
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Complementation of Splicing Activity by a Galectin-3 - U1 snRNP Complex on Beads
In This Article
Summary
This article describes the experimental procedures for (a) depletion of U1 snRNP from nuclear extracts, with concomitant loss of splicing activity; and (b) reconstitution of splicing activity in the U1-depleted extract by galectin-3 - U1 snRNP particles bound to beads covalently coupled with anti-galectin-3 antibodies.
Abstract
Classic depletion-reconstitution experiments indicate that galectin-3 is a required splicing factor in nuclear extracts. The mechanism of incorporation of galectin-3 into the splicing pathway is addressed in this paper. Sedimentation of HeLa cell nuclear extracts on 12%-32% glycerol gradients yields fractions enriched in an endogenous ~10S particle that contains galectin-3 and U1 snRNP. We now describe a protocol to deplete nuclear extracts of U1 snRNP with concomitant loss of splicing activity. Splicing activity in the U1-depleted extract can be reconstituted by the galectin-3 - U1 snRNP particle trapped on agarose beads covalently coupled with anti-galectin-3 antibodies. The results indicate that the galectin-3 - U1 snRNP - pre-mRNA ternary complex is a functional E complex leading to intermediates and products of the splicing reaction and that galectin-3 enters the splicing pathway through its association with U1 snRNP. The scheme of using complexes affinity- or immuno-selected on beads to reconstitute splicing activity in extracts depleted of a specific splicing factor may be generally applicable to other systems.
Introduction
Production of most eukaryotic messenger RNAs (mRNAs) involves removal of introns and ligation of exons in a nuclear process termed pre-mRNA splicing1. Two classes of RNA-protein complexes (RNPs) direct the processing of pre-messenger RNA into mature mRNA via spliceosomal complexes. One class, nascent pre-messenger RNPs, is formed co-transcriptionally by the binding of heterogeneous nuclear RNP proteins and other RNA-binding proteins, including some members of the SR family, yielding hnRNP complexes2. The second class, uracil-rich small nuclear RNPs (U snRNPs with U1, U2, U4, U5, and U6 snRNAs) is associated with U-specific and core proteins3,4. The U snRNPs interact in an ordered fashion with specific regions of pre-messenger RNPs in a dynamic remodeling pathway as introns are excised and exons are ligated to produce mature mRNPs5. Many additional nuclear proteins participate in these processing events6.
Galectin-1 (Gal1) and galectin-3 (Gal3) are two proteins that are required factors in the splicing pathway as shown by depletion-reconstitution studies7,8. Removal of both galectins from splicing competent nuclear extracts (NE) abolishes spliceosome assembly and splicing activity at an early step. Addition of either galectin to such a doubly depleted NE restores both activities. Gal1 and Gal3 are components of active spliceosomes as evidenced by specific immunoprecipitation of pre-mRNA, splicing intermediates, and mature mRNA by antiserum specific for either Gal1 or Gal39. Importantly, Gal3 associates with endogenous U snRNA containing particles in the NE outside the splicing pathway as shown by precipitation of snRNPs by anti-Gal3 antisera10. Finally, silencing of Gal3 in HeLa cells alters splicing patterns of numerous genes11.
In NE pre-incubated to disassemble preformed spliceosomes12, snRNPs are found in multiple complexes sedimenting in glycerol gradients from 7S to greater than 60S. Although glycerol gradient fractionation is a common technique for the isolation of spliceosomal complexes and components (see references13,14,15 for example), we have extended this method by characterizing specific fractions using antibody immunoprecipitations. An snRNP sedimenting at 10S contains only U1 snRNA along with Gal3. Immunoprecipitation of the 10S fraction with antisera specific for Gal3 or U1 snRNP co-precipitates both U1 and Gal3 indicating some of the U1 snRNP monoparticles are bound to Gal310. As U1 snRNP is the first complex that binds to pre-mRNP in spliceosomal assembly1,5, this step represents a potential entry site for Gal3 into the splicing pathway. On this basis, we showed that 10S Gal3-U1 snRNP monoparticles bound to anti-Gal3 containing beads restored splicing activity to a U1 snRNP depleted NE, establishing this complex as one mechanism by which Gal3 is recruited into the spliceosomal pathway16. This contrasts with attempts to isolate spliceosomes at specific stages in the splicing reaction and cataloging the associated factors17,18. In such studies, the presence of certain factors at some time point is ascertained but not the mechanism by which they were loaded.
We had previously described in detail the preparation of NE, the splicing substrate, the assembly of the splicing reaction mixture, and the analysis of products in our documentation of the role of galectins in pre-mRNA splicing19. We now describe the experimental procedures for fractionation of nuclear extracts to obtain a fraction enriched in Gal3 - U1 snRNP complex and for immuno-selection of the latter complex to reconstitute splicing activity in a U1-depleted nuclear extract.

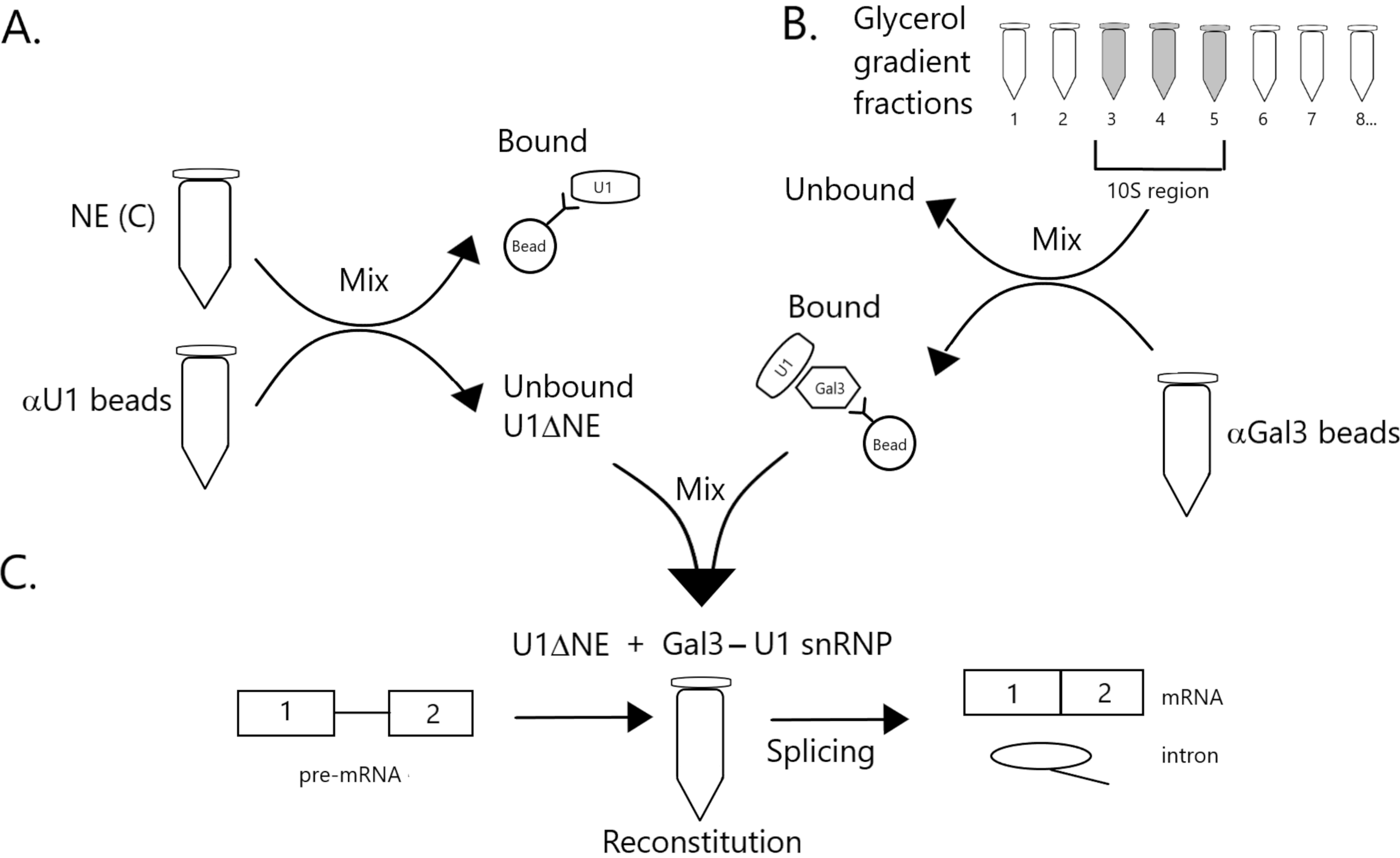
Figure 1: Schematic diagram illustrating the complementation of splicing activity in nuclear extract depleted of U1 snRNP by a Gal3-U1 snRNP complex on beads. (A) NE in Buffer C (NE(C)) is incubated with Protein A-Sepharose beads covalently coupled with anti-U1 snRNP (αU1 beads). The unbound fraction is depleted of U1 snRNP (U1ΔNE). (B) NE in Buffer D (NE(D)) is fractionated over a 12%-32% glycerol gradient by ultracentrifugation. Fractions corresponding to the 10S region (fractions 3-5) are combined and mixed with beads covalently coupled with anti-Gal3 antibodies (αGal3 beads). The material bound to the beads contains a Gal3-U1 snRNP monoparticle. (C) The Gal3-U1 snRNP complex from Part (B) is mixed with U1ΔNE from Part (A) in a splicing assay using 32P-labeled MINX pre-mRNA substrate and the intermediates and products of the splicing reaction are analyzed by gel electrophoresis and autoradiography. Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. Notes on general procedures
- Ensure that all chemicals (buffer components, enzymes, etc.) are kept free of ribonuclease (RNase). Sequester all commercially purchased reagent bottles from general lab use. Wear gloves for all steps of the experimental procedure. Use only glassware and utensils that have been baked (see step 1.2 below) and solutions that have been pre-treated (see step 1.3 below).
- Bake all glassware (beakers, flasks, bottles, pipettes, etc.) for a minimum of 4 h at 177 °C. Wrap other utensils (spatulas, stir-bars, etc.) in aluminum foil before baking under the same conditions.
- Prepare a 0.1% (vol/vol) solution of diethylpyrocarbonate (DEPC) in double-distilled water (ddH2O). Using a magnetic stir-bar, stir this solution overnight and then autoclave. Use this DEPC-treated H2O to make all solutions containing Tris; then, filter sterilize using a bottle top vacuum filter. Use regular ddH2O to prepare all other solutions (without Tris); then, treat with DEPC (0.1%, vol/vol) and autoclave.
NOTE: The buffers used in the following set of experimental procedures are listed alphabetically in Table 1.
| Name of buffer | Composition |
| Borate buffer | 0.2 M sodium borate, pH 9 |
| Buffer C | 20 mM HEPES, pH 7.9, 25% (vol/vol) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 0.5 mM dithiothreitol (DTT) |
| Buffer D | 10 mM HEPES, pH 7.9, 20% (vol/vol) glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT |
| 60%D | 60% Buffer D and 40% H2O |
| Ethanolamine | 0.2 M ethanolamine, pH 8 |
| HEPES binding buffer | 20 mM HEPES, pH 7.9 |
| HEPES wash buffer | 20 mM HEPES, pH 7.9, 0.5 M NaCl |
| RNA loading buffer | 90% formamide, 20 mM EDTA, pH 8, 0.05% (w/v) bromophenol blue |
| SDS-PAGE buffer | 25 mM Tris, 169 mM glycine, 0.1% sodium dodecyl sulfate (SDS), pH 8.8 |
| SDS sample buffer | 62.5 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.1% (w/v) bromophenol blue |
| TBE buffer for RNA gels | 89 mM Tris, 89 mM boric acid, 2.5 mM EDTA, pH 8.3 |
| TE buffer | 10 mM Tris, pH 8, 1 mM EDTA |
| Transfer buffer | 25 mM Tris, 1.92 M glycine, 20% methanol, pH 8.3 |
| T-TBS buffer | 10 mM Tris, 0.5 M NaCl, 0.05% Tween 20, pH 7.5 |
| TX wash buffer | 0.05% Triton X-100 (TX) in 60%D |
Table 1: Name and Composition of Buffers
2. Preparation of NE depleted of U1 snRNP (U1 ΔNE)
- Preparation of anti-U1 beads for immunoadsorption
- Pre-swell 50 mg Protein A-Sepharose CL-4B beads in excess DEPC-treated H2O to produce approximately 200 µL of swollen beads and then wash in HEPES wash buffer.
- For this wash and all subsequent washes, pellet the beads by centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s) and remove the unbound wash using a micropipettor and discard.
- Mix 150 µL of washed beads with 150 µL of human autoimmune serum specific for U1 snRNP (volume of antibody to volume of beads in a ratio of 1:1).
- Adjust, on the basis of the total volume of (~300 µL from step 2.1.3 above), the mixture to 20 mM HEPES, pH 7.9, corresponding to conditions of HEPES binding buffer; incubate this mixture with continuous rocking at room temperature for 60 min.
- Wash the beads bound with antibodies with 1 mL of borate buffer (0.2 M sodium borate, pH 9) and resuspend in 1 mL of the same borate buffer.
- To covalently couple the antibody bound to the Protein A-Sepharose beads, add dimethylpimelimidate to a final concentration of 20 mM and incubate at room temperature with rocking for 60 min.
- Wash the beads with 1 mL of borate buffer.
- To block any unreacted cross-linking reagent, add 1 mL of 0.2 M ethanolamine (pH 8) and incubate at room temperature with rocking for 60 min.
- Wash the antibody-coupled beads, hereafter designated as anti-U1 beads, twice with 0.5 mL of TX wash buffer (0.05% Triton X-100 in 60% D).
- Depletion of U1 snRNP from NE (see Figure 1A)
NOTE: The procedure for preparing NE from HeLa cells was initially developed by Dignam et al.20. We have described the materials and detailed methods for the preparation of NE for splicing assays19 (see steps 2.1 and 3.1 of that reference). NE, as initially prepared is in Buffer C and will hereafter be designated as NE(C). NE(C) dialyzed against and equilibrated with Buffer D will be designated as NE(D).- Incubate 200 µL of NE(C) with 100 µL of anti-U1 beads from step 2.1.9 above.
- Add 5 µL of RNasin to the mixture.
- Rotate the microtube head-over-tail at 4 °C for 1 h.
- Pellet the mixture by centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s) and collect the unbound material (U1ΔNE) using a Hamilton syringe.
- Dialyze the entire volume of U1ΔNE, along with a separate 50 µL aliquot of the original nondepleted NE(C), in separate compartments of a microdialyzer, with stirring, for 75 min against 60% D using a dialysis membrane with 8 K molecular weight cutoff.
- Immediately after dialysis, divide these preparations (U1ΔNE and NE in 60% D) into 20 µL aliquots; then snap freeze in a dry ice/ethanol bath and store at -80 °C.
- Analysis of the RNA and protein content of U1ΔNE and material bound on anti-U1 beads
- After the removal of the unbound material (U1ΔNE) (step 2.2.4), wash the material bound to the anti-U1 beads by adding 0.5 mL of TX wash buffer. Pellet the mixture by centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s) and remove the supernatant using a micropipettor and discard.
- Repeat the wash steps 2.3.1 twice.
- Remove the material bound to the anti-U1 beads by adding 100 µL of 2x SDS sample buffer to 100 µL of the beads and incubating for 10 min at room temperature.
- Pellet the mixture by centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s); remove the supernatant by Hamilton syringe, and snap freeze in a dry ice/ethanol bath. Store at -80 °C.
- Compare the nondepleted NE, the depleted NE (U1ΔNE), and the material bound to the beads (removed from the beads by SDS sample buffer as described in steps 2.3.3 and 2.3.4 above). Follow steps 2.3.6-2.3.8 for RNA analysis or steps 2.3.9-2.3.10 for protein analysis.
- For each sample, extract the RNA with 200 µL of phenol-chloroform (50:50, v/v); then extract again with 180 µL of chloroform-isoamyl alcohol (25:1, v/v). Following extraction, add 300 µL of cold 200-proof ethanol, invert to mix, and store the precipitated RNA overnight at -20 °C.
- Centrifuge the ethanol-precipitated RNA (12,000 x g for 10 min at 4 °C). Wash the pellets with 150 µL of cold 70% ethanol. Centrifuge again (12,000 x g) at 4 °C for 15 min. Remove the supernatant using a micropipettor and dry the pellets in a speed vac for 10-15 min without heat.
- Resuspend the dried RNA pellet in 10 µL of RNA loading buffer, gently vortex, heat to 75-85 °C for 90 s, and then incubate on ice for 2 min. Separate the snRNAs by gel electrophoresis (2 h at 16 mA) through 13% polyacrylamide - 8.3 M urea gels and then either stain with ethidium bromide or subject to northern blotting10,16.
- Load the protein samples, in SDS sample buffer from step 2.3.5, onto 12.5% polyacrylamide gels and electrophorese at 200 V for approximately 45-50 min in SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) buffer.
- Transfer the separated proteins to nitrocellulose membrane at 400 mA for 2 h in transfer buffer. After transfer, block the membrane by incubating overnight in T-TBS containing 10% non-fat dry milk. Then, immunoblot the membrane to reveal specific proteins8,21.
3. Immunoprecipitation of 10S fractions of glycerol gradients by anti-Gal3
- Preparation of anti-Gal3 beads for immunoadsorption
NOTE: The derivation and characterization of rabbit polyclonal antisera against Gal3 for rabbit #2421 and for rabbit #4910 have been described previously.- Use preimmune serum from rabbit #49 as control.
- For the preparation of anti-Gal3 beads follow the procedure previously described for the preparation of anti-U1 beads (step 2.1), with the exception that corresponding to step 2.1.3, the ratio of antiserum (e.g., anti-Gal3, #49) to beads is 3:1.
- Just before use, wash the antibody-coupled beads, hereafter designated as anti-Gal3 beads, twice with 0.5 mL of TX wash buffer. Remove the supernatant, first with a micropipettor to get most of the liquid out and then with a Hamilton syringe to get the liquid out of the beads; discard.
- Immunoprecipitaton of glycerol gradient fractions by anti-Gal3 (see Figure 1B)
- Fractionate NE(D) over a 12%-32% glycerol gradient10. Combine and mix glycerol gradient fractions 3, 4, and 5 (numbered from the top of the gradient), which are near the 10S region of the gradient.
- Prepare two samples, each with 150 µL aliquot of combined gradient fractions 3-5 (step 3.2.1), and place in 50 µL of anti-Gal3 beads.
- In parallel, prepare two samples each with 150 µL of fraction 1 (containing Gal3 not in complex with U1 snRNP10; step 3.2.1) and place in 50 µL of anti-Gal3 beads.
- As a control, place 150 µL of 60% D in another microtube of 50 µL anti-Gal3 beads.
- Mix gently by tapping the tube, then rotate the microtube head-over-tail at 4°C for 1 h.
- Pellet the mixture by gentle centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s).
- Remove the supernatant (unbound material) using a Hamilton syringe. Do not wash the beads and use immediately for the addition of the splicing reactions (section 4.2).
- Analysis of the RNA and protein content in the unbound and bound material from the anti-Gal3 precipitation of 10S gradient fractions
- For the analysis of components of the bound and unbound material from anti-Gal3 precipitation of the 10S gradient fractions, collect the unbound material (supernatant after step 3.2.6), transfer into a fresh microtube, and freeze at -20 °C.
- Wash the precipitated beads from step 3.2.6 (containing material bound to anti-Gal3) by adding 0.5 mL of TX wash buffer.
- Pellet the mixture by gentle centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s); remove the supernatant using a micropipettor and discard. Repeat the wash steps twice more.
- Add 50 µL of 2X SDS sample buffer to the washed and pelleted anti-Gal3 beads.
- Mix the beads gently and incubate for 10 min at room temperature.
- Pellet the mixture by gentle centrifugation (1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s), collect the supernatant by Hamilton syringe and store in a fresh microtube at -20 °C.
- Compare the unbound material (section 3.3.1) and the bound material (step 3.3.6) of the anti-Gal3 precipitation in terms of RNA and protein components, using procedures as described in steps 2.3.6. to 2.3.10, respectively.
4. Assembly of splicing reaction and analysis of products
- Preparation of the splicing substrate
NOTE: The pre-mRNA substrate, designated MINX, contains two exon sequences and one intron sequence from Adenovirus22. The MINX DNA sequence in the plasmid is under the control of T3, T7, or SP6 RNA polymerase promoters. The materials and detailed methods for the linearization of the MINX plasmid DNA with BamHI restriction endonuclease, transcription by SP6 RNA polymerase in the presence of α-32P[GTP] and the purification of 32P-labeled MINX for splicing assays are described previously19 (see steps 2.2 and 3.2 of that reference).- Store the radiolabeled MINX as an ethanol precipitate at -20 °C; use the labeled splicing substrate within 4-6 weeks post transcription.
- Just before use, centrifuge the ethanol precipitated 32P-labeled MINX at 12,000 x g for 10 min at 4 °C; remove the supernatant with a micropipettor and discard.
- Add 150 µL of 70% ethanol and centrifuge at 12,000 x g for 15 min at 4 °C. Discard the supernatant and dry the pellet in speed vac with no heat for 15 min.
- Rehydrate the pellet in 50 µL of DEPC water. Spot 2 µL on each of two GF/C filters; immerse the filters in cold 5% trichloroacetic acid (TCA) for 10 min. Rinse with cold 5% TCA, followed by 180-proof ethanol on a vacuum flask. Air dry the filters and subject to scintillation counting in 4 mL of Safety-Solve.
- Dilute 32P-labeled MINX in 60% D to 104 cpm/µL for the splicing assay.
- Assembly of the splicing reaction (see Figure 1C)
- Assemble, on ice, splicing reactions in a total volume of 24 µL (8 µL U1ΔNE (from step 2.2.6), 3.5 mM MgCl2, 1.5 mM ATP, 20 mM creatine phosphate, 0.5 mM DTT, 20 units RNasin, 4 µL 32P-labeled MINX splicing substrate (104 cpm/µL), 60% D) and add to each tube of beads from section 3.2.7. Assemble an identical set of splicing reactions in a total volume of 24 µL but without U1ΔNE and add to each tube of beads from step 3.2.7.
- Prepare a control splicing reaction in a 12 µL total volume (4 µL NE(D), 3.5 mM MgCl2, 1.5 mM ATP, 20 mM creatine phosphate, 0.5 mM DTT, 20 units RNasin, 2 µL 32P-labeled MINX splicing substrate (104 cpm/µL), 60% D).
- Mix the tubes gently by tapping and rotate end-over-tail at 30°C for 90 min. Pellet the mixture by gentle centrifugation at 1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s.
- Stop the reaction and elute the proteins off the beads by adding 24 µL of 2x SDS sample buffer to the tubes containing beads, and 12 µL of 2x SDS sample buffer to the control tube containing NE but no beads. Heat the tubes at 100 °C for 7 min.
- Centrifuge the tubes gently at 1,000 x g in a swinging bucket rotor at 4 °C for 10-15 s.
- Transfer the supernatants (elutions) to fresh microtubes: approximately 48 µL from the bead tubes and 24 µL from the NE control tube.
- Add Proteinase K (20 mg/mL) to digest and solubilize the proteins: add 5 µL to the 48 µL elution from beads and add 2.5 µL to the 24 µL NE control.
- Incubate the tubes at 37°C for 40 min.
- Gently centrifuge the tubes at 1,000 x g in a swinging bucket rotor at 4 °C for 10 s.
- Dilute the bead elutions with 39.5 µL TE and 10 µL 3 M sodium acetate. Dilute the NE control with 63.5 µL TE and 10 µL 3 M sodium acetate.
- Extract and analyze the RNA as described below (section 4.3).
- Analysis of products of the splicing reaction
- Extract the RNAs in each sample with phenol-chloroform, followed by chloroform-isoamyl alcohol; precipitate the RNAs with ethanol, centrifuge, wash the pellets, remove the supernatant and dry the pellets following the same procedure as described in steps 2.3.6 and 2.3.7.
- Resuspend the dried RNA pellet in 10 µL of RNA loading buffer, gently vortex, heat to 75-85 °C for 90 s, and then incubate on ice for 2 min.
- Prepare 20 mL of a solution containing 13% polyacrylamide (bisacrylamide:acrylamide, 1.9:50 [wt/wt]) in 8.3 M urea; cast gels 15 cm in length using this solution.
- Once the gel is cast, electrophorese it (without any samples loaded) at 400 V for 20 min using TBE as the running buffer. After this step, wash the wells with TBE running buffer.
- Load the RNA samples, in RNA loading buffer, and electrophorese with TBE running buffer at 400 V for 3.5 to 4 h. After electrophoresis, remove the urea by immersing and rotating the gel in distilled water for 10 min.
- Vacuum dry the gel on 3 M filter paper, first for 2 h 15 min at 80 °C and then for 30 min without heat to slowly cool it. Subject the dried gel to autoradiography on film to detect the positions of migration of the radioactive components.
Results
NE depleted of U1 snRNP (U1ΔNE from Section 2.2.6) and Gal3 - U1 snRNP complexes from the 10S region of the glycerol gradient immunoprecipitated by anti-Gal3 (step 3.2.7) were mixed in a splicing reaction. This reaction mixture contained U1 snRNA (Figure 2A, lane 3), as well as the U1-specific protein, U1-70K (Figure 2B, lane 3). As expected, the anti-Gal3 precipitated Gal3 (Figure 2B, lane...
Discussion
This report provides the experimental details that document a Gal3 - U1 snRNP complex trapped on anti-Gal3 coated beads can bind to pre-mRNA substrate and this ternary complex can restore splicing activity to an U1 snRNP-depleted NE. Gal3 is one member of a family of proteins originally isolated on the basis of its galactose-specific carbohydrate-binding activity23. Early immunofluorescence and subcellular fractionation studies provided the initial hint of an association of Gal3 with components of...
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work has been supported by National Science Foundation Grant MCB-0092919 and Michigan State University Intramural Research Grant 09-CDFP-2001 (to RJP) and by National Institutes of Health Grant GM-38740 and Michigan AgBioResearch Project MICL02455 (to JLW).
The MINX pre-mRNA substrate used in the splicing assays was a kind gift from Dr. Susan Berget (Baylor College of Medicine, Houston, TX, USA).
Materials
| Name | Company | Catalog Number | Comments |
| anti-U1 snRNP | The Binding Site | Hu ENA-RNP #33471 | human autoimmune serum specific for U1 snRNP |
| bottle top vacuum filter | Fisher Scientific | Corning 431153 (0.22 μm; PES 150 ml) | for filtering solutions containing Tris |
| centrifuge | International Equipment Company | IEC Model PR-6 | for pelletting Sepharose beads in immunoprecipitation |
| diethylpyrocarbonate (DEPC) | Sigma-Aldrich | 159220-5G | for treatment of water used in preparation of all solutions |
| dimethylpimelimidate (DMP) | Sigma-Aldrich | 80490-5G | for cross-linking antibody to Sepharose beads |
| electrophoresis cell | BioRad Laboratories, Inc | Mini-Protean II | for SDS-PAGE separation of proteins |
| ethanolamine | Sigma-Aldrich | 411000-100ml | for blocking after the cross-linking reaction |
| gel electrophoresis system | Hoefer, Inc | HSI SE 500 Series | for separating snRNAs by gel electrophoresis |
| gel slab dryer | BioRad | Model 224 | for drying gel slabs for autoradiography |
| Hybond ECL membrane | GE Healthcare | RPN3032D (0.2 μm; 30 cm x 3 m) | for immunoblotting of proteins on membrane |
| microdialyzer (12 x 100 μl sample capacity) | Pierce | Microdialyzer System 100 | for exchanging the buffer of nuclear extract |
| microdialyzer membranes (8K cutoff) | Pierce | 66310 | for exchanging the buffer of nuclear extract |
| non-fat dry milk | Spartan Stores | Spartan Instant Non-fat Dry Milk | |
| Protein A Sepharose CL-4B | Millipore-Sigma | GE 17-0780-01 | for coupling antibody to beads |
| Proteinase K | Millipore-Sigma | P2308-5mg | for stopping the splicing reaction to isolate the RNAs |
| RNasin | Promega | N2111 | for inhibiting ribonuclease activity |
| rocker/rotator | Lab Industries, Inc | Labquake Shaker 400-110 | for mixing protein solutions in coupling reactions and in immunoprecipitation |
| Safety-Solve | Research Products International Corp. | No. 111177 | scintillation counting cocktail for determination of radioactivity in splicing substrate |
| scintillation counter | Beckman Instruments | LS6000SC | scintillation counter for determination of radioactivity |
| speed vaccum concentrator | Savant | SVC 100H | for drying ethanol-precipitated RNA pellets |
| Transphor electrophoresis unit | Hoefer, Inc | Hoefer TE Series Transphor | for protein transfer from SDS-PAGE to blotting membrane |
References
- Hoskins, A. A., Moore, M. J. The spliceosome: a flexible, reversible macromolecular machine. Trends In Biochemical Sciences. 37, 179-188 (2012).
- Choi, Y. D., Grabowski, P., Sharp, P. A., Dreyfuss, G. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science. 231, 1534-1539 (1986).
- Lerner, M., Steitz, J. A. Snurps and scyrps. Cell. 25, 298-300 (1981).
- Maniatis, T., Reed, R. The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature. 325, 673-678 (1987).
- Hoskins, A. A., et al. Ordered and dynamic assembly of single spliceosomes. Science. 331, 1289-1295 (2011).
- Coppin, L., Leclerc, J., Vincent, A., Porchet, N., Pigny, P. Messenger RNA life-cycle in cancer: emerging role of conventional and non-conventional RNA-binding proteins. International Journal of Molecular Sciences. 19, 650-676 (2018).
- Dagher, S. F., Wang, J. L., Patterson, R. J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 92, 1213-1217 (1995).
- Vyakarnam, A., Dagher, S. F., Wang, J. L., Patterson, R. J. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and Cellular Biology. 17, 4730-4737 (1997).
- Wang, W., Park, J. W., Wang, J. L., Patterson, R. J. Immunoprecipitation of spliceosomal RNAs by antisera to galectin-1 and galectin-3. Nucleic Acids Research. 34, 5166-5174 (2006).
- Haudek, K. C., Voss, P. G., Locascio, L. E., Wang, J. L., Patterson, R. J. A mechanism for incorporation of galectin-3 into the spliceosome through its association with U1 snRNP. Biochemistry. 48, 7705-7712 (2009).
- Fritsch, K., et al. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 16, 502-511 (2016).
- Conway, G. C., Krainer, A. R., Spector, D. L., Roberts, R. J. Multiple splicing factors are released from endogenous complexes during in vitro pre-mRNA splicing. Molecular and Cellular Biology. 9, 5273-5280 (1989).
- Dery, K. J., Yean, S. L., Lin, R. J. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods in Molecular Biology. 488, 41-63 (2008).
- Yoshimoto, R., Kataoka, N., Okawa, K., Ohno, M. Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Research. 37, 891-902 (2009).
- Malca, H., Shomron, N., Ast, G. The U1 snRNP base pairs with the 5' splice site within a penta-snRNP complex. Molecular and Cellular Biology. 23, 3442-3455 (2003).
- Haudek, K. C., Voss, P. G., Wang, J. L., Patterson, R. J. A 10S galectin-3 - snRNP complex assembles into active spliceosomes. Nucleic Acids Research. 44, 6391-6397 (2016).
- Rappsilber, J., Ryder, U., Lamond, A. I., Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 12, 1231-1245 (2002).
- Jurica, M. S., Moore, M. J. Capturing splicing complexes to study structure and mechanism. Methods. 28, 336-345 (2002).
- Patterson, R. J., Haudek, K. C., Voss, P. G., Wang, J. L. Examination of the role of galectins in pre-mRNA splicing. Methods in Molecular Biology. 1207, 431-449 (2015).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 11, 1475-1489 (1983).
- Agarwal, N., Sun, Q., Wang, S. Y., Wang, J. L. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. Journal of Biological Chemistry. 268, 14932 (1993).
- Zillmann, M., Zapp, M. I., Berget, S. M. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Molecular and Cellular Biology. 8, 814-821 (1988).
- Barondes, S. H., et al. Galectins: a family of animal β-galactoside-binding proteins. Cell. 76, 597-598 (1994).
- Laing, J. G., Wang, J. L. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 27, 5329-5334 (1988).
- Vyakarnam, A., Lenneman, A. J., Lakkides, K. M., Patterson, R. J., Wang, J. L. A comparative nuclear localization study of galectin-1 with other splicing components. Experimental Cell Research. 242, 419-428 (1998).
- Michaud, S., Reed, R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Development. 5, 2534-2546 (1991).
- Chiu, Y. -. F., et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Molecular and Cellular Biology. 29, 5671-5678 (2009).
- Krishnan, R., et al. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nature Structural and Molecular Biology. 20, 1450-1457 (2013).
- Gray, R. M., et al. Distinct effects on splicing of two monoclonal antibodies directed against the amino-terminal domain of galectin-3. Archives of Biochemistry and Biophysics. 475, 100-108 (2008).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved