
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Microscopia elettronica a scansione a blocchi seriali (SBEM) per lo studio delle spine dendritiche
In questo articolo
Riepilogo
La microscopia elettronica a scansione a blocchi seriali (SBEM) viene applicata per l'immagine e l'analisi delle spine dendritiche nell'ippocampo murino.
Abstract
La microscopia elettronica tridimensionale (3D EM) offre la possibilità di analizzare i parametri morfologici delle spine dendritiche con risoluzione su scala nanometrica. Inoltre, alcune caratteristiche delle spine dendritiche, come il volume della colonna vertebrale e la densità post-sinaptica (PSD) (che rappresenta la parte post-sinaptica della sinapsi), la presenza di terminale presinaptico e il reticolo endoplasmatico liscio o la forma atipica di PSD (ad esempio, spine multi-innervate), possono essere osservate solo con EM 3D. Utilizzando la microscopia elettronica a scansione a blocchi seriali (SBEM) è possibile ottenere dati EM 3D in modo più semplice e riproducibile rispetto a quando si esegue il sezionamento seriale tradizionale. Qui mostriamo come preparare campioni di ippocampo di topo per l'analisi SBEM e come questo protocollo può essere combinato con lo studio di immunofluorescenza delle spine dendritiche. La lieve perfusione di fissazione ci consente di eseguire studi di immunofluorescenza con microscopia ottica su una metà del cervello, mentre l'altra metà è stata preparata per SBEM. Questo approccio riduce il numero di animali da utilizzare per lo studio.
Introduzione
La maggior parte delle sinapsi eccitatorie nel sistema nervoso centrale si trovano su spine dendritiche - piccole sporgenze di una membrana neuronale. Queste sporgenze formano compartimenti biochimici confinati che controllano la trasduzione del segnale intracellulare. La plasticità strutturale delle spine dendritiche e delle sinapsi è strettamente correlata ai cambiamenti funzionali nell'efficacia sinaptica che sono alla base di processi importanti come l'apprendimento e la memoria1,2. È importante notare che la microscopia elettronica (EM) è l'unica tecnica che consente di determinare se una colonna vertebrale dendritica ha un input presinaptico. La risoluzione EM è necessaria anche per studiare dettagli ultrastrutturali come la forma di una densità postsinaptica (PSD), che rappresenta una parte postsinaptica di una sinapsi, o le dimensioni di una colonna vertebrale dendritica, nonché le dimensioni e la forma di un bouton assonale. Inoltre, con EM è possibile visualizzare le sinapsi e l'ambiente circostante.
Grazie ai progressi delle tecnologie di imaging e calcolo è possibile ricostruire interi circuiti neurali. Le tecniche di microscopia elettronica a volume, come la microscopia elettronica a trasmissione a sezione seriale (ssTEM), la microscopia elettronica a scansione a blocchi seriali (SBEM) e la microscopia elettronica a scansione a fascio ionico focalizzato (FIB-SEM) sono comunemente utilizzate per le ricostruzioni di circuiti neuronali3.
Nei nostri studi, il metodo SBEM è impiegato con successo per studiare la plasticità strutturale delle spine dendritiche e delle PSD in campioni di ippocampo di topo e fette di cervello organotipico 4,5. Lo SBEM si basa sull'installazione di un ultramicrotomo in miniatura all'interno della camera del microscopio elettronico a scansione6,7,8,9. La parte superiore del blocco campione viene tagliata, quindi il campione viene tagliato a una profondità specificata dall'ultramicrotomo, rivelando una nuova faccia del blocco, che viene nuovamente tagliata e quindi il processo viene ripetuto8. Di conseguenza, rimane solo l'immagine di una faccia di blocco mentre la fetta che è stata tagliata viene persa come detriti. Ecco perché SBEM è chiamata una tecnica distruttiva, il che significa che non è possibile immaginare di nuovo lo stesso posto. Tuttavia, il vantaggio dei metodi distruttivi on-block è che non soffrono di problemi di deformazione e perdita di sezione che possono influire in modo significativo sulla qualità dei dati e sull'analisi dei dati3. Inoltre, SBEM dà la possibilità di visualizzare un campo visivo relativamente ampio (> 0,5 mm × 0,5 mm) ad alta risoluzione3.
Per utilizzare SBEM, i campioni devono essere preparati secondo un protocollo dedicato e altamente contrastante a causa del rilevatore di elettroni retrodatterato utilizzato per l'acquisizione di immagini. Mostriamo qui come eseguire la preparazione del campione secondo il protocollo basato su una procedura sviluppata da Deerinck10 (National Center for Microscopy and Imaging Research (NCMIR) metodo), utilizzando macchie ridotte di osmio-tiocarboidrazide-osmio (rOTO) sviluppate nel 19808,11. Inoltre, introduciamo un approccio di fissazione in due fasi, con lieve perfusione di fissazione che consente di utilizzare lo stesso cervello sia per studi di immunofluorescenza con microscopia ottica e SBEM.
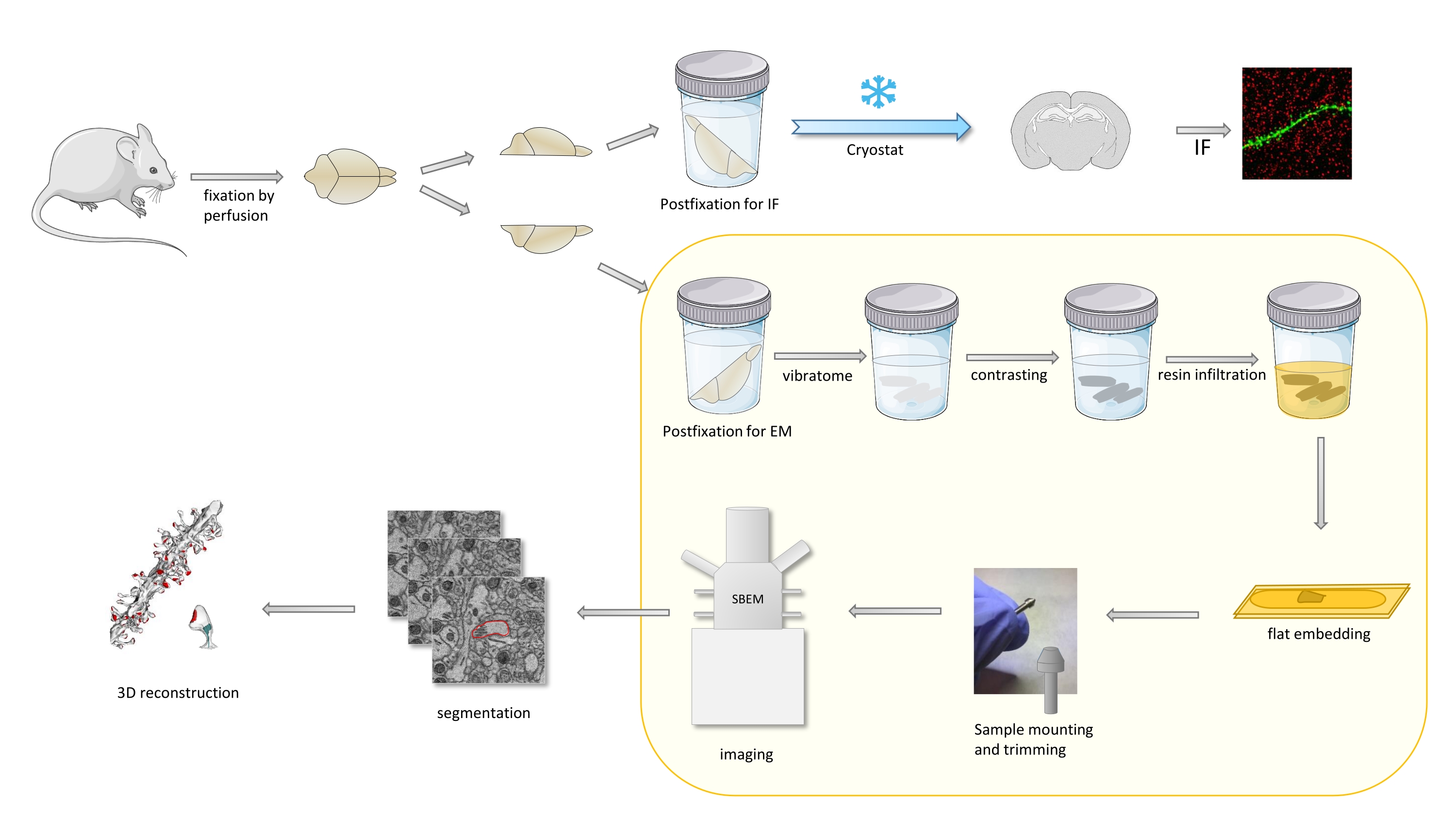
Nel protocollo un cervello di topo viene fissato principalmente con un lieve fissativo, e poi tagliato a metà, e un emisfero viene postfisso e preparato per l'immunofluorescenza (IF), mentre l'altro per gli studi EM (Figura 1).

Figura 1. Rappresentazione schematica del flusso di lavoro per la preparazione delle spine dendritiche per l'analisi con SBEM. I topi sono stati sacrificati e perfusi con un lieve fissativo primario. Il cervello è stato tagliato a metà e un emisfero è stato postfisso con fissativo dedicato all'immunofluorescenza (IF), crioprotetto, affettato usando un criostato ed elaborato per studi IF, mentre l'altro emisfero è stato postfisso con fissativo EM, affettato con il vibratoma e preparato per gli studi EM. Le fette di cervello per gli studi SBEM sono state contrastate, piatte incorporate nella resina, quindi una regione CA1 dell'ippocampo è stata montata sul perno e tagliata con SBEM (Figura 1). La parte del protocollo evidenziata in una casella gialla è stata presentata nel video. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Protocollo
La ricerca è stata condotta in conformità con le linee guida dell'Istituto Nencki e il permesso del Comitato Etico Locale. Gli studi sono stati effettuati conformemente alla direttiva del Consiglio delle Comunità europee del 24 novembre 1986 (86/609/CEE), alla legge polacca sulla protezione degli animali e approvati dal primo comitato etico locale di Varsavia. Tutti gli sforzi sono stati fatti per ridurre al minimo il numero di animali utilizzati e la loro sofferenza.
ATTENZIONE: Tutte le procedure descritte di seguito devono essere eseguite in una cappa aspirante da laboratorio. A causa della natura pericolosa dei reagenti utilizzati. Sono richieste misure di sicurezza personali come guanti, camice da laboratorio, occhiali di sicurezza e mascherina.
1. Preparazione del fissativo per la perfusione (2% wt/vol paraformaldeide (PFA) e 0,5% vol/vol glutaraldeide (GA) in tampone fosfato 0,1 M (PB), pH 7,4)
NOTA: Preparare la soluzione fissativa lo stesso giorno in cui verrà utilizzata e non conservarla più a lungo di 3 ore. In caso di mancanza di tempo, preparare il 2% di PFA in 0,1 M PB il giorno prima, conservarlo a 4 °C e aggiungere GA fresco poco prima della perfusione.
- Prendere 400 ml di acqua distillata sterile doppia (ddH2O) e riscaldarla a 60 °C usando una piastra calda di agitazione. Quindi aggiungere 20 g di PFA. Aggiungere gocce di 1 M NaOH fino a quando il PFA non è completamente sciolto e lasciare raffreddare la miscela.
- Aggiungere 500 mL di 0,2 M PB (pH 7,4).
- Filtrare la soluzione per rimuovere eventuali depositi e raffreddarla a 4 °C.
- Poco prima della perfusione, aggiungere 20 ml di GA al 25% alla soluzione e quindi ricaricare il volume con ddHda 2O a 1 L.
2. Preparazione del fissativo postperfusione per SBEM (2% wt/vol PFA e 2,5% vol/vol GA in 0,1 M PB, pH 7,4)
- Prendi 50 ml del fissativo per la perfusione (2% PFA e 0,5% GA in 0,1 M PB).
- Aggiungere 5 ml di GA al 25%.
3. Preparazione del fissativo postperfusione per la colorazione IF (4% PFA in soluzione salina tamponata con fosfato (PBS))
- Sciogliere una compressa da 1x PBS (pH 7,4) in acqua purificata e deionizzata (H2O) secondo le istruzioni del produttore.
- Utilizzare una piastra riscaldante per riscaldare la soluzione a 60 °C e aggiungere 40 g di PFA.
- Aggiungere gocce di 1 M NaOH fino a quando il PFA è completamente sciolto e lasciare raffreddare la miscela.
- Regolare il pH della soluzione a 7,5 con 1 M HCl, quindi ricaricare il volume con H2O a 1 L.
- Filtrare la soluzione per rimuovere eventuali depositi.
4. Perfusione transcardica di animali
NOTA: Tutti i rifiuti PFA e GA devono essere raccolti e stoccati per lo smaltimento secondo le normative locali. L'anestesia e la perfusione devono seguire le normative locali. Nel protocollo descritto sono stati utilizzati topi Thy1-GFP(M) femmine di 3 mesi di 3 mesi e 20±1 mese (Thy1-GFP +/-)12 che esprimono proteine di fluorescenza verde (GFP) in una popolazione scarsamente distribuita di neuroni glutammatergici, ma qualsiasi altro può essere usato pure. Gli animali sono stati allevati come eterozigoti con il background C57BL / 6J nella Animal House dell'Istituto di biologia sperimentale di Nencki.
- Prima della perfusione anestetizzare un topo mediante somministrazione di una miscela di ketamina/xilazina (fino a 90 mg/kg di peso corporeo ketamina e 10 mg/kg di peso corporeo xilazina) tramite iniezione intraperitoneale (ago calibro 27).
- Valutare se la profondità dell'anestesia è sufficiente controllando il riflesso agli stimoli del dolore (pizzicamento) e il riflesso corneale (strabismo).
- Dopo 20 minuti eseguire un'iniezione intraperitoneale (ago calibro 27) di pentobarbital di sodio (50 mg/kg di peso corporeo).
- Perfondere un topo secondo un protocollo di chirurgia di perfusione descritto da Gage et al.13 (vedi punto 4; Figura 5-6). Utilizzando una pompa di perfusione iniziare con un tampone fosfato 0,1 M, pH 7,4 (30 mL) per 3 minuti e continuare con 2% PFA e 0,5% GA in 0,1 M PB, pH 7,4 per 6 minuti (80 mL).
- Sezionare delicatamente il cervello dal cranio e dividerlo a metà (vedi Figura 9-10 in Gage et al., 201213). Mettere un pezzo in un flaconcino contenente fissativo per SBEM e il secondo nel flaconcino con il 4% di PFA/PBS per la colorazione IF.
- Mantenere gli emisferi nel fissativo a 4 °C durante la notte.
5. Preparazione di fette di cervello per microscopia elettronica
- Scegliere le impostazioni del vibrato (velocità di corsa della lama: 0,075 mm/s, frequenza di taglio: 80 Hz).
- Posizionare la camera della fetta nel supporto, attaccarla al vibratoma e circondarla di ghiaccio. Quindi posizionare una lama di rasoio nel supporto della lama del vibratome.
- Usa un cucchiaio o un oggetto simile e posiziona il cervello refrigerato (superficie dorsale in alto) su una superficie di taglio dura (ad esempio, un coperchio di vetro per piatti di Petri). Per preparare una fetta coronale dell'ippocampo fare un taglio perpendicolare tra l'emisfero cerebrale e il cervelletto con una lama di rasoio o bisturi, rimuovendo così il cervelletto. Il bulbo olfattivo può anche essere rimosso.
- Applicare la colla cianoacrilata sulla piattaforma asciutta del vibratoma.
- Raccogli il cervello con una pinna e asciugalo accuratamente su carta da filtro.
- Incollare l'emisfero alla piattaforma vicino alla lama da taglio con la punta rostrale verso l'alto. Collegare la piattaforma al supporto e riempirla immediatamente con PB 0,1 M ghiacciato, pH 7,4. Se il taglio perpendicolare è fatto correttamente, l'emisfero si alimenterà dritto fornendo un angolo di 90 ° necessario per effettuare un taglio coronale simmetrico contenente l'ippocampo. Assicurare che il cervello sia coperto di PB.
- Posizionare la lama del vibratomo davanti all'emisfero e abbassarla sul lato coronale dell'emisfero. Abbassare ulteriormente la lama a 400 μm nella direzione caudale e iniziare a affettare. Continuare a affettare fino a quando le prime due fette sono completamente separate dal blocco di tessuto.
- Ritrarre la lama e abbassare altri 100 μm, quindi affettare di nuovo.
- Quando l'ippocampo diventa visibile (usa l'atlante del cervello del topo Paxinos e Franklin, 200414) raccogli le fette con il piccolo pennello o la pipetta Pasteur di plastica allargata.
- Trasferire le fette in una piastra a 12 pozzetti riempita con PB freddo da 0,1 M, pH 7,4.
- Sezionare l'ippocampo in una capsula di Vetro di Petri riempita con 0,1 M PB, pH 7,4 usando una lama di rasoio o un bisturi e metterlo in fiale di vetro con lo stesso tampone fosfato.
NOTA: Per la conservazione a lungo termine delle fette supplemento 0,1 M PB con 0,05% di azide di sodio (NaN3).
6. Preparazione del campione di cervello per l'immuno colorazione
- Dopo la fissazione notturna in soluzione di PFA/PBS al 4% in una cappa aspirante, mettere il tessuto cerebrale nella soluzione di crioconservazione (30% di saccarosio in PBS con 0,05% NaN3) e tenerlo a 4 °C per 2 giorni (fino ad affondamento).
- Preparare una soluzione antigelo (15% saccarosio/30% glicole etilenico/0,05% NaN3/PBS).
- Impostare la temperatura dell'armadio del criostato a -19 °C e assicurarsi che la temperatura sia raggiunta prima di procedere ulteriormente. Durante il sezionamento, assicurarsi che la temperatura dell'armadio rimanga compresa tra -18 °C e -20 °C.
- Usa un cucchiaio o un oggetto simile e posiziona il cervello refrigerato (superficie dorsale in alto) su una superficie di taglio dura (ad esempio, un coperchio di vetro per piatti di Petri). Per preparare una fetta coronale dell'ippocampo fare un taglio perpendicolare tra l'emisfero cerebrale e il cervelletto con una lama di rasoio o bisturi, rimuovendo così il cervelletto.
- Selezionare un disco campione pre-raffreddato, coprirlo con un mezzo per il congelamento su un ripiano liberatore e usando una pinna fissare l'emisfero al disco con una punta rostrale verso l'alto e attendere che il campione sia completamente congelato. Inserire il disco campione in una testa campione.
- Installare una lama in un supporto lama all'interno della criocamacca e tagliare fette spesse 40 μm.
- Trasferire le fette con un piccolo pennello sulla piastra a 96 pozzetti riempita con soluzione antigelo fredda e srotolare delicatamente le sezioni (raccogliere una fetta dopo ogni giro di affettatura per evitare che si perdano nella camera delle fette).
NOTA: cervelli o sezioni possono essere conservati in una soluzione antigelo a -20 °C per lungo tempo.
7. Immunostaining delle fette di cervello
NOTA: tutte le fasi di colorazione sono state eseguite in una piastra a 24 pozzetti su uno shaker della piattaforma.
- Lavare le fette con PBS tre volte, ogni volta per 6 minuti.
- Incubare le fette in 300 μL di soluzione bloccante (5% di siero d'asino normale (NDS)/0,3% Triton X-100) per 1 ora con un delicato scuotimento su un rotatore.
- Incubare le fette con l'anticorpo primario contro PSD-95 diluito 1:500 in 5% NDS/0,3% Triton X-100/PBS (300 μL per pozzet) durante la notte a 4 °C. La concentrazione finale dell'anticorpo primario è di 2 μg/mL.
- Lavare le fette con 0,3% Triton X-100/PBS a temperatura ambiente (RT) tre volte, ogni volta per 6 minuti.
- Incubare fette con l'anticorpo secondario diluito 1:500 in 300 μL di 0,3% Triton X-100/PBS per 90 minuti. La concentrazione finale dell'anticorpo secondario è di 4 μg/mL.
- Lavare le fette con PBS tre volte, ogni volta per 6 minuti.
- Montare le fette su diapositive utilizzando un mezzo di montaggio e quindi chiuderle con una diapositiva di copertura.
- Esaminare i campioni utilizzando un microscopio confocale (obiettivo olio da 63 ×, NA 1.4, dimensione dei pixel 0,13 μm × 0,13 μm).
NOTA: Per la conservazione a lungo termine: conservare i campioni a 4 °C, proteggerli dalla luce.
8. Preparazione del campione SBEM
ATTENZIONE: A causa della natura pericolosa dei reagenti utilizzati, tutte le procedure descritte di seguito devono essere eseguite in una cappa aspirante da laboratorio. Prima di utilizzare queste sostanze chimiche leggere attentamente le schede di sicurezza dei materiali fornite dai produttori e chiedere al responsabile della sicurezza le norme locali per garantire la manipolazione sicura e lo smaltimento dei rifiuti.
- Campione a contrasto
NOTA: i lavaggi delle fette e l'incubazione a temperatura ambiente devono essere effettuati con un lieve scuotimento (ad esempio, su uno shaker a piattaforma). È stata utilizzata acqua degassata in autoclave.- Lavare le fette con PB freddo 0,1 M, pH 7,4 cinque volte, ogni volta per 3 minuti.
- Preparare una miscela 1:1 di teossido di osmio acquoso al 4% e ferrocianuro di potassio al 3% (1:1 vol). Il prodotto finale diventerà marrone. Immergere i campioni in questa miscela, metterli sul ghiaccio e da questa fase in poi è importante proteggerli dalla luce. Incubarli con un leggero scuotimento per 1 ora.
- Nel frattempo preparare una soluzione di tiocarboidrazide (TCH). Mescolare 10 mL di H2O (ddH2O) e 0,1 g di TCH e metterlo in forno a 60 °C per 1 ora. È importante ruotare la soluzione di volta in volta (ad esempio, ogni 10 minuti). Quando è pronto, raffreddarlo a temperatura ambiente.
- Lavare le fette con ddH2O cinque volte, ogni volta per 3 minuti.
- Filtrare la soluzione TCH utilizzando un filtro a siringa da 0,22 μm e immergere il campione in soluzione filtrata. Le fette diventerà nera. Incubarli per 20 minuti a temperatura ambiente.
- Lavare i campioni con ddH2O cinque volte, ogni volta per 3 minuti.
- Incubare i campioni con una soluzione acquosa al 2% di OsO4 per 30 minuti a temperatura ambiente.
- Lavare i campioni con ddH2O cinque volte, ogni volta per 3 minuti.
- Collocare i campioni in acetato di uranile acquoso filtrato all'1% e incubarli a 4 °C durante la notte. Utilizzare un filtro a siringa da 0,22 μm per la filtrazione.
- Preparare la soluzione di acido L-aspartico mescolando 0,4 g di acido L-aspartico e 80 ml di ddH2O, regolare il pH a 3,8 per una più facile dissoluzione e quindi ricaricare con acqua a 100 ml.
- Il giorno dopo, inizia con la preparazione dell'aspartato15 di Walton. Miscelare 0,066 g di nitrato di piombo con 10 mL di soluzione di acido L-aspartico (punto 8.1.10) preriscaldato a 60 °C e regolare il pH a 5,5 (misurato a 60 °C) con 1 M NaOH. Chiudere il flaconcino con aspartato di piombo e lasciarlo a 60 °C per 30 minuti a bagnomaria. La soluzione dovrebbe essere chiara. Se diventa torbido, deve essere scartato e ne deve essere preparato uno nuovo.
- Nel frattempo, lavare i campioni con ddH2O degassato cinque volte, ogni volta per 3 minuti. Quindi tenerli in forno a 60 °C per 30 minuti.
- Immergere i campioni nella soluzione di aspartato di piombo appena preparata e incubarli nel forno impostato a 60 °C per 20 minuti.
- Lavare i campioni con ddH2O degassato cinque volte, ogni volta per 3 minuti.
- Disidratazione e incorporamento di resina
- Preparare la resina epossidica. Pesare gli ingredienti (33,3 g di componente A/M, 33,3 g di componente B e 1 g di componente D) e mescolare bene la resina (ad esempio, agitarla su uno shaker rotante in un tubo da 15 ml) per almeno 30 minuti prima di aggiungere 16 gocce di acceleratore DMP 30. Mescolare di nuovo per altri 10 minuti.
NOTA: Pesare gli ingredienti sotto la cappa aspirante. Questa quantità di componenti fornisce circa 60 ml di resina ciò che è sufficiente per 15 fiale con campioni. È possibile preparare meno o conservare il resto della resina in una siringa a 4 °C e utilizzarla il giorno successivo. Ricordarsi di sigillare la punta della siringa. - Preparare flaconcini con diluizioni gradevoli di etanolo (30%, 50%, 70%, 90%, 100%, 100% etanolo in acqua e 100% essiccato su un setaccio molecolare).
- Mescolare la resina con etanolo al 100% in proporzione 1:1 per ottenere il 50% di resina. Mescolare bene.
- Disidratare i campioni per 5 minuti in ogni diluizione di etanolo a partire dal 30% e terminando con etanolo anidro al 100% essiccato su setaccio molecolare. Ricorda, i campioni non devono mai asciugarsi completamente.
- Infiltrare i campioni prima in resina al 50% per 30 minuti, poi in resina al 100% per un'ora e poi di nuovo in resina al 100% durante la notte. Eseguire tutte le fasi di infiltrazione con scuotimento lento costante.
- Il giorno successivo, posizionare i campioni in resina fresca al 100% per un'ora e quindi incorporarli tra i fogli di incorporamento in fluoropolimero. Sgrassare pezzi di foglio di incorporamento con etanolo e incorporare i campioni tra due strati di esso, usando due vetrini come supporto. Cerca di evitare bolle d'aria in resina su o vicino al campione.
- Polimerizzare i campioni in forno a 70 °C per almeno 48 ore.
- Preparare la resina epossidica. Pesare gli ingredienti (33,3 g di componente A/M, 33,3 g di componente B e 1 g di componente D) e mescolare bene la resina (ad esempio, agitarla su uno shaker rotante in un tubo da 15 ml) per almeno 30 minuti prima di aggiungere 16 gocce di acceleratore DMP 30. Mescolare di nuovo per altri 10 minuti.
- Rifilatura e montaggio
- Separare i fogli di incorporamento e tagliare un pezzo del campione incorporato (circa 1 mm x 1 mm) con una lama di rasoio. Trasferiscilo in un parafilm. Ciò ridurrà al minimo il pericolo di perdere il campione a causa dell'elettrostatica.
- Prendi un perno di alluminio che è stato sgrassato con etanolo. Mescolare bene una resina epossidica conduttiva e utilizzarne una piccola quantità per montare il campione sul perno. Polimerizzare la resina epossidica conduttiva a 70 °C per 10 minuti.
- Tagliare ogni lato del blocco campione con il coltello diamantato e quindi lucidare la faccia del blocco fino a quando il tessuto non è esposto.
NOTA: In questa fase, confermare la presenza di una regione di interesse raccogliendo alcune sezioni ed eseguendo la colorazione blu toluidina16 o controllando al microscopio elettronico. - Ridurre il più possibile le dimensioni del campione. Quindi mantecare il campione sul perno con vernice conduttiva e polimerizzarlo (per 24 ore a temperatura ambiente o in forno a 65 °C per 40 minuti).
- Per ridurre al minimo gli artefatti di carica, sputter rivestire i campioni con un sottile strato di oro o oro / palladio.
9. Imaging SBEM
- Posizionare il perno con un campione nella camera del microscopio elettronico a scansione a blocchi seriale. Allineare il campione al coltello. Chiudere la camera e impostare i parametri.
- Raccogli una pila di immagini all'ingrandimento desiderato, alla dimensione dei pixel, allo spessore della fetta, alla tensione accelerata (EHT), all'apertura, alla pressione, ecc.
NOTA: i parametri dipendono dal campione e dall'obiettivo dell'esperimento. Le impostazioni di imaging iniziali esemplari sono incluse nella tabella dei materiali.
Ricostruzioni 10.3D
NOTA: Per i passaggi indicati di seguito utilizziamo software ad accesso aperto, ad esempio FijiJ17 (ImageJ versione 1.49b), Microscopy Image Browser (MIB)18 e Reconstruct19, ma è possibile utilizzare vari altri software.
- Convertire i file di micrografia digitale (dm) in formato TIFF. Per prima cosa importa la sequenza di immagini (in FijiJ: File > Importa > Sequenza di immagini > Scegli formato a 8 bit) e poi salva come TIFF (in FijiJ: File > Salva con nome > Sequenza di immagini > Scegli Tiff).
- Regola la luminosità e il contrasto della pila di immagini (in FijiJ: Image > Adjust > Brightness/Contrast) e, se necessario, denoise (usa il plugin DenoisEM per FijJ20 : Plugin > DenoiseEM > Denoise).
- Allineare lo stack (in FijiJ: Plugins > StagReg o MIB: Dataset > Alignment Tools).
- Segmentare spine dendritiche e sinapsi (nel software MIB o Reconstruct sono disponibili tutorial completi (vedere la tabella dei materiali per i dettagli).
Risultati
Utilizzando il metodo sopra descritto ad alto contrasto, è possibile ottenere immagini a buona risoluzione del tessuto cerebrale del topo. Un ampio campo visivo fornito dalla tecnica SBEM facilita la selezione precisa della regione di interesse. La grande immagine della regione CA1 dell'ippocampo è stata presa per misurare la lunghezza dello strato radiato (SR) (Figura 2A) e per impostare l'imaging precisamente al centro (Figura 2B). Successivamente, ...
Discussione
Ci sono molte varianti del metodo NCMIR primario descritto da Deerinck nel 201010. I principi di base rimangono gli stessi ma, a seconda del tipo di materiale studiato, vengono implementate lievi modifiche. È stato descritto in precedenza che diverse resine possono essere utilizzate per incorporare campioni per SBEM e ad esempio nel caso di piante, Spurr's è la resina di scelta grazie alla sua bassa viscosità che consente una migliore infiltrazione attraverso le pareti cellulari
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
L'imaging SBEM, l'imaging al microscopio ottico e la preparazione del campione al microscopio elettronico sono stati eseguiti con l'uso dell'attrezzatura del Laboratorio di imaging tissutale e funzionale che funge da struttura centrale di imaging presso l'Istituto di biologia sperimentale di Nencki.
Per la preparazione della Figura 1 è stata utilizzata l'immagine di un mouse (Souris_02) e di una fiala del https://smart.servier.com/.
Questo lavoro è stato sostenuto dal National Science Centre (Polonia) Grant Opus (UMO-2018/31/B/NZ4/01603) assegnato a KR.
Materiali
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade,  98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands | 50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands | 10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
Riferimenti
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693 (2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. . NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. J. . The mouse brain in stereotaxic coordinates. , (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771 (2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084 (2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. . Biological specimen preparation for transmission electron microscopy. , (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati