
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
半乳糖凝集素-3 - U1 snRNP复合物在珠子上的剪接活性的补充
摘要
本文描述了(a)核提取物中U1 snRNP的消耗,伴随剪接活性的丧失;(b)通过与珠子共价偶联的微乳凝集素-3 - U1 snRNP颗粒与抗半乳糖凝集素-3抗体共价结合,在U1耗尽的提取物中重建剪接活性。
摘要
经典的耗竭重建实验表明,半乳糖凝集素-3是核提取物中必需的剪接因子。本文探讨了半乳糖凝集素-3掺入剪接途径的机制。HeLa细胞核提取物在12%-32%的甘油梯度上沉淀产生富含含有半乳糖凝集素-3和U1 snRNP的内源性〜10S颗粒的馏分。我们现在描述了一种方案,用于消耗U1 snRNP的核提取物,同时伴随剪接活性的丧失。U1耗尽提取物中的剪接活性可以通过捕获在琼脂糖珠上的半乳糖凝集素-3 -U1 snRNP颗粒与抗半乳糖凝集素-3抗体共价偶联来重建。结果表明,半乳糖凝集素-3 - U1 snRNP - 前mRNA三元复合物是导致剪接反应中间体和产物的功能性E复合物,半乳糖凝集素-3通过其与U1 snRNP的结合进入剪接途径。使用在磁珠上选择的复合物亲和力或免疫力来重建特定剪接因子耗尽的提取物中的剪接活性的方案通常可能适用于其他系统。
引言
大多数真核信使RNA(mRNA)的产生涉及在称为前mRNA剪接的核过程中去除内含子和连接外显子1。两类RNA-蛋白质复合物(RNPs)通过剪接体复合物指导前信使RNA加工成成熟的mRNA。一类是新生的前信使RNPs,由异质核RNP蛋白和其他RNA结合蛋白(包括SR家族的一些成员)结合共转录形成,产生hnRNP复合物2。第二类富含尿嘧啶的小核RNPs(U snRNPs与U1,U2,U4,U5和U6 snRNA)与U特异性和核心蛋白相关3,4。U snRNPs以有序的方式与信使前RNP的特定区域在动态重塑途径中相互作用,因为内含子被切除并且外显子被连接以产生成熟的mRNPs5。许多其他核蛋白参与这些处理事件6。
如耗竭-重建研究所示,半乳糖凝集素-1(Gal1)和半乳糖凝集素-3(Gal3)是剪接途径中必需因子的两种蛋白质7,8。从剪接中去除两种半乳糖凝集素,可在早期阶段消除剪接体组装和剪接活性。将任一半凝集素添加到这种双耗竭的NE中可以恢复两种活性。Gal1 和 Gal3 是活性剪接体的组分,其前 mRNA 的特异性免疫沉淀、剪接中间体和成熟 mRNA 通过 Gal1 或 Gal39 特异性抗血清证明了这一点。重要的是,Gal3与剪接途径外的NE中含有的内源性U snRNA结合,如抗Gal3抗血清沉淀snRNPs所示10。最后,HeLa细胞中Gal3的沉默改变了许多基因的剪接模式11。
