
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Microscopía electrónica de barrido serial de cara de bloque (SBEM) para el estudio de espinas dendríticas
En este artículo
Resumen
La microscopía electrónica de barrido serial de cara de bloque (SBEM) se aplica para obtener imágenes y analizar espinas dendríticas en el hipocampo murino.
Resumen
La microscopía electrónica tridimensional (3D EM) da la posibilidad de analizar parámetros morfológicos de espinas dendríticas con resolución a nanoescala. Además, algunas características de las espinas dendríticas, como el volumen de la columna vertebral y la densidad postsináptica (PSD) (que representa la parte postsináptica de la sinapsis), la presencia de terminal presináptico y el retículo endoplásmico liso o la forma atípica de PSD (por ejemplo, espinas multiinervadas), se pueden observar solo con EM 3D. Mediante el empleo de la microscopía electrónica de barrido de cara de bloque serie (SBEM) es posible obtener datos EM 3D más fácilmente y de una manera más reproducible que cuando se realiza la sección en serie tradicional. Aquí mostramos cómo preparar muestras de hipocampo de ratón para el análisis de SBEM y cómo este protocolo se puede combinar con el estudio de inmunofluorescencia de espinas dendríticas. La perfusión de fijación leve nos permite realizar estudios de inmunofluorescencia con microscopía de luz en una mitad del cerebro, mientras que la otra mitad fue preparada para SBEM. Este enfoque reduce el número de animales que se utilizarán para el estudio.
Introducción
La mayoría de las sinapsis excitatorias en el sistema nervioso central se encuentran en las espinas dendríticas, pequeñas protuberancias de una membrana neuronal. Estas protuberancias forman compartimentos bioquímicos confinados que controlan la transducción de señales intracelulares. La plasticidad estructural de las espinas dendríticas y las sinapsis está estrechamente relacionada con los cambios funcionales en la eficacia sináptica que subyacen a procesos tan importantes como el aprendizaje y la memoria1,2. Es importante tener en cuenta que la microscopía electrónica (EM) es la única técnica que permite determinar si una columna dendrítica tiene una entrada presináptica. La resolución EM también es necesaria para estudiar detalles ultraestructurales como la forma de una densidad postsináptica (PSD), que representa una parte postsináptica de una sinapsis, o las dimensiones de una columna dendrítica, así como el tamaño y la forma de un bouton axonal. Además, con EM es posible visualizar las sinapsis y su entorno.
Gracias a los avances en las tecnologías de imagen y computación es posible reconstruir circuitos neuronales completos. Las técnicas de microscopía electrónica de volumen, como la microscopía electrónica de transmisión de sección serie (ssTEM), la microscopía electrónica de barrido de cara de bloque serie (SBEM) y la microscopía electrónica de barrido de haz de iones enfocado (FIB-SEM) se utilizan comúnmente para reconstrucciones de circuitos neuronales3.
En nuestros estudios, el método SBEM se emplea con éxito para investigar la plasticidad estructural de las espinas dendríticas y las PSD en muestras del hipocampo del ratón y cortes cerebrales organotípicos 4,5. El SBEM se basa en la instalación de un ultramicrotomo en miniatura dentro de la cámara del microscopio electrónico de barrido6,7,8,9. Se toma una imagen de la parte superior del bloque de muestra, y luego el ultramicrotomo corta la muestra a una profundidad especificada, revelando una nueva cara de bloque, que se vuelve a obtener una imagen y luego se repite el proceso8. Como resultado, solo queda la imagen de una cara de bloque, mientras que la rebanada que se ha cortado se pierde como escombros. Es por eso que SBEM se llama una técnica destructiva, lo que significa que no es posible obtener imágenes del mismo lugar nuevamente. Sin embargo, la ventaja de los métodos destructivos en bloque es que no sufren problemas de deformación y pérdida de sección que pueden afectar significativamente la calidad de los datos y el análisis de datos3. Además, SBEM ofrece la posibilidad de obtener imágenes de un campo de visión relativamente grande (> 0,5 mm × 0,5 mm) a alta resolución3.
Para emplear SBEM, las muestras deben prepararse de acuerdo con un protocolo dedicado y altamente contrastante debido al detector de electrones retrodibuerados utilizado para adquirir imágenes. Mostramos aquí cómo realizar la preparación de muestras de acuerdo con el protocolo basado en un procedimiento desarrollado por Deerinck10 (método del Centro Nacional de Microscopía e Investigación de Imágenes (NCMIR)), utilizando tinciones reducidas de osmio-tiocarbohidrazida-osmio (rOTO) desarrolladas en la década de 19808,11. Además, introducimos un enfoque de fijación en dos pasos, con perfusión de fijación leve que permite utilizar el mismo cerebro tanto para estudios de inmunofluorescencia con microscopía de luz como para SBEM.
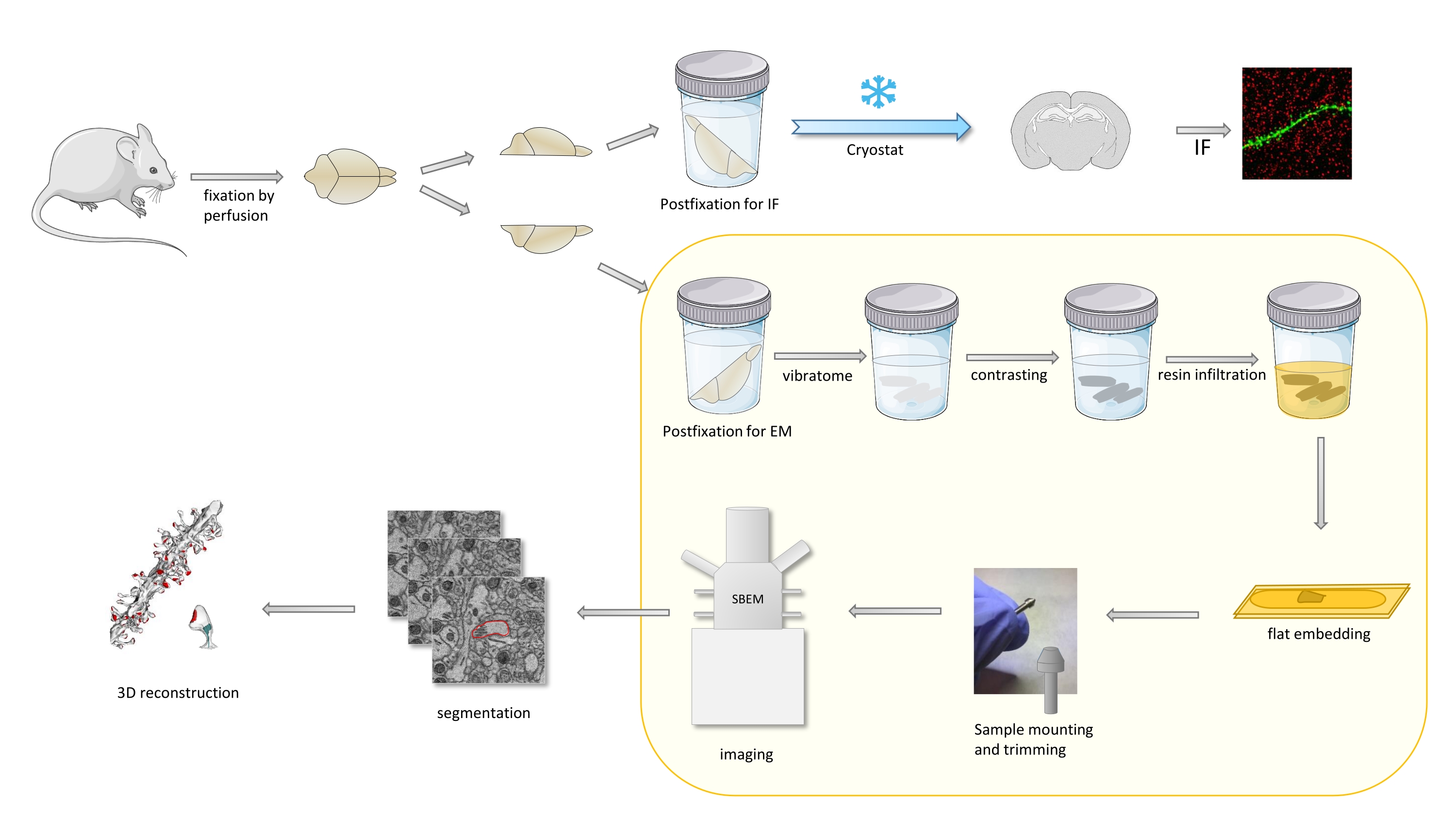
En el protocolo, un cerebro de ratón se fija principalmente con un fijador suave, y luego se corta en mitades, y un hemisferio se posfija y se prepara para la inmunofluorescencia (IF), mientras que el otro para los estudios EM(Figura 1).

Figura 1. Representación esquemática del flujo de trabajo para la preparación de espinas dendríticas para el análisis con SBEM. Los ratones fueron sacrificados y perfundidos con un fijador primario suave. El cerebro se cortó en mitades, y un hemisferio se colocó con fijador dedicado a la inmunofluorescencia (IF), crioprotegido, se cortó en rodajas con un criostato y se procesó para estudios de FI, mientras que el otro hemisferio se posfijó con fijador EM, se cortó con el vibrátomo y se preparó para los estudios em. Las rebanadas cerebrales para los estudios de SBEM se contrastaron, se incrustaron de forma plana en resina, luego se montó una región CA1 del hipocampo en el pasador y se tomaron imágenes con SBEM(Figura 1). La parte del protocolo resaltada en un cuadro amarillo se presentó en el video. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Protocolo
La investigación se realizó de conformidad con las directrices del Instituto Nencki y el permiso del Comité De Ética Local. Los estudios se llevaron a cabo de conformidad con la Directiva del Consejo de las Comunidades Europeas de 24 de noviembre de 1986 (86/609/CEE), Ley de Protección animal de Polonia y aprobados por el primer Comité de Ética Local en Varsovia. Se hicieron todos los esfuerzos para minimizar el número de animales utilizados y su sufrimiento.
PRECAUCIÓN: Todos los procedimientos descritos a continuación deben llevarse a cabo en una campana extractora de humos de laboratorio. Debido a la naturaleza peligrosa de los reactivos utilizados. Se requieren medidas de seguridad personal como guantes, bata de laboratorio, gafas de seguridad y una máscara facial.
1. Preparación del fijador para perfusión (2% en peso/vol paraformaldehído (PFA) y 0,5% vol/vol glutaraldehído (GA) en tampón de fosfato (PB) de 0,1 M, pH 7,4)
NOTA: Prepare la solución fijadora el mismo día en que se usará y no la almacene por más de 3 horas. En caso de escasez de tiempo, prepare el PFA al 2% en 0,1 M PB el día anterior, guárdelo a 4 °C y agregue GA fresco poco antes de la perfusión.
- Tome 400 ml de agua destilada doble estéril (ddH2O) y caliéle a 60 °C con una placa caliente de agitación. Luego agregue 20 g de PFA. Agregue gotas de 1 M de NaOH hasta que el PFA se disuelva por completo y deje que la mezcla se enfríe.
- Añadir 500 mL de 0,2 M PB (pH 7,4).
- Filtre la solución para eliminar cualquier depósito y enfríelo a 4 °C.
- Justo antes de la perfusión, agregue 20 ml de GA al 25% a la solución y luego rellene el volumen con ddH2O a 1 L.
