
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Complémentation de l’activité d’épissage par un complexe Galectin-3 - U1 snRNP sur perles
Dans cet article
Résumé
Cet article décrit les procédures expérimentales pour (a) l’épuisement du PNSNS U1 à partir d’extraits nucléaires, avec perte concomitante de l’activité d’épissage; et b) reconstitution de l’activité d’épissage dans l’extrait appauvri en U1 par des particules de galectine-3 -U1 snRNP liées à des billes couplées de manière covalente à des anticorps anti-galectine-3.
Résumé
Les expériences classiques d’épuisement-reconstitution indiquent que la galectine-3 est un facteur d’épissage requis dans les extraits nucléaires. Le mécanisme d’incorporation de la galectine-3 dans la voie d’épissage est abordé dans cet article. La sédimentation d’extraits nucléaires de cellules HeLa sur des gradients de glycérol de 12% à 32% donne des fractions enrichies en une particule endogène ~10S qui contient de la galectine-3 et du snRNP U1. Nous décrivons maintenant un protocole visant à épuiser les extraits nucléaires de SnRNP U1 avec perte concomitante d’activité d’épissage. L’activité d’épissage dans l’extrait appauvri en U1 peut être reconstituée par la particule de galectine-3 - U1 snRNP piégée sur des billes d’agarose couplées de manière covalente à des anticorps anti-galectine-3. Les résultats indiquent que le complexe ternaire pré-ARNm galectine-3 - U1 snRNP - est un complexe fonctionnel E conduisant à des intermédiaires et des produits de la réaction d’épissage et que la galectine-3 pénètre dans la voie d’épissage par son association avec U1 snRNP. Le schéma consistant à utiliser des complexes d’affinité ou d’immuno-sélection sur des billes pour reconstituer l’activité d’épissage dans des extraits appauvris d’un facteur d’épissage spécifique peut être généralement applicable à d’autres systèmes.
Introduction
La production de la plupart des ARN messagers eucaryotes (ARNm) implique l’élimination des introns et la ligature des exons dans un processus nucléaire appelé épissage pré-ARNm1. Deux classes de complexes ARN-protéine (RNP) dirigent le traitement de l’ARN pré-messager en ARNm mature via des complexes splicéosomiques. Une classe, les RNP pré-messagers naissants, est formée co-transcriptionnellement par la liaison de protéines RNP nucléaires hétérogènes et d’autres protéines de liaison à l’ARN, y compris certains membres de la famille SR, produisant des complexes hnRNP2. Les petits RNP nucléaires de deuxième classe, riches en uraciles (SNNP U avec des SNRNA U1, U2, U4, U5 et U6) sont associés à des protéines spécifiques à l’U et au noyau3,4. Les SNNP U interagissent de manière ordonnée avec des régions spécifiques des RNP pré-messagers dans une voie de remodelage dynamique à mesure que les introns sont excisés et que les exons sont ligaturés pour produire des ARNm matures5. De nombreuses protéines nucléaires supplémentaires participent à ces événements de traitement6.
La galectine-1 (Gal1) et la galectine-3 (Gal3) sont deux protéines qui sont des facteurs requis dans la voie d’épissage, comme le montrent les études d’épuisement-reconstitution7,8. L’élimination des deux galectines de l’épissage d’extraits nucléaires compétents (NE) abolit l’assemblage et l’épissage des épissages à un stade précoce. L’ajout de l’une ou l’autre galectine à un NE doublement appauvri restaure les deux activités. Gal1 et Gal3 sont des composants des spliceosomes actifs, comme en témoigne l’immunoprécipitation spécifique du pré-ARNm, des intermédiaires d’épissage et de l’ARNm mature par antisérum spécifique pour Gal1 ou Gal39. Il est important de noter que Gal3 s’associe à des particules endogènes contenant de l’ARN U dans le NE en dehors de la voie d’épissage, comme le montre la précipitation des snRNP par l’antisérum anti-Gal310. Enfin, le silence de Gal3 dans les cellules HeLa modifie les schémas d’épissage de nombreux gènes11.
Dans le NE pré-incubé pour désassembler les spliceosomes préformés12, les snRNP se retrouvent dans de multiples complexes sédimentant dans des gradients de glycérol de 7S à plus de 60S. Bien que le fractionnement du gradient de glycérol soit une technique courante pour l’isolement des complexes et composants splicéosomiques (voir références13,14,15 par exemple), nous avons étendu cette méthode en caractérisant des fractions spécifiques à l’aide d’immunoprécipitations d’anticorps. Un snRNP sédimentant à 10S ne contient que de l’ARNS U1 avec Gal3. L’immunoprécipitation de la fraction 10S avec des antisérums spécifiques pour Gal3 ou U1 snRNP co-précipite à la fois U1 et Gal3 indiquant que certaines des monoparticules SnRNP U1 sont liées à Gal310. Comme U1 snRNP est le premier complexe qui se lie au pré-mRNP dans l’assemblage splicéosomal1,5, cette étape représente un site d’entrée potentiel pour Gal3 dans la voie d’épissage. Sur cette base, nous avons montré que les monoparticules de snRNP 10S Gal3-U1 liées à des perles contenant des anti-Gal3 ont restauré l’activité d’épissage à un NE appauvri en U1 snRNP, établissant ce complexe comme un mécanisme par lequel Gal3 est recruté dans la voie splicéosomale16. Cela contraste avec les tentatives d’isoler les spliceosomes à des stades spécifiques de la réaction d’épissage et de catalogage des facteurs associés17,18. Dans de telles études, la présence de certains facteurs à un moment donné est déterminée, mais pas le mécanisme par lequel ils ont été chargés.
Nous avions précédemment décrit en détail la préparation du NE, le substrat d’épissage, l’assemblage du mélange réactionnel d’épissage et l’analyse des produits dans notre documentation sur le rôle des galectines dans l’épissage pré-ARNm19. Nous décrivons maintenant les procédures expérimentales de fractionnement d’extraits nucléaires pour obtenir une fraction enrichie en complexe snRNP Gal3 - U1 et pour l’immuno-sélection de ce dernier complexe afin de reconstituer l’activité d’épissage dans un extrait nucléaire appauvri en U1.

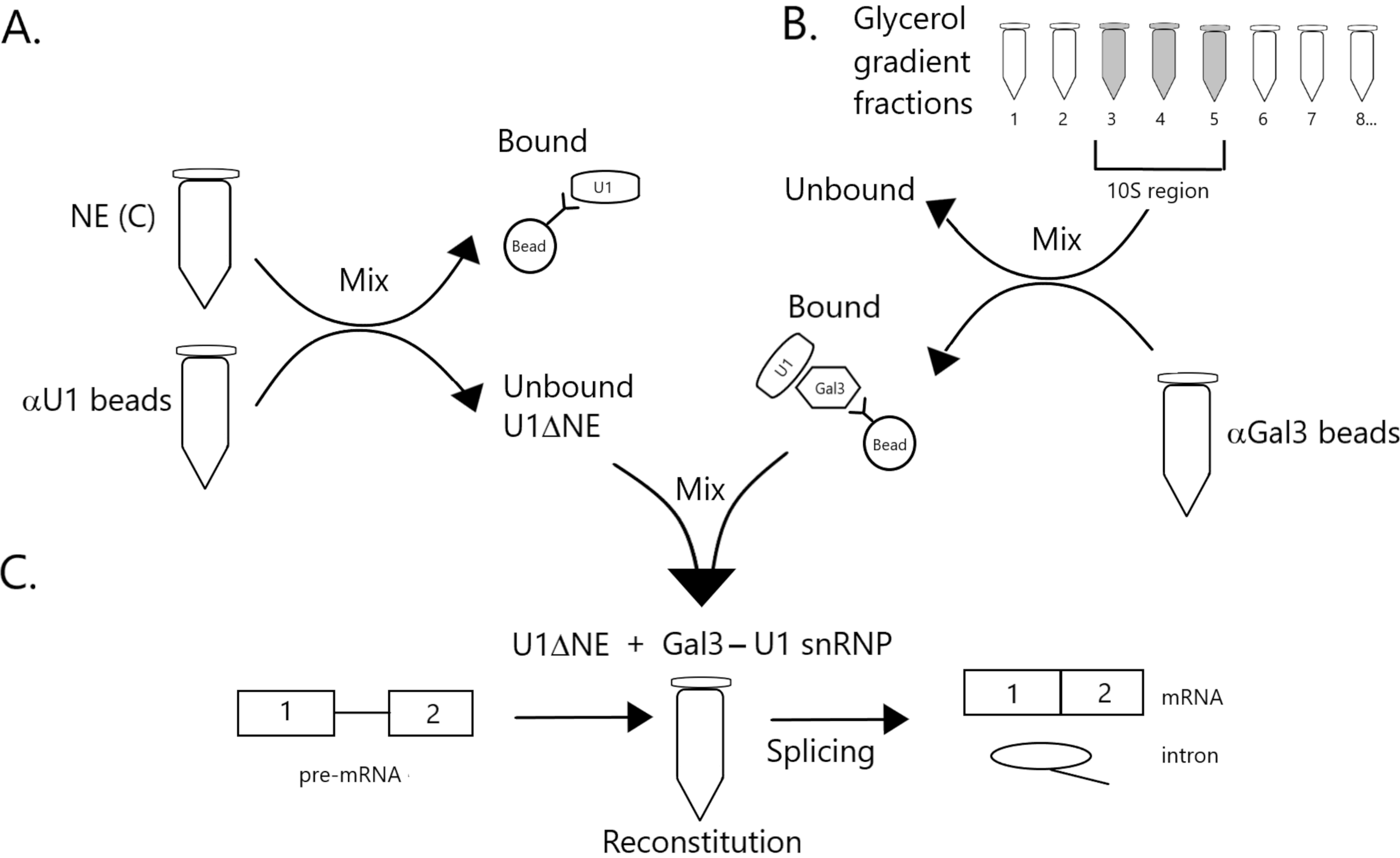
Figure 1 : Schéma illustrant la complémentarité de l’activité d’épissage dans l’extrait nucléaire appauvri en U1 snRNP par un complexe snRNP Gal3-U1 sur des billes. (A) NE dans le tampon C (NE(C)) est incubé avec des billes de protéine A-sépharose couplées de manière covalente avec des billes anti-U1 snRNP (αU1). La fraction non liée est appauvrie en U1 snRNP (U1ΔNE). (B) LE NE dans le tampon D (NE(D)) est fractionné sur un gradient de glycérol de 12 % à 32 % par ultracentrifugation. Les fractions correspondant à la région 10S (fractions 3-5) sont combinées et mélangées avec des billes couplées de manière covalente avec des anticorps anti-Gal3 (billes αGal3). Le matériau lié aux perles contient une monoparticule Gal3-U1 snRNP. (C) Le complexe snRNP Gal3-U1 de la partie (B) est mélangé avec U1ΔNE de la partie (A) dans un essai d’épissage utilisant un substrat pré-ARNm MINX marqué 32P et les intermédiaires et produits de la réaction d’épissage sont analysés par électrophorèse sur gel et autoradiographie. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
1. Notes sur les procédures générales
- S’assurer que tous les produits chimiques (composants tampons, enzymes, etc.) sont exempts de ribonucléase (RNase). Séquestrer tous les flacons de réactif achetés dans le commerce à partir d’une utilisation générale en laboratoire. Portez des gants pour toutes les étapes de la procédure expérimentale. N’utilisez que de la verrerie et des ustensiles cuits au four (voir l’étape 1.2 ci-dessous) et des solutions prétraitées (voir l’étape 1.3 ci-dessous).
- Cuire toute la verrerie (béchers, flacons, bouteilles, pipettes, etc.) pendant au moins 4 h à 177 °C. Envelopper les autres ustensiles (spatules, agitateurs, etc.) dans du papier d’aluminium avant de cuire dans les mêmes conditions.
- Préparer une solution à 0,1 % (vol/vol) de diéthylpyrocarbonate (DEPC) dans de l’eau double distillée (ddH2O). À l’aide d’une barre d’agitation magnétique, remuez cette solution pendant la nuit, puis autoclavez. Utilisez ce H2O traité au DEPC pour fabriquer toutes les solutions contenant du Tris; ensuite, filtrer stériliser à l’aide d’un filtre à vide à bouchon de bouteille. Utilisez régulièrement du ddH2O pour préparer toutes les autres solutions (sans Tris); ensuite, traiter avec du DEPC (0,1%, vol/vol) et un autoclave.
REMARQUE : Les tampons utilisés dans l’ensemble suivant de procédures expérimentales sont énumérés par ordre alphabétique dans le tableau 1.
| Nom de la mémoire tampon | Composition |
| Tampon de borate | Borate de sodium 0,2 M, pH 9 |
| Tampon C | 20 mM HEPES, pH 7,9, 25 % (vol/vol) de glycérol, 0,42 M NaCl, 1,5 mM MgCl2, 0,2 mM EDTA, 0,5 mM de fluorure de phénylméthylsulfonyle (PMSF), 0,5 mM de dithiothréitol (TNT) |
| Tampon D | 10 mM HEPES, pH 7,9, 20 % (vol/vol) de glycérol, 0,1 M KCl, 0,2 mM EDTA, 0,5 mM PMSF, 0,5 mM TNT |
| 60 %D | 60% De tampon D et 40% de H2O |
| Éthanolamine | 0,2 M éthanolamine, pH 8 |
| Tampon de liaison HEPES | 20 mM HEPES, pH 7,9 |
| Tampon de lavage HEPES | 20 mM HEPES, pH 7,9, 0,5 M NaCl |
| Tampon de chargement d’ARN | Formamide à 90 %, EDTA à 20 mM, pH 8, 0,05 % (p/v) de bleu de bromophénol |
| Tampon SDS-PAGE | 25 mM de Tris, 169 mM de glycine, 0,1 % de dodécylsulfate de sodium (FDS), pH 8,8 |
| Exemple de tampon SDS | 62,5 mM Tris, pH 6,8, 2 % SDS, 10 % glycérol, 5 % 2-mercaptoéthanol, 0,1 % (p/v) de bleu de bromophénol |
| Tampon TBE pour gels d’ARN | 89 mM Tris, 89 mM d’acide borique, 2,5 mM d’EDTA, pH 8,3 |
| Tampon TE | 10 mM Tris, pH 8, 1 mM EDTA |
| Tampon de transfert | 25 mM de Tris, 1,92 M de glycine, 20 % de méthanol, pH 8,3 |
| Tampon T-TBS | 10 mM Tris, 0,5 M NaCl, 0,05 % Tween 20, pH 7,5 |
| Tampon de lavage TX | 0,05 % Triton X-100 (TX) en 60 %D |
Tableau 1 : Nom et composition des tampons
2. Préparation du NE appauvri en U1 snRNP (U1 ΔNE)
- Préparation de billes anti-U1 pour l’immunoadsorption
- Pré-gonfler 50 mg de perles de protéine A-sépharose CL-4B en excès de H2O traité par DEPC pour produire environ 200 μL de billes gonflées, puis laver dans un tampon de lavage HEPES.
- Pour ce lavage et tous les lavages ultérieurs, abreuver les billes par centrifugation (1 000 x g dans un rotor de godet oscillant à 4 °C pendant 10-15 s) et retirer le lavage non lié à l’aide d’un micropipetteur et jeter.
- Mélanger 150 μL de billes lavées avec 150 μL de sérum auto-immun humain spécifique pour U1 snRNP (volume d’anticorps par rapport au volume de perles dans un rapport de 1:1).
- Ajuster, sur la base du volume total de (~300 μL à partir de l’étape 2.1.3 ci-dessus), le mélange à 20 mM HEPES, pH 7,9, correspondant aux conditions du tampon de liaison HEPES; incuber ce mélange avec un balancement continu à température ambiante pendant 60 min.
- Laver les billes liées avec des anticorps avec 1 mL de tampon de borate (0,2 M de borate de sodium, pH 9) et remettre en suspension dans 1 mL du même tampon de borate.
- Pour coupler de manière covalente l’anticorps lié aux billes de protéine A-sépharose, ajouter le diméthylpimélimidate à une concentration finale de 20 mM et incuber à température ambiante avec un basculement pendant 60 min.
- Laver les billes avec 1 mL de tampon de borate.
- Pour bloquer tout réactif de réticulation non réagi, ajouter 1 mL d’éthanolamine de 0,2 M (pH 8) et incuber à température ambiante avec bascule pendant 60 min.
- Laver les billes couplées aux anticorps, ci-après désignées comme perles anti-U1, deux fois avec 0,5 mL de tampon de lavage TX (0,05 % triton X-100 dans 60 % D).
- Épuisement du PNNSN SNN U1 du NE (voir figure 1A)
REMARQUE: La procédure de préparation du NE à partir de cellules HeLa a été initialement développée par Dignam et al.20. Nous avons décrit les matériaux et les méthodes détaillées de préparation du NE pour les essais d’épissage19 (voir les étapes 2.1 et 3.1 de cette référence). NE, tel que préparé initialement, est dans le tampon C et sera désigné ci-après comme NE(C). NE(C) dialysé contre et équilibré avec le tampon D sera désigné comme NE(D).- Incuber 200 μL de NE(C) avec 100 μL de billes anti-U1 de l’étape 2.1.9 ci-dessus.
- Ajouter 5 μL de RNasin au mélange.
- Faire pivoter le microtube tête-dessus queue à 4 °C pendant 1 h.
- Pastuler le mélange par centrifugation (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s) et recueillir le matériau non lié (U1ΔNE) à l’aide d’une seringue Hamilton.
- Dialysez le volume entier d’U1ΔNE, ainsi qu’une aliquote séparée de 50 μL du NE(C) non épuisé d’origine, dans des compartiments séparés d’un microdialyseur, sous agitation, pendant 75 min contre 60% D à l’aide d’une membrane de dialyse avec une coupure de poids moléculaire de 8 K.
- Immédiatement après la dialyse, diviser ces préparations (U1ΔNE et NE dans 60% D) en aliquotes de 20 μL; puis congeler dans un bain de glace carbonique/éthanol et conserver à -80 °C.
- Analyse de la teneur en ARN et en protéines de l’U1ΔNE et du matériel lié sur les billes anti-U1
- Après le retrait du matériau non lié (U1ΔNE) (étape 2.2.4), lavez le matériau lié aux billes anti-U1 en ajoutant 0,5 mL de tampon de lavage TX. Mettez le mélange en granulés par centrifugation (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10 à 15 s) et retirez le surnageant à l’aide d’un micropipetteur et jetez-le.
- Répétez deux fois les étapes de lavage 2.3.1.
- Retirez le matériau lié aux billes anti-U1 en ajoutant 100 μL de tampon d’échantillon SDS 2x à 100 μL des billes et en incubant pendant 10 min à température ambiante.
- Pastillez le mélange par centrifugation (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s); retirer le surnageant à l’aide de la seringue Hamilton et le congeler dans un bain de glace carbonique et d’éthanol. Conserver à -80 °C.
- Comparez le NE non épuisé, le NE appauvri (U1ΔNE) et le matériau lié aux billes (retiré des billes par le tampon d’échantillon SDS comme décrit aux étapes 2.3.3 et 2.3.4 ci-dessus). Suivez les étapes 2.3.6 à 2.3.8 pour l’analyse de l’ARN ou les étapes 2.3.9 à 2.3.10 pour l’analyse des protéines.
- Pour chaque échantillon, extraire l’ARN avec 200 μL de phénol-chloroforme (50:50, v/v); puis extraire à nouveau avec 180 μL d’alcool chloroforme-isoamylique (25:1, v/v). Après l’extraction, ajouter 300 μL d’éthanol froid résistant à 200, inverser pour mélanger et conserver l’ARN précipité pendant la nuit à -20 °C.
- Centrifuger l’ARN précipité à l’éthanol (12 000 x g pendant 10 min à 4 °C). Laver les granulés avec 150 μL d’éthanol froid à 70 %. Centrifuger à nouveau (12 000 x g) à 4 °C pendant 15 min. Retirez le surnageant à l’aide d’un micropipeteur et séchez les granulés dans un aspirateur de vitesse pendant 10 à 15 minutes sans chaleur.
- Remettre en suspension la pastille d’ARN séchée dans 10 μL de tampon de chargement d’ARN, tourbillonner doucement, chauffer à 75-85 °C pendant 90 s, puis incuber sur de la glace pendant 2 min. Séparer les SNRNA par électrophorèse sur gel (2 h à 16 mA) à l’aide de gels d’urée à 13 % de polyacrylamide - 8,3 M, puis soit les tacher avec du bromure d’éthidium, soit les soumettre à un bâchage nordique10,16.
- Chargez les échantillons de protéines, dans le tampon d’échantillon SDS de l’étape 2.3.5, sur des gels de polyacrylamide à 12,5% et de l’électrophorèse à 200 V pendant environ 45 à 50 minutes dans un tampon SDS-PAGE (électrophorèse sur gel de polyacrylamide de dodécylsulfate de sodium).
- Transférer les protéines séparées dans la membrane de nitrocellulose à 400 mA pendant 2 h dans un tampon de transfert. Après le transfert, bloquez la membrane en incubant pendant la nuit dans du T-TBS contenant 10% de lait sec non gras. Ensuite, immunoblotez la membrane pour révéler des protéines spécifiques8,21.
3. Immunoprécipitation de fractions 10S de gradients de glycérol par anti-Gal3
- Préparation de billes anti-Gal3 pour l’immunoadsorption
NOTE: La dérivation et la caractérisation de l’antisérum polyclonal de lapin contre Gal3 pour le lapin #2421 et pour le lapin #4910 ont été décrites précédemment.- Utilisez le sérum préimmun du lapin #49 comme témoin.
- Pour la préparation des billes anti-Gal3, suivez la procédure décrite précédemment pour la préparation des perles anti-U1 (étape 2.1), à l’exception que correspondant à l’étape 2.1.3, le rapport entre l’antisérum (par exemple, anti-Gal3, #49) et les perles est de 3:1.
- Juste avant utilisation, laver les billes couplées aux anticorps, ci-après désignées comme perles anti-Gal3, deux fois avec 0,5 mL de tampon de lavage TX. Retirez le surnageant, d’abord avec un micropipeteur pour extraire la majeure partie du liquide, puis avec une seringue Hamilton pour extraire le liquide des perles; jeter.
- Immunoprécipitaton des fractions du gradient glycérol par anti-Gal3 (voir Figure 1B)
- Fractionner NE(D) sur un gradient de glycérol de 12 % à 32 %10. Combinez et mélangez les fractions de gradient de glycérol 3, 4 et 5 (numérotées à partir du haut du gradient), qui sont proches de la région 10S du gradient.
- Préparer deux échantillons, chacun avec 150 μL aliquote de fractions de gradient combinées 3-5 (étape 3.2.1), et placer dans 50 μL de billes anti-Gal3.
- En parallèle, préparer deux échantillons contenant chacun 150 μL de fraction 1 (contenant Du Gal3 non en complexe avec U1 snRNP10 ; étape 3.2.1) et placer dans 50 μL de billes anti-Gal3.
- Comme témoin, placer 150 μL de 60% D dans un autre microtube de 50 μL de billes anti-Gal3.
- Mélanger doucement en tapotant le tube, puis faire pivoter le microtube tête-dessus queue à 4°C pendant 1 h.
- Pastillez le mélange par centrifugation douce (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s).
- Retirez le surnageant (matériau non lié) à l’aide d’une seringue Hamilton. Ne pas laver les billes et utiliser immédiatement pour l’ajout des réactions d’épissage (rubrique 4.2).
- Analyse de la teneur en ARN et en protéines dans le matériau non lié et lié à partir de la précipitation anti-Gal3 des fractions de gradient 10S
- Pour l’analyse des composants du matériau lié et non lié à partir de la précipitation anti-Gal3 des fractions de gradient 10S, recueillir le matériau non lié (surnageant après l’étape 3.2.6), le transférer dans un microtube frais et congeler à -20 °C.
- Laver les billes précipitées de l’étape 3.2.6 (contenant un matériau lié à l’anti-Gal3) en ajoutant 0,5 mL de tampon de lavage TX.
- Pastillez le mélange par centrifugation douce (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s); retirer le surnageant à l’aide d’un micropipetteur et jeter. Répétez les étapes de lavage deux fois de plus.
- Ajouter 50 μL de tampon d’échantillon SDS 2X aux billes anti-Gal3 lavées et granulées.
- Mélanger doucement les perles et incuber pendant 10 min à température ambiante.
- Pastuler le mélange par centrifugation douce (1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s), recueillir le surnageant par seringue Hamilton et le conserver dans un microtube frais à -20 °C.
- Comparer le matériau non lié (section 3.3.1) et le matériau lié (étape 3.3.6) de la précipitation anti-Gal3 en termes d’ARN et de composants protéiques, en utilisant les procédures décrites à l’étape 2.3.6. à 2.3.10, respectivement.
4. Assemblage de la réaction d’épissage et analyse des produits
- Préparation du substrat d’épissage
REMARQUE: Le substrat pré-ARNm, désigné MINX, contient deux séquences d’exons et une séquence d’introns d’Adénovirus22. La séquence d’ADN MINX dans le plasmide est sous le contrôle de promoteurs de l’ARN polymérase T3, T7 ou SP6. Les matériaux et les méthodes détaillées pour la linéarisation de l’ADN plasmidique MINX avec l’endonucléase de restriction BamHI, la transcription par l’ARN polymérase SP6 en présence de α-32P [GTP] et la purification du MINX marqué 32P pour les essais d’épissage sont décrits précédemment19 (voir les étapes 2.2 et 3.2 de cette référence).- Conserver le MINX radiomarqué sous forme de précipité d’éthanol à -20 °C; utiliser le substrat d’épissage étiqueté dans les 4 à 6 semaines suivant la transcription.
- Juste avant utilisation, centrifuger l’éthanol précipité MINX marqué 32P à 12 000 x g pendant 10 min à 4 °C; retirer le surnageant à l’aide d’un micropipetteur et jeter.
- Ajouter 150 μL d’éthanol à 70 % et centrifuger à 12 000 x g pendant 15 min à 4 °C. Jetez le surnageant et séchez la pastille dans un aspirateur de vitesse sans chaleur pendant 15 min.
- Réhydrater la pastille dans 50 μL d’eau DEPC. Spot 2 μL sur chacun des deux filtres GF/C; immerger les filtres dans de l’acide trichloroacétique (TCA) froid à 5% pendant 10 min. Rincer à froid 5% de TCA, suivi d’éthanol à 180 épreuves sur une fiole sous vide. Sécher les filtres à l’air libre et les soumettre à la scintillation en 4 mL de Safety-Solve.
- Diluer le MINX marqué au 32P dans 60 % D à 104 cpm/μL pour le dosage d’épissage.
- Assemblage de la réaction d’épissage (voir Figure 1C)
- Assembler, sur glace, les réactions d’épissage dans un volume total de 24 μL (8 μL U1ΔNE (à partir de l’étape 2.2.6), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de phosphate de créatine, 0,5 mM DTT, 20 unités RNasin, 4 μL de substrat d’épissage MINX marqué 32P (104 cpm/μL), 60 % D) et ajouter à chaque tube de billes de la section 3.2.7. Assembler un ensemble identique de réactions d’épissage dans un volume total de 24 μL mais sans U1ΔNE et ajouter à chaque tube des billes de l’étape 3.2.7.
- Préparer une réaction d’épissage témoin dans un volume total de 12 μL (4 μL NE(D), 3,5 mM MgCl2, 1,5 mM ATP, 20 mM de phosphate de créatine, 0,5 mM de TNT, 20 unités de RNasin, substrat d’épissage MINX marqué 2 μL 32P (104 cpm/μL), 60 % D).
- Mélanger doucement les tubes en tapotant et faire pivoter l’extrémité sur la queue à 30°C pendant 90 min. Pastillez le mélange par centrifugation douce à 1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10-15 s.
- Arrêtez la réaction et éliminez les protéines des billes en ajoutant 24 μL de tampon d’échantillon SDS 2x aux tubes contenant des billes, et 12 μL de tampon d’échantillon SDS 2x au tube témoin contenant NE mais pas de perles. Chauffer les tubes à 100 °C pendant 7 min.
- Centrifugez doucement les tubes à 1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10 à 15 s.
- Transférer les surnageants (élutions) dans des microtubes frais : environ 48 μL des tubes de billes et 24 μL du tube de contrôle NE.
- Ajouter la protéinase K (20 mg/mL) pour digérer et solubiliser les protéines : ajouter 5 μL à l’élution de 48 μL des billes et ajouter 2,5 μL au témoin NE de 24 μL.
- Incuber les tubes à 37°C pendant 40 min.
- Centrifugez doucement les tubes à 1 000 x g dans un rotor à godet oscillant à 4 °C pendant 10 s.
- Diluer les élutions des billes avec 39,5 μL TE et 10 μL 3 M d’acétate de sodium. Diluer le témoin NE avec 63,5 μL TE et 10 μL 3 M d’acétate de sodium.
- Extraire et analyser l’ARN comme décrit ci-dessous (rubrique 4.3).
- Analyse des produits de la réaction d’épissage
- Extraire les ARN de chaque échantillon avec du phénol-chloroforme, suivi d’alcool chloroforme-isoamylique; précipiter les ARN avec de l’éthanol, centrifuger, laver les granulés, retirer le surnageant et sécher les granulés selon la même procédure que celle décrite aux étapes 2.3.6 et 2.3.7.
- Remettre en suspension la pastille d’ARN séchée dans 10 μL de tampon de chargement d’ARN, tourbillonner doucement, chauffer à 75-85 °C pendant 90 s, puis incuber sur de la glace pendant 2 min.
- Préparer 20 mL d’une solution contenant 13 % de polyacrylamide (bisacrylamide:acrylamide, 1,9:50 [wt/wt]) dans 8,3 M d’urée; gels coulés de 15 cm de longueur en utilisant cette solution.
- Une fois le gel coulé, électrophorèsez-le (sans aucun échantillon chargé) à 400 V pendant 20 min en utilisant TBE comme tampon de fonctionnement. Après cette étape, lavez les puits avec un tampon de fonctionnement TBE.
- Chargez les échantillons d’ARN, dans un tampon de chargement d’ARN, et électrophorèse avec tampon de fonctionnement TBE à 400 V pendant 3,5 à 4 h. Après électrophorèse, retirer l’urée en immergeant et en faisant pivoter le gel dans de l’eau distillée pendant 10 min.
- Sécher le gel sous vide sur du papier filtre 3 M, d’abord pendant 2 h 15 min à 80 °C puis pendant 30 min sans chaleur pour le refroidir lentement. Soumettre le gel séché à une autoradiographie sur film pour détecter les positions de migration des composants radioactifs.
Résultats
L’épuisement ne-ne des complexes U1 snRNP (U1ΔNE de la section 2.2.6) et Gal3 - U1 snRNP de la région 10S du gradient glycérol immunoprécipité par anti-Gal3 (étape 3.2.7) a été mélangé dans une réaction d’épissage. Ce mélange réactionnel contenait de l’ARNN U1 (figure 2A, voie 3), ainsi que la protéine spécifique U1, U1-70K (figure 2B, voie 3). Comme prévu, l’anti-Gal3 a précipité Gal3 (
Discussion
Ce rapport fournit les détails expérimentaux qui documentent un complexe snRNP Gal3 - U1 piégé sur des billes enrobées anti-Gal3 peut se lier au substrat pré-ARNm et ce complexe ternaire peut restaurer l’activité d’épissage à un NE appauvri en U1 snRNP. Gal3 est un membre d’une famille de protéines isolées à l’origine sur la base de son activité de liaison aux glucides spécifiques au galactose23 . Les premières études d’immunofluorescence et de fractionnement subcellulair...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail a été soutenu par la subvention MCB-0092919 de la National Science Foundation et la subvention de recherche intra-muros 09-CDFP-2001 de la Michigan State University (à RJP) et par la subvention GM-38740 des National Institutes of Health et le projet MICL02455 du Michigan AgBioResearch (à JLW).
Le substrat pré-ARNm MINX utilisé dans les essais d’épissage était un cadeau aimable du Dr Susan Berget (Baylor College of Medicine, Houston, TX, États-Unis).
matériels
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
Références
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932 (1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -. F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.