在预孵育以拆卸预制的剪接体12的NE中,snRNPs存在于从7S到大于60S的甘油梯度沉淀的多个复合物中。虽然甘油梯度分馏是分离剪接体复合物和组分的常用技术(例如参见参考文献13,14,15 ),但我们通过使用抗体免疫沉淀表征特定组分来扩展该方法。在10S下沉淀的snRNP仅含有U1 snRNA和Gal3。10S级分的免疫沉淀与对Gal3或U1 snRNP特异性的抗血清共沉淀U1和Gal3均表明一些U1 snRNP单颗粒与Gal310结合。由于U1 snRNP是第一个与剪接体组装中的前mRNP结合的复合物1,5,因此该步骤代表了Gal3进入剪接途径的潜在入口位点。在此基础上,我们表明与含有抗Gal3的珠子结合的10S Gal3-U1 snRNP单颗粒恢复了U1 snRNP耗尽的NE的剪接活性,将该复合物建立为Gal3被招募到剪接体途径中的一种机制16。这与在剪接反应的特定阶段分离剪接体并编目相关因子的尝试形成鲜明对比17,18。在此类研究中,确定了某些因素在某个时间点的存在,但不确定它们被加载的机制。
我们之前在关于半乳糖凝集素在前mRNA剪接中的作用的文献中详细描述了NE的制备,剪接底物,剪接反应混合物的组装以及产物的分析19。我们现在描述了核提取物的分馏以获得富含Gal3 - U1 snRNP复合物的馏分以及后一种复合物的免疫选择以在U1耗尽的核提取物中重建剪接活性的实验程序。

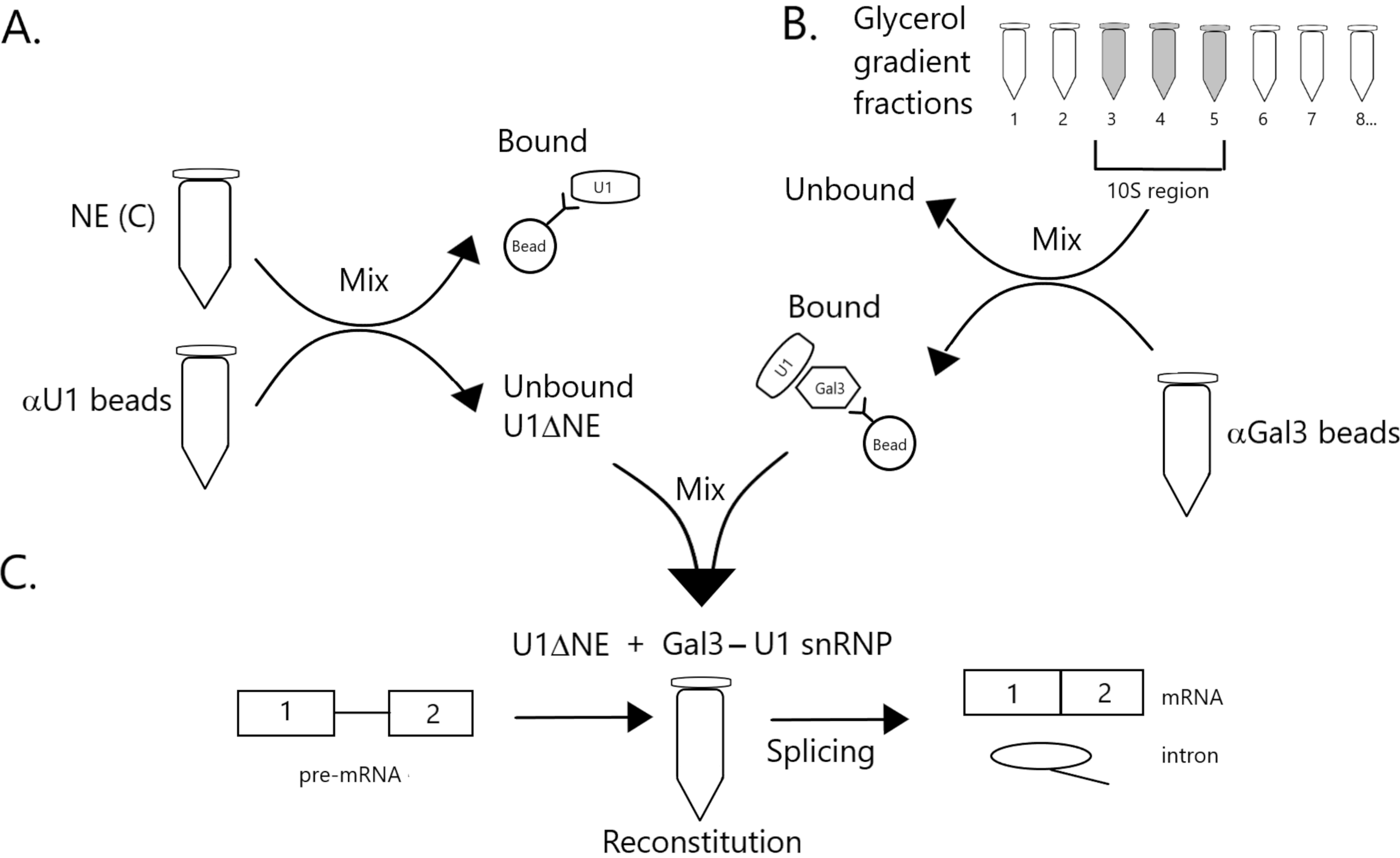
图1:示意图,说明在珠子上用Gal3-U1 snRNP复合物耗尽U1 snRNP的核提取物中剪接活性的互补性。未结合的分数耗尽U1 snRNP(U1ΔNE)。(B)缓冲液D(NE(D))中的NE通过超速在12%-32%的甘油梯度上分馏。将对应于10S区域的馏分(级分3-5)与与抗Gal3抗体(αGal3微球)共价偶联的微球混合。与磁珠结合的材料含有Gal3-U1 snRNP单颗粒。(C)使用32P标记的MINX前mRNA底物在剪接测定中将来自(B)部分的Gal3-U1 snRNP配合物与来自(A)部分的U1ΔNE混合,并通过凝胶电泳和放射自显像分析剪接反应的中间体和产物。请点击此处查看此图的放大版本。
{kind=link}
研究方案
1. 关于一般程序的说明
- 确保所有化学物质(缓冲液成分,酶等)均不含核糖核酸酶(RNase)。隔离所有商业购买的试剂瓶,使其免受一般实验室使用。在实验过程的所有步骤中都要戴上手套。仅使用已经烘烤的玻璃器皿和器皿(见下面的步骤1.2)和经过预处理的溶液(见下面的步骤1.3)。
- 在177°C下烘烤所有玻璃器皿(烧杯,烧瓶,瓶子,移液器等)至少4小时。 在相同条件下烘烤之前,将其他器皿(刮刀,搅拌棒等)包裹在铝箔中。
- 在双蒸馏水(ddH 2 O)中制备0.1%(体积/体积)的焦碳酸二乙酯(DEPC)溶液。使用磁性搅拌棒,将此溶液搅拌过夜,然后高压灭菌。使用这种DEPC处理的H2O来制造所有含有Tris的溶液;然后,使用瓶顶真空过滤器进行过滤灭菌。使用常规的ddH2O制备所有其他溶液(不含Tris);然后,用DEPC(0.1%,体积/体积)和高压灭菌器处理。
注:以下实验步骤中使用的缓冲液按字母顺序列于 表1中。
| 缓冲区的名称 | 组成 |
| 硼酸盐缓冲液 | 0.2 M 硼酸钠,pH 值 9 |
| 缓冲液 C | 20 mM HEPES, pH 7.9, 25% 甘油, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM 苯基甲基磺酰氟 (PMSF), 0.5 mM 二硫代甲状腺醇 (DTT) |
| 缓冲区 D | 10 mM HEPES, pH 7.9, 20% 甘油, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT |
| 60%D | 60% 缓冲液 D 和 40% H2O |
| 乙醇胺 | 0.2 甲醇胺,pH值8 |
| HEPES结合缓冲液 | 20 mM HEPES, pH 7.9 |
| 高效清洗缓冲液 | 20 mM HEPES, pH值 7.9, 0.5 M NaCl |
| RNA 上样缓冲液 | 90% 甲酰胺, 20 mM EDTA, pH 8, 0.05% 溴酚蓝 |
| SDS-PAGE 缓冲区 | 25 mM Tris, 169 mM 甘氨酸, 0.1% 十二烷基硫酸钠 (SDS), pH 8.8 |
| SDS样品缓冲液 | 62.5 mM Tris, pH 6.8, 2% SDS, 10% 甘油, 5% 2-巯基乙醇, 0.1% (w/v) 溴酚蓝 |
| 用于RNA凝胶的TBE缓冲液 | 89 mM Tris, 89 mM 硼酸, 2.5 mM EDTA, pH 8.3 |
| TE 缓冲液 | 10 mM Tris, pH 8, 1 mM 乙二胺四乙酸酯 |
| 转印缓冲液 | 25 mM Tris, 1.92 M 甘氨酸, 20% 甲醇, pH值 8.3 |
| T-TBS 缓冲液 | 10 mM Tris, 0.5 M 氯化钠, 0.05% 吐温 20, pH值 7.5 |
| TX清洗缓冲液 | 0.05% 海卫一 X-100 (TX) 在 60%D |
表 1: 缓冲液的名称和组成
2. U1 snRNP(U1 ΔNE)耗尽的NE的制备
- 免疫吸收用抗U1微球的制备
- 将50毫克蛋白质A-Sepharose CL-4B微球预膨胀在过量的DEPC处理的H2 O中,以产生约200μL肿胀的珠子,然后在HEPES洗涤缓冲液中洗涤。
- 对于该洗涤和所有后续洗涤,通过离心(在4°C下摆动的斗式转子中1,000× g 沉淀10-15 s)沉淀珠子,并使用微量移液器除去未结合的洗涤并丢弃。
- 将150μL洗涤的微球与150μL对U1 snRNP(抗体体积与珠子体积的比例为1:1)特异性的人自身免疫血清混合。
- 调节,在总体积(从上述步骤2.1.3开始的〜300μL)的基础上,将混合物调整为20mM HEPES,pH 7.9,对应于HEPES结合缓冲液的条件;将这种混合物在室温下连续摇动60分钟。
- 用1mL硼酸盐缓冲液(0.2M硼酸钠,pH 9)洗涤与抗体结合的磁珠,并重悬于1mL相同的硼酸盐缓冲液中。
- 为了共价偶联与蛋白A-琼脂糖珠结合的抗体,将二甲基吡美亚胺酸酯加入至终浓度为20mM,并在室温下摇动孵育60分钟。
- 用1 mL硼酸盐缓冲液洗涤珠子。
- 为了阻断任何未反应的交联试剂,加入1mL0.2 M乙醇胺(pH 8),并在室温下摇孵育60分钟。
- 用0.5mL TX洗涤缓冲液(0.05%Triton X-100在60%D中)洗涤抗体偶联微球(以下称为抗U1微球)洗涤两次。
- NE中U1 snRNP的耗尽(见 图1A)
注意:从HeLa细胞制备NE的程序最初是由Dignam等人开发的。我们已经描述了制备用于剪接测定的NE的材料和详细方法19 (参见该参考文献的步骤2.1和3.1)。NE,最初准备在缓冲区C中,此后将被指定为NE(C)。与缓冲液D透析并与缓冲液D平衡的NE(C)将被指定为NE(D)。- 将 200 μL NE(C) 与上述步骤 2.1.9 中的 100 μL 抗 U1 微球孵育。
- 向混合物中加入5μL雷诺辛。
- 将微管头朝下旋转4°C1小时。
- 通过离心(在4°C下摆动桶转子中1,000× g ,持续10-15秒)沉淀混合物,并使用Hamilton注射器收集未结合的材料(U1ΔNE)。
- 使用具有8 K分子量截止值的透析膜,在微透析器的单独隔室中透析整个体积的U1ΔNE以及单独的50μL等分试样的原始未破损NE(C),搅拌75分钟,对60%D。
- 透析后立即将这些制剂(U1ΔNE和NE在60%D中)分成20μL等分试样;然后在干冰/乙醇浴中快速冷冻并储存在-80°C。
- 分析抗U1ΔNE的RNA和蛋白质含量以及抗U1微球上的物质
- 除去未结合材料(U1ΔNE)(步骤2.2.4)后,通过加入0.5mL的TX洗涤缓冲液洗涤与抗U1珠结合的材料。通过离心(在4°C下摆动桶转子中1,000×g10-15秒)沉淀混合物,并使用微量移液器除去上清液并丢弃。
- 重复洗涤步骤2.3.1两次。
- 通过向100μL微珠中加入100μL2x SDS样品缓冲液并在室温下孵育10分钟,除去与抗U1微球结合的材料。
- 通过离心(在4°C下摆动斗式转子中1,000× g )沉淀混合物10-15 s);通过汉密尔顿注射器除去上清液,并在干冰/乙醇浴中快速冷冻。储存在-80°C。
- 比较未消除的NE,耗尽的NE(U1ΔNE)和与磁珠结合的材料(如上述步骤2.3.3和2.3.4所述,通过SDS样品缓冲液从磁珠中除去)。RNA 分析遵循步骤 2.3.6-2.3.8 或蛋白质分析步骤 2.3.9-2.3.10。
- 对于每个样品,用200μL苯酚氯仿(50:50,v / v)提取RNA;然后用180μL氯仿异戊醇(25:1,v / v)再次提取。提取后,加入300μL冷200耐乙醇,倒置混合,并将沉淀的RNA在-20°C下储存过夜。
- 离心乙醇沉淀的RNA(在4°C下12,000× g10 分钟)。用150μL冷70%乙醇洗涤沉淀。再次(12,000× g)在4°C下离心15分钟。使用微量吸塑剂除去上清液,并在高速真空中干燥沉淀10-15分钟,无需加热。
- 将干燥的RNA沉淀重悬于10μLRNA上样缓冲液中,轻轻涡旋,加热至75-85°C90秒,然后在冰上孵育2分钟。通过凝胶电泳(2小时在16 mA下)通过13%聚丙烯酰胺 - 8.3M尿素凝胶分离snRNA,然后用溴化乙锭染色或进行北方印迹10,16。
- 将蛋白质样品从步骤2.3.5开始的SDS样品缓冲液中加载到12.5%聚丙烯酰胺凝胶中,并在SDS-PAGE(十二烷基硫酸钠聚丙烯酰胺凝胶电泳)缓冲液中以200 V电泳约45-50分钟。
- 将分离的蛋白质转移到400mA的硝酸纤维素膜中,在转印缓冲液中2小时。转移后,通过在含有10%脱脂奶粉的T-TBS中孵育过夜来阻断膜。然后,免疫印迹膜以显示特定的蛋白质8,21。
3. 抗Gal3对甘油梯度10S级分的免疫沉淀
- 免疫吸收用抗Gal3微球的制备
注:兔#2421和兔#4910的兔多克隆抗血清对Gal3的推导和表征已经描述过了。- 使用来自兔子#49的免疫前血清作为对照。
- 对于抗Gal3微球的制备,请遵循前面描述的制备抗U1微珠的程序(步骤2.1),除了对应于步骤2.1.3之外,抗血清(例如,抗Gal3,#49)与微珠的比例为3:1。
- 在使用前,用0.5mL TX洗涤缓冲液洗涤抗体偶联微球(以下称为抗Gal3微球)两次。除去上清液,首先用微量移液器将大部分液体取出,然后用Hamilton注射器将液体从珠子中取出;丢弃。
- 抗Gal3对甘油梯度级分的免疫沉淀(见 图1B)
- 在12%-32%的甘油梯度上分馏NE(D)10。组合并混合甘油梯度级分数3,4和5(从梯度顶部开始编号),它们靠近梯度的10S区域。
- 准备两个样品,每个样品用150μL等分试样的组合梯度级分3-5(步骤3.2.1),并置于50μL抗Gal3微球中。
- 同时,用150μL馏分1(含有与U1 snRNP10不复合的Gal3;步骤3.2.1)分别制备两个样品,并置于50μL抗Gal3微球中。
- 作为对照,将150μL60%D置于另一个50μL抗Gal3微珠的微管中。
- 通过敲击管轻轻混合,然后在4°C下将微管头对尾旋转1小时。
- 通过轻柔离心(在4°C下摆动的斗式转子中1,000× g )沉淀混合物10-15秒)。
- 使用汉密尔顿注射器去除上清液(未粘合的材料)。不要清洗珠子,并立即用于添加拼接反应(第4.2节)。
- 分析10S梯度级分抗Gal3沉淀中未结合和结合材料中的RNA和蛋白质含量
- 为了分析从10S梯度级分的反Gal3沉淀中结合和未结合材料的组分,收集未结合的材料(步骤3.2.6之后的上清液),转移到新鲜的微管中,并在-20°C下冷冻。
- 通过加入0.5mL的TX洗涤缓冲液来洗涤步骤3.2.6(含有与抗Gal3结合的材料)中的沉淀珠子。
- 通过温和的离心(在4°C下摆动的斗式转子中1,000× g )沉淀混合物10-15 s);使用微量移液器除去上清液并丢弃。再重复两次洗涤步骤。
- 将50μL2X SDS样品缓冲液加入洗涤和沉淀的抗Gal3微球中。
- 轻轻混合珠子,在室温下孵育10分钟。
- 通过温和的离心(在4°C下摆动的水桶转子中1,000× g ,10-15秒)沉淀混合物,通过Hamilton注射器收集上清液并储存在-20°C的新鲜微管中。
- 使用步骤2.3.6中描述的程序,根据RNA和蛋白质组分比较抗Gal3沉淀的未结合材料(第3.3.1节)和结合材料(步骤3.3.6)。分别为 2.3.10。
4. 拼接反应的组装和产品的分析
- 拼接基板的制备
注:前mRNA底物,指定为MINX,包含两个来自腺病毒的外显子序列和一个内含子序列22。质粒中的MINX DNA序列由T3,T7或SP6 RNA聚合酶启动子控制。前面描述了使用BamHI限制性内切酶线性化MINX质粒DNA的材料和详细方法,在α-32P[GTP]存在下通过SP6 RNA聚合酶转录以及纯化 32P标记的MINX用于剪接测定19 (参见该参考文献的步骤2.2和3.2)。- 将放射性标记的MINX作为乙醇沉淀物储存在-20°C;在转录后4-6周内使用标记的剪接基板。
- 在使用前,在4°C下以 12,000× g 离心沉淀的32P标记的MINX乙醇10分钟;用微量移液器除去上清液并丢弃。
- 加入150μL70%乙醇,并在4°C下以12,000× g 离心15分钟。 弃去上清液,并在高速真空中干燥沉淀,不加热15分钟。
- 将沉淀在50μLDEPC水中重新水合。在两个GF / C过滤器中每个滤点上点2μL;将过滤器浸入冷的5%三氯乙酸(TCA)中10分钟。用冷的5%TCA冲洗,然后在真空瓶上加入180°C的乙醇。空气干燥过滤器,并在4 mL的安全解决方案中进行闪烁计数。
- 将 32P 标记的 MINX 稀释在 60% D 至 104 cpm/μL 中,用于剪接测定。
- 拼接反应的组装(见 图1C)
- 在冰上组装总体积为24μL(8μL U1ΔNE(来自步骤2.2.6),3.5mM MgCl2, 1.5mM ATP,20 mM磷酸肌酸,0.5mM DTT,20单位RNasin,4μL 32P标记的MINX剪接底物(104 cpm / μL),60%D)的剪接反应,并加入到3.2.7部分的每管珠中。组装一组相同的剪接反应,总体积为24μL,但没有U1ΔNE,并从步骤3.2.7中加入到每个珠管中。
- 在12μL总体积(4μL NE(D),3.5mM MgCl2,1.5mM ATP,20mM磷酸肌酸,0.5mM DTT,20单位RNasin,2μL 32P标记的MINX剪接底物(104 cpm / μL),60%D中制备对照剪接反应。
- 轻轻搅拌管子,轻轻敲击,并在30°C下旋转尾部90分钟。通过在4°C下摆动的斗式转子中以1,000× g 的轻轻离心将混合物沉淀10-15 s。
- 停止反应并将蛋白质从磁珠上洗脱出来,方法是向含有磁珠的管中加入24μL2x SDS样品缓冲液,向含有NE但没有微珠的对照管中加入12μL的2x SDS样品缓冲液。将管子在100°C下加热7分钟。
- 在4°C的摆动斗式转子中以1,000 x g 轻轻离心管10-15 s。
- 将上清液(洗脱液)转移到新鲜的微管中:来自微球管约48μL,来自NE控制管约24μL。
- 加入蛋白酶K(20mg / mL)以消化和溶解蛋白质:向从微珠中洗脱的48μL加入5μL,向24μLNE对照中加入2.5μL。
- 将试管在37°C孵育40分钟。
- 在4°C的摆动斗式转子中以1,000 x g 轻轻离心管10 s。
- 用39.5μLTE和10μL3M乙酸钠稀释珠洗脱。用63.5μL TE和10μL3M乙酸钠稀释NE对照。
- 提取和分析RNA,如下所述(第4.3节)。
- 拼接反应产物分析
- 用苯酚 - 氯仿提取每个样品中的RNA,然后提取氯仿 - 异戊醇;用乙醇沉淀RNA,离心,洗涤沉淀,除去上清液并按照步骤2.3.6和2.3.7中描述的相同步骤干燥沉淀。
- 将干燥的RNA沉淀重悬于10μLRNA上样缓冲液中,轻轻涡旋,加热至75-85°C90秒,然后在冰上孵育2分钟。
- 在8.3M尿素中制备20毫升含有13%聚丙烯酰胺(双丙烯酰胺:丙烯酰胺,1.9:50 [重量/重量])的溶液;使用该溶液浇注15厘米长的凝胶。
- 一旦凝胶被浇注,使用TBE作为电泳缓冲液在400 V下电泳(没有任何样品)20分钟。在此步骤之后,用TBE运行缓冲液清洗孔。
- 将RNA样品加载到RNA上样缓冲液中,并用TBE电泳缓冲液在400 V下电泳3.5至4小时。电泳后,通过将凝胶浸入蒸馏水中并旋转10分钟来除去尿素。
- 在3M滤纸上真空干燥凝胶,首先在80°C下2小时15分钟,然后在不加热的情况下30分钟以缓慢冷却。对干燥的凝胶进行胶片自显影检查,以检测放射性成分的迁移位置。
结果
将U1 snRNP(U1ΔNE来自2.2.6节)和来自抗Gal3免疫沉淀的甘油梯度10S区域的Gal3 -U1 snRNP复合物的NE耗尽(步骤3.2.7)在剪接反应中混合。该反应混合物含有U1 snRNA(图2A,泳道3)以及U1特异性蛋白U1-70K(图2B,泳道3)。正如预期的那样,反Gal3沉淀了Gal3(图2B,通道3)。这些组分(U1 snRNA,U1-70K蛋白和Gal3)在免疫?...
讨论
本报告提供了实验细节,记录了被困在抗Gal3包被珠上的Gal3 - U1 snRNP复合物可以与前mRNA底物结合,并且这种三元复合物可以恢复U1 snRNP耗尽的NE的剪接活性。.早期的免疫荧光和亚细胞分馏研究提供了 Gal3 与剪接机制组分关联的初步线索:在核斑点中与 snRNPs 的 Sm 核多肽以及富含丝氨酸和精氨酸的 (SR) 家族的调味因子24,25 进行共定位,...
披露声明
作者没有什么可透露的。
致谢
这项工作得到了美国国家科学基金会拨款MCB-0092919和密歇根州立大学校内研究拨款09-CDFP-2001(到RJP)以及美国国立卫生研究院拨款GM-38740和密歇根AgBioResearch项目MICL02455(到JLW)的支持。
剪接测定中使用的MINX前mRNA底物是Susan Berget博士(美国德克萨斯州休斯顿贝勒医学院)的亲切礼物。
材料
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
参考文献
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932 (1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -. F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。