2. Preparación del fijador postperfusión para SBEM (2% wt/vol PFA y 2,5% vol/vol GA en 0,1 M PB, pH 7,4)
- Tomar 50 ml del fijador para perfusión (2% de PFA y 0,5% de GA en 0,1 M de PB).
- Agregue 5 ml de 25% de GA.
3. Preparación de fijador postperfusión para tinción de IF (4% de PFA en solución salina tamponada con fosfato (PBS))
- Disuelva una tableta de 1x PBS (pH 7.4) en agua purificada y desionizada (H2O) de acuerdo con las instrucciones del fabricante.
- Utilice una placa caliente de agitación para calentar la solución a 60 °C y agregue 40 g de PFA.
- Agregue gotas de 1 M de NaOH hasta que el PFA se disuelva completamente y deje que la mezcla se enfríe.
- Ajuste el pH de la solución a 7.5 con 1 M HCl y luego rellene el volumen con H2O a 1 L.
- Filtre la solución para eliminar cualquier depósito.
4. Perfusión transcárdica de animales
NOTA: Todos los desechos de PFA y GA deben recolectarse y almacenarse para su eliminación de acuerdo con las regulaciones locales. La anestesia y la perfusión deben seguir las regulaciones locales. En el protocolo descrito se utilizaron ratones adultos de 3 meses de edad y 20±1 mes de edad Thy1-GFP(M) (Thy1-GFP +/-)12 que expresan proteína de fluorescencia verde (GFP) en una población escasamente distribuida de neuronas glutamatérgicas, pero también se puede usar cualquier otra. Los animales fueron criados como heterocigotos con el fondo C57BL / 6J en la Casa animal del Instituto Nencki de Biología Experimental.
- Antes de la perfusión, anestesiar a un ratón mediante la administración de una mezcla de ketamina/xilazina (hasta 90 mg/kg de ketamina de peso corporal y 10 mg/kg de xilazina de peso corporal) mediante inyección intraperitoneal (aguja calibre 27).
- Evalúe si la profundidad de la anestesia es suficiente comprobando los estímulos reflejos al dolor (pellizco) y el reflejo corneal (entrecerrar los ojos).
- Después de 20 minutos realizar una inyección intraperitoneal (aguja calibre 27) de pentobarbital sódico (50 mg/kg de peso corporal).
- Perfundir un ratón según un protocolo de cirugía de perfusión descrito por Gage et al.13 (ver punto 4; Figura 5-6). Usando una bomba de perfusión comience con un tampón de fosfato de 0.1 M, pH 7.4 (30 mL) durante 3 minutos y continúe con 2% de PFA y 0.5% de GA en 0.1 M PB, pH 7.4 durante 6 minutos (80 mL).
- Diseccionar suavemente el cerebro del cráneo y dividirlo por la mitad (ver Figura 9-10 en Gage et al., 201213). Coloque una pieza en un vial que contenga fijador para SBEM y la segunda en el vial con 4% de PFA/PBS para la tinción de IF.
- Mantenga los hemisferios en el fijador a 4 °C durante la noche.
5. Preparación de cortes cerebrales para microscopía electrónica
- Elija la configuración del vibratomo (velocidad de desplazamiento de la cuchilla: 0.075 mm / s, frecuencia de corte: 80 Hz).
- Coloque la cámara de rebanada en el soporte, conéctela al vibratomo y rodéela con hielo. Luego coloque una cuchilla de afeitar en el soporte de la cuchilla vibratome.
- Use una cuchara u objeto similar y coloque el cerebro frío (superficie dorsal hacia arriba) sobre una superficie de corte resistente (por ejemplo, una tapa de placa de Petri de vidrio). Para preparar una rebanada coronal del hipocampo hacer un corte perpendicular entre el hemisferio cerebral y el cerebelo con una cuchilla de afeitar o bisturí, eliminando así el cerebelo. El bulbo olfativo también se puede eliminar.
- Aplique pegamento de cianoacrilato en la plataforma seca del vibratomo.
- Recoge el cerebro con forrceps y séquelo cuidadosamente en papel de filtro.
- Pegue el hemisferio a la plataforma cerca de la cuchilla de corte con la punta rostral hacia arriba. Conecte la plataforma al soporte e inmediatamente llénelo con 0.1 M PB helado, pH 7.4. Si el corte perpendicular se realiza correctamente, el hemisferio se mantendrá recto proporcionando el ángulo de 90 ° requerido para hacer un corte coronal simétrico que contenga el hipocampo. Asegúrese de que el cerebro esté cubierto de PB.
- Coloque la hoja del vibratomo delante del hemisferio y bajéela hacia el lado coronal del hemisferio. Baje la cuchilla a 400 μm más en la dirección caudal y comience a cortar. Continúe cortando hasta que las dos primeras rebanadas estén completamente separadas del bloque de tejido.
- Retraiga la cuchilla y baje otros 100 μm, luego vuelva a cortar.
- Cuando el hipocampo se hace visible (use el atlas cerebral de ratón Paxinos y Franklin, 200414)recoja las rodajas con el pequeño pincel o la pipeta Pasteur de plástico ensanchado.
- Transfiera las rodajas a una placa de 12 pozos llena de PB frío de 0.1 M, pH 7.4.
- Diseccionar el hipocampo en una placa de Petri de vidrio llena de 0.1 M PB, pH 7.4 usando una cuchilla de afeitar o bisturí, y ponerlo en viales de vidrio con el mismo tampón de fosfato.
NOTA: Para el almacenamiento a largo plazo de las rebanadas, complemente 0.1 M PB con azida de sodio al 0.05% (NaN3).
6. Preparación de muestras cerebrales para la inmunotinción
- Después de la fijación durante la noche en una solución de PFA/PBS al 4% en una campana extractora de humos, coloque el tejido cerebral en la solución de criopreservación (sacarosa al 30% en PBS con NaN3 al 0,05%)y manténgalo a 4 °C durante 2 días (hasta que se hunda).
- Preparar una solución anticongelante (15% sacarosa/30% etilenglicol/0,05% NaN3/PBS).
- Ajuste la temperatura del gabinete del criostato a -19 ° C y asegúrese de que se alcance la temperatura antes de continuar. Durante la sección, asegúrese de que la temperatura del gabinete permanezca entre -18 ° C y -20 ° C.
- Use una cuchara u objeto similar y coloque el cerebro frío (superficie dorsal hacia arriba) sobre una superficie de corte resistente (por ejemplo, una tapa de placa de Petri de vidrio). Para preparar una rebanada coronal del hipocampo hacer un corte perpendicular entre el hemisferio cerebral y el cerebelo con una cuchilla de afeitar o bisturí, eliminando así el cerebelo.
- Seleccione un disco de muestra preenfriado, cúbralo con un medio para congelar en un estante liberador y use forzadores fije el hemisferio al disco con una punta rostral hacia arriba y espere hasta que el espécimen esté completamente congelado. Inserte el disco de la muestra en una cabeza de muestra.
- Instale una cuchilla en un soporte de cuchilla dentro de la criocámara y corte rodajas de 40 μm de grosor.
- Transfiera las rodajas con un pincel pequeño a la placa de 96 pozos llena de solución anticongelante fría y desenrolle suavemente las secciones (recoja una rebanada después de cada ronda de corte para evitar que se pierdan en la cámara de rebanadas).
NOTA: Los cerebros o secciones se pueden almacenar en una solución anticongelante a -20 ° C durante mucho tiempo.
7. Inmunotinción de cortes cerebrales
NOTA: Todos los pasos de tinción se realizaron en una placa de 24 pozos en un agitador de plataforma.
- Lave las rodajas con PBS tres veces, cada vez durante 6 minutos.
- Incubar rodajas en 300 μL de solución bloqueadora (5% de suero normal de burro [NDS]/0,3% Tritón X-100) durante 1 hora con agitación suave en un rotador.
- Incubar las rodajas con el anticuerpo primario contra PSD-95 diluido 1:500 en 5% NDS/0,3% Triton X-100/PBS (300 μL por pozo) durante la noche a 4 °C. La concentración final del anticuerpo primario es de 2 μg/ml.
- Lave las rodajas con Triton X-100/PBS al 0,3% a temperatura ambiente (RT) tres veces, cada vez durante 6 minutos.
- Incubar rodajas con el anticuerpo secundario diluido 1:500 en 300 μL de Tritón X-100/PBS al 0,3% durante 90 minutos. La concentración final del anticuerpo secundario es de 4 μg/mL.
- Lave las rodajas con PBS tres veces, cada vez durante 6 minutos.
- Monte las rodajas en diapositivas con un medio de montaje y luego ciéntelas con una diapositiva de cubierta.
- Examinar las muestras utilizando un microscopio confocal (63 × objetivo de aceite, NA 1,4, tamaño de píxel 0,13 μm × 0,13 μm).
NOTA: Para el almacenamiento a largo plazo: mantenga las muestras a 4 °C, protéjalas de la luz.
8. Preparación de muestras SBEM
PRECAUCIÓN: Debido a la naturaleza peligrosa de los reactivos utilizados, todos los procedimientos descritos a continuación deben llevarse a cabo en una campana extractora de humos de laboratorio. Antes de usar estos productos químicos, lea detenidamente las hojas de datos de seguridad de materiales proporcionadas por los fabricantes y pregunte al oficial de seguridad sobre las reglas locales para garantizar un manejo seguro y la eliminación de desechos.
- Muestra de contraste
NOTA: Los lavados de rodajas y la incubación a temperatura ambiente deben llevarse a cabo con agitación leve (por ejemplo, en un agitador de plataforma). Se utilizó agua desgasificación en autoclave.- Lavar las rodajas con 0,1 M pb frío, pH 7,4 cinco veces, cada vez durante 3 minutos.
- Prepare una mezcla 1:1 de tetróxido de osmio acuoso al 4% y ferrocianuro de potasio al 3% (1:1 por vol). El producto final se volverá marrón. Sumergir las muestras en esta mezcla, colocarlas sobre hielo, y a partir de esta etapa es importante protegerlas de la luz. Incubarlos con un suave agitación durante 1 hora.
- Mientras tanto, prepare una solución de tiocarbohidrazida (TCH). Mezclar 10 ml de H2O (ddH2 O) de doble destilación y 0,1 g de TCH y colocarlo en un horno a 60 °C durante 1 hora. Es importante girar la solución de vez en cuando (por ejemplo, cada 10 minutos). Cuando esté listo, enfríelo a temperatura ambiente.
- Lavar las rodajas con ddH2O cinco veces, cada vez durante 3 minutos.
- Filtre la solución de TCH con un filtro de jeringa de 0,22 μm y sumerja la muestra en una solución filtrada. Las rodajas se volverán negras. Incubarlos durante 20 minutos a temperatura ambiente.
- Lave las muestras con ddH2O cinco veces, cada vez durante 3 minutos.
- Incubar las muestras con una solución acuosa al 2% de OsO4 durante 30 minutos a temperatura ambiente.
- Lave las muestras con ddH2O cinco veces, cada vez durante 3 minutos.
- Coloque las muestras en acetato de uranilo acuoso al 1% filtrado e incube a 4 °C durante la noche. Utilice un filtro de jeringa de 0,22 μm para la filtración.
- Prepare la solución de ácido L-aspártico mezclando 0,4 g de ácido L-aspártico y 80 ml de ddH2O, ajuste el pH a 3,8 para una disolución más fácil y luego rellene con agua a 100 ml.
- Al día siguiente, comience con la preparación del aspartato15 de Walton. Mezclar 0,066 g de nitrato de plomo con 10 ml de solución de ácido L-aspártico (punto 8.1.10) precalentada a 60 °C y ajustar el pH a 5,5 (medido a 60 °C) con 1 M de NaOH. Cierre el vial con aspartato de plomo y déjelo a 60 °C durante 30 minutos en un baño de agua. La solución debe ser clara. Si se vuelve turbio, debe descartarse y se debe preparar uno nuevo.
- Mientras tanto, lave las muestras con ddH2O desgasificación cinco veces, cada vez durante 3 minutos. A continuación, manténgalos en el horno a 60 °C durante 30 minutos.
- Sumergir las muestras en la solución de aspartato de plomo recién preparada e incubarlas en el horno a 60 °C durante 20 minutos.
- Lave las muestras con ddH2O desgasesado cinco veces, cada vez durante 3 minutos.
- Deshidratación e incrustación de resina
- Preparar resina epoxi. Pesar los ingredientes (33,3 g de componente A/M, 33,3 g de componente B y 1 g de componente D) y mezclar bien la resina (por ejemplo, agitarla en un agitador rotativo en un tubo de 15 ml) durante al menos 30 minutos antes de agregar 16 gotas de acelerador DMP 30. Revuelva de nuevo durante otros 10 minutos.
NOTA: Pesa los ingredientes debajo de la campana de humos. Esta cantidad de componentes da aproximadamente 60 ml de resina, lo que es suficiente para 15 viales con muestras. Puede preparar menos o almacenar el resto de la resina en una jeringa a 4 °C y utilizarla al día siguiente. Recuerde sellar la punta de la jeringa. - Preparar viales con diluciones graduadas de etanol (30%, 50%, 70%, 90%, 100%, 100% etanol en agua y 100% secado en un tamiz molecular).
- Mezclar la resina con etanol al 100% en proporción 1:1 para obtener un 50% de resina. Mézclalo bien.
- Deshidratar las muestras durante 5 minutos en cada dilución de etanol a partir del 30% y terminando con etanol 100% anhidro secado en tamiz molecular. Recuerde, las muestras nunca deben secarse por completo.
- Infiltrar las muestras primero en resina al 50% durante 30 minutos, luego en resina 100% durante una hora, y luego nuevamente en resina al 100% durante la noche. Realice todos los pasos de infiltración con agitación lenta constante.
- Al día siguiente, coloque las muestras en resina fresca 100% durante una hora y luego insértelas entre las láminas de incrustación de fluoropolímeros. Desengrase trozos de lámina de incrustación con etanol, y planplan las muestras entre dos capas de la misma, utilizando dos portaobjetos de vidrio como soporte. Trate de evitar las burbujas de aire en la resina en o cerca de la muestra.
- Curar las muestras en el horno a 70 °C durante al menos 48 horas.
- Preparar resina epoxi. Pesar los ingredientes (33,3 g de componente A/M, 33,3 g de componente B y 1 g de componente D) y mezclar bien la resina (por ejemplo, agitarla en un agitador rotativo en un tubo de 15 ml) durante al menos 30 minutos antes de agregar 16 gotas de acelerador DMP 30. Revuelva de nuevo durante otros 10 minutos.
- Recorte y montaje
- Separe las hojas de incrustación y corte un trozo de la muestra incrustada (aproximadamente 1 mm x 1 mm) con una cuchilla de afeitar. Transfiéralo a una película parafilm. Esto minimizará el peligro de perder la muestra debido a la electrostática.
- Tome un pasador de aluminio que haya sido desengrasado con etanol. Mezcle bien un epoxi conductor y use una pequeña cantidad de él para montar la muestra en el pasador. Curar epoxi conductor a 70 °C durante 10 minutos.
- Recorte cada lado del bloque de muestra con el cuchillo de diamante y luego pula la cara del bloque hasta que el tejido quede expuesto.
NOTA: En este paso, confirme la presencia de una región de interés recolectando algunas secciones y realizando tinción azul toluidina16 o verificando bajo el microscopio electrónico. - Reduzca el tamaño de la muestra tanto como sea posible. Luego moele la muestra al pasador con pintura conductora y cújela (durante 24 horas a temperatura ambiente o en el horno a 65 ° C durante 40 minutos).
- Para minimizar los artefactos de carga, cubra las muestras con una capa delgada de oro u oro / paladio.
9. Imágenes SBEM
- Coloque el pasador con una muestra en la cámara del microscopio electrónico de barrido de cara de bloque serie. Alinee la muestra con el cuchillo. Cierre la cámara y establezca los parámetros.
- Recopile una pila de imágenes con el aumento deseado, el tamaño de píxel, el grosor de la rebanada, el voltaje acelerado (EHT), la apertura, la presión, etc.
NOTA: Los parámetros dependen de la muestra y del objetivo del experimento. En la tabla de materiales se incluyen ajustes de imagen iniciales ejemplares.
10.3D reconstrucciones
NOTA: Para los pasos mencionados a continuación, utilizamos software de acceso abierto, por ejemplo, FijiJ17 (ImageJ versión 1.49b), Microscopy Image Browser (MIB)18 y Reconstruct19, pero se puede emplear otro software.
- Convierta archivos de micrografía digital (dm) a formato TIFF. Primero importe la secuencia de imágenes (en FijiJ: File > Import > Image sequence > Choose 8 Bit Format)y luego guárdela como TIFF (en FijiJ: File > Save As > Image Sequence > Choose Tiff).
- Ajuste el brillo y el contraste de la pila de imágenes (en FijiJ: Image > Adjust > Brightness/Contrast)y, si es necesario, denóselo (use el complemento DenoisEM para FijJ20: Plugins > DenoiseEM > Denoise).
- Alinear la pila (en FijiJ: Plugins > StagReg o MIB: Dataset > Alignment Tools).
- Segmentar espinas dendríticas y sinapsis (en el software MIB o Reconstruct, hay tutoriales completos disponibles (consulte la Tabla de materiales para obtener más detalles).
Resultados
Utilizando el método descrito anteriormente de alto contraste, se pueden obtener imágenes de buena resolución del tejido cerebral del ratón. Un gran campo de visión proporcionado por la técnica SBEM facilita la selección precisa de la región de interés. La imagen grande de la región CA1 del hipocampo se tomó para medir la longitud del estrato radiatum (SR)(Figura 2A)y para establecer la imagen con precisión en el centro(Figura 2B). A continu...
Discusión
Hay muchas variaciones del método NCMIR primario descrito por Deerinck en 201010. Los principios básicos siguen siendo los mismos pero, dependiendo del tipo de material estudiado, se implementan ligeros cambios. Anteriormente se describió que se pueden utilizar diferentes resinas para incrustar especímenes para SBEM y por ejemplo en el caso de las plantas, Spurr's es la resina de elección debido a su baja viscosidad que permite una mejor infiltración a través de las paredes celulares
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Las imágenes SBEM, las imágenes de microscopía de luz y la preparación de muestras de microscopía electrónica se realizaron con el uso del equipo del Laboratorio de Imágenes de Tejido y Función que sirve como una instalación central de imágenes en el Instituto Nencki de Biología Experimental.
Para la preparación de la Figura 1, se utilizó la imagen de un ratón (Souris_02) y un vial del https://smart.servier.com/.
Este trabajo fue apoyado por el Centro Nacional de Ciencias (Polonia) Grant Opus (UMO-2018/31/B/NZ4/01603) otorgado a KR.
Materiales
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade,  98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands | 50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands | 10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
Referencias
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693 (2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. . NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. J. . The mouse brain in stereotaxic coordinates. , (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771 (2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084 (2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. . Biological specimen preparation for transmission electron microscopy. , (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados