывать.
- Иммунопреципитатон фракций градиента глицерина анти-Gal3 (см. Рисунок 1B)
- Фракционировать NE(D) более 12%-32% градиента глицерина10. Смешайте и смешайте фракции градиента глицерина 3, 4 и 5 (пронумерованные из верхней части градиента), которые находятся вблизи области 10S градиента.
- Подготовьте два образца, каждый со 150 мкл аликвотой комбинированных фракций градиента 3-5 (стадия 3.2.1), и поместите в 50 мкл шариков анти-Gal3.
- Параллельно готовят два образца по 150 мкл фракции 1 (содержащие Gal3, не в комплексе с U1 snRNP10; стадия 3.2.1) и помещают в 50 мкл бусины против Gal3.
- В качестве контроля поместите 150 мкл 60% D в другую микропробирку из 50 мкл анти-Gal3 шариков.
- Осторожно перемешайте, постукивая по трубке, затем поверните микротрубку головой над хвостом при 4°C в течение 1 ч.
- Гранулирование смеси путем щадящей центрифугации (1 000 х г в качающемся роторе ковша при 4 °C в течение 10-15 с).
- Удалите супернатант (несвязанный материал) с помощью шприца Гамильтона. Не мойте шарики и используйте сразу для добавления реакций сращивания (раздел 4.2).
- Анализ содержания РНК и белка в несвязанном и связанном материале из осаждения анти-Gal3 фракций градиента 10S
- Для анализа компонентов связанного и несвязанного материала из осаждения анти-Gal3 фракций градиента 10S собирают несвязанный материал (супернатант после стадии 3.2.6), переносят в свежую микропробирку и замораживают при -20 °C.
- Промыть осажденные шарики с этапа 3.2.6 (содержащего материал, связанный с анти-Gal3), добавив 0,5 мл буфера промывки TX.
- Гранулирование смеси путем щадящего центрифугирования (1 000 х г в качающемся роторе ковша при 4 °C в течение 10-15 с); удалить супернатант с помощью микропипеттера и выбросить. Повторите шаги стирки еще дважды.
- Добавьте 50 мкл буфера образцов 2X SDS в промытые и гранулированные шарики против Gal3.
- Аккуратно перемешайте шарики и высиживайте в течение 10 минут при комнатной температуре.
- Гранулируйте смесь щадящим центрифугированием (1000 х г в качающемся роторе ведра при 4 °C в течение 10-15 с), соберите супернатант шприцем Гамильтона и храните в свежей микротрубке при -20 °C.
- Сравните несвязанный материал (раздел 3.3.1) и связанный материал (этап 3.3.6) осаждения анти-Gal3 в терминах РНК и белковых компонентов, используя процедуры, описанные в шагах 2.3.6. до пункта 2.3.10, соответственно.
4. Сборка реакции сращивания и анализ продуктов
- Подготовка сращивающей подложки
ПРИМЕЧАНИЕ: Субстрат пре-мРНК, обозначенный MINX, содержит две последовательности экзонов и одну интронную последовательность из Аденовируса22. Последовательность ДНК MINX в плазмиде находится под контролем промоторов РНК-полимеразы T3, T7 или SP6. Материалы и подробные способы линеаризации плазмидной ДНК MINX эндонуклеазой рестрикции BamHI, транскрипции полимеразой РНК SP6 в присутствии α-32P[GTP] и очистки меченой 32P MINX для сплайсинговых анализов описаны ранее19 (см. шаги 2.2 и 3.2 этой ссылки).- Хранить радиомеченный MINX в виде этанолового осадка при -20 °C; использовать меченый сращивающий субстрат в течение 4-6 недель после транскрипции.
- Непосредственно перед использованием центрифугируйте осажденный этанолом 32P-меченый MINX при 12 000 х г в течение 10 мин при 4 °C; удалить супернатант с помощью микропипеттера и выбросить.
- Добавьте 150 мкл 70% этанола и центрифугу при 12 000 х г в течение 15 мин при 4 °C. Выбросьте супернатант и высушите гранулы в вакууме без нагрева в течение 15 мин.
- Регидратируйте гранулы в 50 мкл воды DEPC. Пятно 2 мкл на каждом из двух фильтров GF/C; погрузить фильтры в холодную 5% трихлоруксусную кислоту (ТЦА) на 10 мин. Промыть холодным 5% TCA, а затем 180-стойким этанолом на вакуумной колбе. Воздух сушит фильтры и подвергает сцинтилляционному подсчету в 4 мл Safety-Solve.
- Разбавляйте 32P-меченый MINX в 60% D до 104 cpm/μL для анализа сплайсинга.
- Сборка реакции сращивания (см. рисунок 1С)
- Собирают на льду реакции сращивания в общем объеме 24 мкл (8 мкл U1ΔNE (со стадии 2.2.6), 3,5 мМ MgCl2, 1,5 мМ АТФ, 20 мМ креатинфосфата, 0,5 мМ DTT, 20 единиц РНКазина, 4 мкл 32P-меченого сварочного субстрата MINX (104 cpm/μL), 60% D) и добавляют в каждую трубку шарики из секции 3.2.7. Соберите идентичный набор реакций сплайсинга в общем объеме 24 мкл, но без U1ΔNE, и добавьте в каждую трубку шарики из стадии 3.2.7.
- Готовят контрольную реакцию сплайсинга в общем объеме 12 мкл (4 мкл NE(D), 3,5 мМ MgCl2, 1,5 мМ АТФ, 20 мМ креатинфосфата, 0,5 мМ DTT, 20 единиц РНКазина, 2 мкл 32P-меченого сращивающего субстрата MINX (104 ч/мкл), 60% D).
- Осторожно перемешайте трубки, постукивая и поворачивая торец над хвостом при 30°C в течение 90 минут. Гранулируйте смесь щадящей центрифугированием при 1000 х г в качающемся роторе ковша при 4 °С в течение 10-15 с.
- Остановите реакцию и элюируйте белки из шариков, добавив 24 мкл 2x буфера образцов SDS к пробиркам, содержащим шарики, и 12 мкл 2x буфера образцов SDS в контрольную трубку, содержащую NE, но без шариков. Нагревайте трубки при 100 °C в течение 7 мин.
- Осторожно центрифугируйте трубы при 1000 х г в качающемся роторе ковша при 4 °C в течение 10-15 с.
- Перенос супернатантов (элюций) в свежие микротрубки: приблизительно 48 мкл из шариковых пробирок и 24 мкл из контрольной трубки NE.
- Добавьте протеиназу K (20 мг/мл) для переваривания и солюбилизации белков: добавьте 5 мкл к элюированию 48 мкл из шариков и добавьте 2,5 мкл к контроле NE 24 мкл.
- Инкубировать пробирки при 37°C в течение 40 мин.
- Осторожно центрифугируйте трубы при 1 000 х г в качающемся роторе ковша при 4 °C в течение 10 с.
- Разбавить шариковые элюи с 39,5 мкл TE и 10 мкл 3 М ацетата натрия. Разбавить контроль NE 63,5 мкл TE и 10 мкл 3 М ацетата натрия.
- Извлеките и проанализируйте РНК, как описано ниже (раздел 4.3).
- Анализ продуктов реакции сращивания
- Экстрагировать РНК в каждом образце фенол-хлороформом, а затем хлороформом-изоамильным спиртом; осаждение РНК этанолом, центрифуга, промывка гранул, удаление супернатанта и сушка гранул в соответствии с той же процедурой, которая описана на этапах 2.3.6 и 2.3.7.
- Повторно суспендируют высушенную РНК-гранулу в 10 мкл РНК-нагрузочного буфера, осторожно вихрь, нагревают до 75-85 °С в течение 90 с, а затем инкубируют на льду в течение 2 мин.
- Готовят 20 мл раствора, содержащего 13% полиакриламид (бисакриламид:акриламид, 1,9:50 [мас./мас.]) в 8,3 М мочевины; литые гели длиной 15 см с использованием этого раствора.
- После того, как гель отлит, электрофоризируйте его (без загрузки образцов) при 400 В в течение 20 мин, используя TBE в качестве рабочего буфера. После этого шага промывайте скважины с помощью рабочего буфера TBE.
- Загрузите образцы РНК в буфер загрузки РНК и электрофорез с работающим буфером TBE при 400 В в течение 3,5-4 ч. После электрофореза удаляют мочевину, погружая и вращая гель в дистиллированную воду в течение 10 мин.
- Вакуумная сушка геля на фильтровальной бумаге 3 M, сначала в течение 2 ч 15 мин при 80 °C, а затем в течение 30 мин без нагрева, чтобы медленно охладить его. Подвергнуте высушенный гель ауторадиографии на пленке для выявления положений миграции радиоактивных компонентов.
Access restricted. Please log in or start a trial to view this content.
Результаты
NE, обедненные комплексами U1 snRNP (U1ΔNE из раздела 2.2.6) и Gal3 - U1 snRNP из области 10S градиента глицерина, иммунопреципитированного анти-Gal3 (стадия 3.2.7), смешивали в реакции сплайсинга. Эта реакционная смесь содержала u1 snRNA (рисунок 2A, полоса 3), а также U1-специфичес?...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
В этом отчете представлены экспериментальные детали, которые документируют комплекс Gal3 - U1 snRNP, захваченный на бусинах, покрытых анти-Gal3, может связываться с субстратом пре-мРНК, и этот троичный комплекс может восстанавливать активность сплайсинга к обедненному U1 snRNP NE. Gal3 является одни?...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Эта работа была поддержана грантом Национального научного фонда MCB-0092919 и грантом Мичиганского государственного университета 09-CDFP-2001 (для RJP) и грантом Национальных институтов здравоохранения GM-38740 и Мичиганским agBioResearch Project MICL02455 (для JLW).
Субстрат MINX пре-мРНК, используемый в анализах сплайсинга, был добрым подарком от доктора Сьюзан Бергет (Медицинский колледж Бейлора, Хьюстон, Техас, США).
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
Ссылки
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932(1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены