
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Microscopie électronique à balayage en bloc en série (SBEM) pour l’étude des épines dendritiques
Dans cet article
Résumé
La microscopie électronique à balayage en bloc en série (SBEM) est appliquée pour imager et analyser les épines dendritiques de l’hippocampe murin.
Résumé
La microscopie électronique tridimensionnelle (3D EM) donne la possibilité d’analyser les paramètres morphologiques des épines dendritiques avec une résolution à l’échelle nanométrique. En outre, certaines caractéristiques des épines dendritiques, telles que le volume de la colonne vertébrale et la densité post-synaptique (PSD) (représentant la partie post-synaptique de la synapse), la présence de terminal présynaptique et le réticulum endoplasmique lisse ou la forme atypique de PSD (par exemple, les épines multi-innervées), ne peuvent être observées qu’avec l’EM 3D. En utilisant la microscopie électronique à balayage série bloc-face (SBEM), il est possible d’obtenir des données 3D EM plus facilement et de manière plus reproductible que lors de la section série traditionnelle. Nous montrons ici comment préparer des échantillons d’hippocampe de souris pour l’analyse SBEM et comment ce protocole peut être combiné avec une étude d’immunofluorescence des épines dendritiques. La perfusion de fixation légère nous permet d’effectuer des études d’immunofluorescence avec microscopie optique sur une moitié du cerveau, tandis que l’autre moitié a été préparée pour SBEM. Cette approche réduit le nombre d’animaux à utiliser pour l’étude.
Introduction
La plupart des synapses excitatrices du système nerveux central sont situées sur des épines dendritiques - de petites protubérances d’une membrane neuronale. Ces protubérances forment des compartiments biochimiques confinés qui contrôlent la transduction du signal intracellulaire. La plasticité structurelle des épines dendritiques et des synapses est étroitement liée aux changements fonctionnels de l’efficacité synaptique qui sous-tendent des processus aussi importants que l’apprentissage et la mémoire1,2. Il est important de noter que la microscopie électronique (EM) est la seule technique qui permet de déterminer si une colonne vertébrale dendritique a une entrée présynaptique. La résolution EM est également nécessaire pour étudier des détails ultrastructuraux tels que la forme d’une densité postsynaptique (PSD), représentant une partie postsynaptique d’une synapse, ou les dimensions d’une colonne vertébrale dendritique, ainsi que la taille et la forme d’une bouton axonale. De plus, avec EM, il est possible de visualiser les synapses et leur environnement.
Grâce aux progrès des technologies d’imagerie et de calcul, il est possible de reconstruire des circuits neuronaux entiers. Les techniques de microscopie électronique volumique, telles que la microscopie électronique à transmission à section série (ssTEM), la microscopie électronique à balayage série bloc-face (SBEM) et la microscopie électronique à balayage par faisceau d’ions focalisés (FIB-SEM) sont couramment utilisées pour les reconstructions de circuits neuronaux3.
Dans nos études, la méthode SBEM est utilisée avec succès pour étudier la plasticité structurelle des épines dendritiques et des PSD dans des échantillons de l’hippocampe de la souris et des tranches de cerveau organotypiques 4,5. Le SBEM est basé sur l’installation d’un ultramicrotome miniature à l’intérieur de la chambre de microscope électronique à balayage6,7,8,9. Le haut du bloc d’échantillon est imagé, puis l’échantillon est coupé à une profondeur spécifiée par l’ultramicrotome, révélant une nouvelle face de bloc, qui est à nouveau imagée, puis le processus est répété8. En conséquence, seule l’image d’une face de bloc est laissée tandis que la tranche qui a été coupée est perdue sous forme de débris. C’est pourquoi SBEM est appelé une technique destructrice, ce qui signifie qu’il n’est pas possible d’imager à nouveau le même endroit. Cependant, l’avantage des méthodes on-block destructrices est qu’elles ne souffrent pas de problèmes de déformation et de perte de section pouvant affecter de manière significative la qualité des données et l’analyse des données3. De plus, SBEM donne la possibilité d’imager un champ de vision relativement grand (> 0,5 mm × 0,5 mm) à haute résolution3.
Pour utiliser SBEM, les échantillons doivent être préparés selon un protocole dédié et très contrasté en raison du détecteur d’électrons rétrodéclant utilisé pour l’acquisition d’images. Nous montrons ici comment effectuer la préparation des échantillons selon le protocole basé sur une procédure développée par Deerinck10 (méthode du National Center for Microscopy and Imaging Research (NCMIR),en utilisant des taches réduites d’osmium-thiocarbohydrazide-osmium (rOTO) développées dans les années 19808,11. En outre, nous introduisons une approche de fixation en deux étapes, avec une perfusion de fixation légère qui permet d’utiliser le même cerveau à la fois pour les études d’immunofluorescence avec la microscopie optique et SBEM.
Dans le protocole, un cerveau de souris est principalement fixé avec un fixateur léger, puis coupé en deux, et un hémisphère est postfixé et préparé pour l’immunofluorescence (FI), tandis que l’autre pour les études EM (Figure 1).

Graphique 1. Représentation schématique du flux de travail pour la préparation des épines dendritiques pour l’analyse avec SBEM. Les souris ont été sacrifiées et perfusées avec un fixateur primaire léger. Le cerveau a été coupé en deux, et un hémisphère a été postfixé avec un fixateur dédié à l’immunofluorescence (FI), cryoprotégé, tranché à l’aide d’un cryostat et traité pour les études IF, tandis que l’autre hémisphère a été postfixé avec un fixateur EM, tranché avec le vibratome et préparé pour les études EM. Les tranches de cerveau pour les études SBEM ont été contrastées, à plat incorporé dans de la résine, puis une région CA1 de l’hippocampe a été montée sur l’épingle et imagée avec SBEM (Figure 1). La partie du protocole mise en évidence dans une boîte jaune a été présentée dans la vidéo. Veuillez cliquer ici pour voir une version agrandie de cette figure.
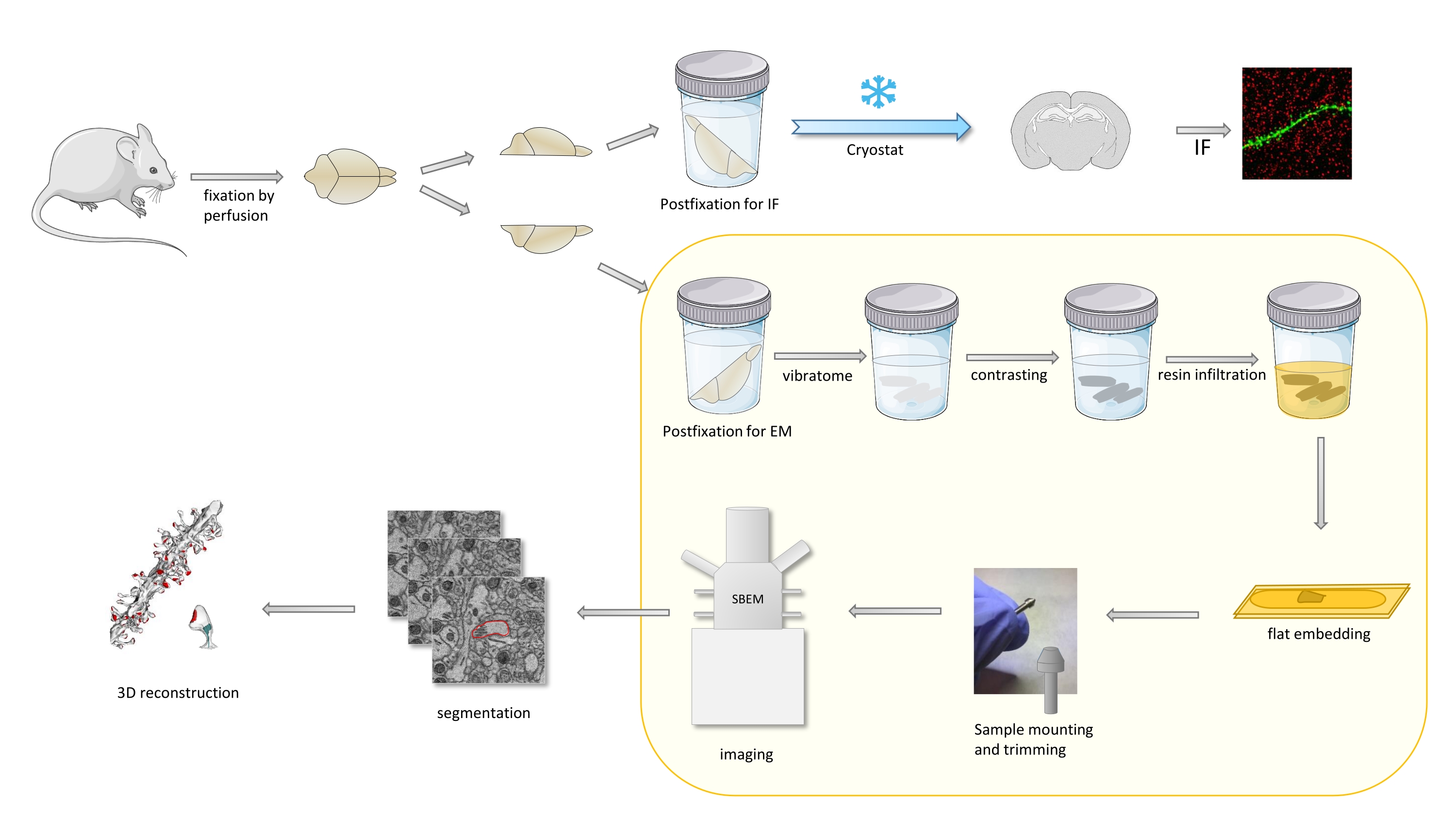{kind=link}
Protocole
La recherche a été réalisée conformément aux directives de l’Institut Nencki et à l’autorisation du comité d’éthique local. Les études ont été réalisées conformément à la directive du Conseil des Communautés européennes du 24 novembre 1986 (86/609/CEE), loi polonaise sur la protection des animaux et approuvées par le premier comité local d’éthique à Varsovie. Tous les efforts ont été faits pour minimiser le nombre d’animaux utilisés et leurs souffrances.
ATTENTION : Toutes les procédures décrites ci-dessous doivent être effectuées dans une hotte de laboratoire. En raison de la nature dangereuse des réactifs utilisés. Des mesures de sécurité personnelle telles que des gants, un manteau de laboratoire, des lunettes de sécurité et un masque facial sont requises.
1. Préparation du fixateur pour perfusion (2 % p/vol de paraformaldéhyde (PFA) et 0,5 % vol/vol de glutaraldéhyde (GA) dans un tampon phosphate (PB) de 0,1 M, pH 7,4)
REMARQUE: Préparez la solution fixative le jour même où elle sera utilisée et ne la conservez pas plus de 3 heures. En cas de manque de temps, préparer 2% de PFA dans 0,1 M PB la veille, le conserver à 4 °C et ajouter de l’AG fraîche peu de temps avant la perfusion.
- Prendre 400 mL d’eau double distillée stérile (ddH2O) et chauffer à 60 °C à l’aide d’une plaque chauffante remuante. Ajouter ensuite 20 g de PFA. Ajouter des gouttes de 1 M de NaOH jusqu’à ce que le PFA soit complètement dissous et laisser refroidir le mélange.
- Ajouter 500 mL de 0,2 M PB (pH 7,4).
- Filtrez la solution pour éliminer tout dépôt et refroidissez-la à 4 °C.
- Juste avant la perfusion, ajouter 20 mL de 25% GA à la solution, puis compléter le volume avec ddH2O à 1 L.
2. Préparation du fixateur post-perfusion pour SBEM (2 % p./vol de PFA et 2,5 % vol/vol d’AG en 0,1 M PB, pH 7,4)
- Prendre 50 mL du fixateur pour la perfusion (2 % de PFA et 0,5 % d’AG dans 0,1 M PB).
- Ajouter 5 mL de 25 % d’AG.
3. Préparation du fixateur post-perfusion pour la coloration IF (4% PFA dans une solution saline tamponnée au phosphate (PBS))
- Dissoudre un comprimé de 1x PBS (pH 7,4) dans une eau purifiée et désionisée (H2O) selon les instructions du fabricant.
- Utilisez une plaque chauffante remuante pour chauffer la solution à 60 °C et ajoutez 40 g de PFA.
- Ajouter des gouttes de 1 M naOH jusqu’à ce que le PFA soit complètement dissous et laisser refroidir le mélange.
- Réglez le pH de la solution à 7,5 avec 1 M HCl puis rechargez le volume avec H2O à 1 L.
- Filtrez la solution pour éliminer les dépôts.
4. Perfusion transcardique d’animaux
REMARQUE: Tous les déchets PFA et GA doivent être collectés et stockés pour élimination conformément aux réglementations locales. L’anesthésie et la perfusion doivent respecter les réglementations locales. Dans le protocole décrit, des souris Thy1-GFP(M) adultes de 3 mois et femelles de 20±1 mois ont été utilisées, mais toute autreexprimant la protéine de fluorescence verte (GFP) dans une population peu distribuée de neurones glutamatergiques a été utilisée, mais toute autre peut également être utilisée. Les animaux ont été élevés en tant qu’hétérozygotes avec le fond C57BL / 6J dans la maison des animaux de l’Institut Nencki de biologie expérimentale.
- Avant la perfusion anesthésier une souris par administration d’un mélange kétamine/xylazine (jusqu’à 90 mg/kg de kétamine de poids corporel et 10 mg/kg de xylazine de poids corporel) par injection intrapéritonéale (aiguille de calibre 27).
- Évaluez si la profondeur de l’anesthésie est suffisante en vérifiant le réflexe aux stimuli douloureux (pincement) et le réflexe cornéen (plissement des voix).
- Après 20 minutes, effectuer une injection intrapéritonéale (aiguille de calibre 27) de pentobarbital de sodium (50 mg / kg de poids corporel).
- Perfuser une souris selon un protocole de chirurgie de perfusion décrit par Gage et al.13 (voir point 4; Figure 5-6). À l’aide d’une pompe de perfusion, commencez avec un tampon phosphate de 0,1 M, pH 7,4 (30 mL) pendant 3 minutes et continuez avec 2 % de PFA et 0,5 % de GA dans 0,1 M PB, pH 7,4 pendant 6 minutes (80 mL).
- Disséquez doucement le cerveau du crâne et divisez-le en deux (voir la figure 9-10 dans Gage et al., 201213). Placez une pièce dans un flacon contenant un fixateur pour SBEM et la seconde dans le flacon avec 4% de PFA / PBS pour la coloration IF.
- Gardez les hémisphères dans le fixateur à 4 °C pendant la nuit.
5. Préparation de tranches de cerveau pour la microscopie électronique
- Choisissez les réglages du vibratome (vitesse de déplacement de la lame : 0,075 mm/s, fréquence de coupe : 80 Hz).
- Placez la chambre à tranches dans le support, fixez-la au vibratome et entourez-la de glace. Placez ensuite une lame de rasoir dans le support de lame vibratome.
- Utilisez une cuillère ou un objet similaire et placez le cerveau réfrigéré (surface dorsale vers le haut) sur une surface de coupe dure (par exemple, un couvercle de boîte de Petri en verre). Pour préparer une tranche coronale de l’hippocampe, faites une coupe perpendiculaire entre l’hémisphère cérébral et le cervelet avec une lame de rasoir ou un scalpel, éliminant ainsi le cervelet. Le bulbe olfactif peut également être retiré.
- Appliquer de la colle cyanoacrylate sur la plate-forme sèche du vibratome.
- Ramassez le cerveau avec une pince et séchez-le soigneusement sur du papier filtre.
- Collez l’hémisphère à la plate-forme près de la lame de coupe avec la pointe rostrale vers le haut. Fixez la plate-forme au support et remplissez-la immédiatement avec du froid glacial de 0,1 M PB, pH 7,4. Si la coupe perpendiculaire est correctement faite, l’hémisphère se tiendra droit en fournissant un angle de 90 ° nécessaire pour faire une coupe coronale symétrique contenant l’hippocampe. Assurez-vous que le cerveau est recouvert de PB.
- Placez la lame du vibratome devant l’hémisphère et abaissez-la sur le côté coronal de l’hémisphère. Abaissez la lame à 400 μm plus loin dans la direction caudale et commencez à trancher. Continuez à trancher jusqu’à ce que les deux premières tranches soient complètement séparées du bloc tissulaire.
- Rétractez la lame et abaissez encore 100 μm, puis coupez à nouveau.
- Lorsque l’hippocampe devient visible (utilisez l’atlas du cerveau de souris Paxinos et Franklin, 200414) collectez les tranches avec le petit pinceau ou la pipette Pasteur en plastique élargie.
- Transférer les tranches dans une assiette de 12 puits remplie de 0,1 M PB froid, pH 7,4.
- Disséquez l’hippocampe dans une boîte de Petri en verre remplie de 0,1 M PB, pH 7,4 à l’aide d’une lame de rasoir ou d’un scalpel, et mettez-le dans des flacons en verre avec le même tampon phosphate.
REMARQUE: Pour le stockage à long terme des tranches, supplémentez 0,1 M PB avec 0,05% d’azide de sodium (NaN3).
6. Préparation d’échantillons de cerveau pour l’immunocoloration
- Après une fixation pendant la nuit dans une solution de PFA/PBS à 4 % dans une hotte aspirante, mettre le tissu cérébral dans la solution de cryoconservation (30 % de saccharose dans le PBS avec 0,05 % deNaN3)et le maintenir à 4 °C pendant 2 jours (jusqu’à ce qu’il soit coulé).
- Préparer une solution d’antigel (15 % de saccharose/30 % d’éthylène glycol/0,05 % de NaN3/PBS).
- Réglez la température de l’armoire du cryostat à -19 °C et assurez-vous que la température est atteinte avant de continuer. Pendant le sectionnement, assurez-vous que la température de l’armoire reste comprise entre -18 °C et -20 °C.
- Utilisez une cuillère ou un objet similaire et placez le cerveau réfrigéré (surface dorsale vers le haut) sur une surface de coupe dure (par exemple, un couvercle de boîte de Petri en verre). Pour préparer une tranche coronale de l’hippocampe, faites une coupe perpendiculaire entre l’hémisphère cérébral et le cervelet avec une lame de rasoir ou un scalpel, éliminant ainsi le cervelet.
- Sélectionnez un disque d’échantillon pré-refroidi, couvrez-le d’un support pour la congélation sur une étagère libérante et, à l’aide d’une pince, fixez l’hémisphère au disque avec une pointe rostrale vers le haut et attendez que l’échantillon soit complètement gelé. Insérez le disque d’échantillon dans une tête d’échantillon.
- Installez une lame dans un porte-lame à l’intérieur de la chambre cryogénique et coupez des tranches de 40 μm d’épaisseur.
- Transférer les tranches avec un petit pinceau dans la plaque de 96 puits remplie de solution antigel froide et dérouler doucement les sections (recueillir une tranche après chaque tour de tranchage pour éviter qu’elles ne se perdent dans la chambre à tranches).
REMARQUE: Les cerveaux ou les sections peuvent être stockés dans une solution d’antigel à -20 ° C pendant une longue période.
7. Immunocoloration des tranches de cerveau
REMARQUE: Toutes les étapes de coloration ont été effectuées dans une plaque de 24 puits sur un agitateur de plate-forme.
- Lavez les tranches avec du PBS trois fois, à chaque fois pendant 6 minutes.
- Incuber des tranches dans 300 μL de solution bloquante (5% de sérum d’âne normal (NDS)/0,3% de Triton X-100) pendant 1 heure en agitant doucement sur un rotateur.
- Incuber les tranches avec l’anticorps primaire contre le PSD-95 dilué à 1:500 dans 5 % de NDS/0,3 % de Triton X-100/PBS (300 μL par puits) pendant la nuit à 4 °C. La concentration finale de l’anticorps primaire est de 2 μg/mL.
- Lavez les tranches avec 0,3 % de Triton X-100/PBS à température ambiante (RT) trois fois, à chaque fois pendant 6 minutes.
- Incuber des tranches avec l’anticorps secondaire dilué à 1:500 dans 300 μL de Triton X-100/PBS à 0,3 % pendant 90 minutes. La concentration finale de l’anticorps secondaire est de 4 μg/mL.
- Lavez les tranches avec du PBS trois fois, à chaque fois pendant 6 minutes.
- Montez les tranches sur les diapositives à l’aide d’un support de montage, puis fermez-les avec une glissière de couverture.
- Examinez les échantillons à l’aide d’un microscope confocal (objectif d’huile de 63 ×, NA 1,4, taille de pixel 0,13 μm × 0,13 μm).
REMARQUE: Pour un stockage à long terme: conservez les échantillons à 4 ° C, protégez-les de la lumière.
8. Préparation de l’échantillon SBEM
ATTENTION: En raison de la nature dangereuse des réactifs utilisés, toutes les procédures décrites ci-dessous doivent être effectuées dans une hotte de laboratoire. Avant d’utiliser ces produits chimiques, lisez attentivement les fiches de données de sécurité fournies par les fabricants et interrogez l’agent de sécurité sur les règles locales pour assurer une manipulation et une élimination des déchets en toute sécurité.
- Exemple contrasté
REMARQUE: Le lavage des tranches et l’incubation à température ambiante doivent être effectués en agitant légèrement (par exemple, sur un agitateur de plate-forme). De l’eau dégazée en autoclave a été utilisée.- Lavez les tranches froides avec 0,1 M PB, pH 7,4 cinq fois, à chaque fois pendant 3 minutes.
- Préparer un mélange 1:1 de tétroxyde d’osmium aqueux à 4 % et de ferrocyanure de potassium à 3 % (1:1 en vol). Le produit final brunisse. Immergez les échantillons dans ce mélange, placez-les sur de la glace et, à partir de ce stade, il est important de les protéger de la lumière. Incubés en les agitant doucement pendant 1 heure.
- Pendant ce temps, préparez une solution de thiocarbohydrazide (TCH). Mélanger 10 mL deH2O(ddH2 O) double distillé et 0,1 g de TCH et le placer dans un four réglé à 60 °C pendant 1 heure. Il est important de faire tourbillonner la solution de temps en temps (par exemple, toutes les 10 minutes). Lorsque vous êtes prêt, refroidissez-le à la température ambiante.
- Laver les tranches avec ddH2O cinq fois, à chaque fois pendant 3 minutes.
- Filtrer la solution TCH à l’aide d’un filtre à seringue de 0,22 μm et immerger l’échantillon dans une solution filtrée. Les tranches deviendront noires. Incubés pendant 20 minutes à température ambiante.
- Lavez les échantillons avec ddH2O cinq fois, à chaque fois pendant 3 minutes.
- Incuber les échantillons avec une solution aqueuse à 2% d’OsO4 pendant 30 minutes à température ambiante.
- Lavez les échantillons avec ddH2O cinq fois, à chaque fois pendant 3 minutes.
- Placer les échantillons dans de l’acétate d’uranyle aqueux filtré à 1 % et les incuber à 4 °C pendant la nuit. Utilisez un filtre à seringue de 0,22 μm pour la filtration.
- Préparer la solution d’acide L-aspartique en mélangeant 0,4 g d’acide L-aspartique et 80 mL de ddH2O, ajuster le pH à 3,8 pour faciliter la dissolution, puis compléter avec de l’eau à 100 mL.
- Le lendemain, commencez par la préparation de Walton aspartate15. Mélanger 0,066 g de nitrate de plomb avec 10 mL de solution d’acide L-aspartique (point 8.1.10) préchauffé à 60 °C et ajuster le pH à 5,5 (mesuré à 60 °C) avec 1 M de NaOH. Fermer le flacon avec de l’aspartate de plomb et le laisser à 60 °C pendant 30 minutes au bain-marie. La solution doit être claire. S’il devient trouble, il doit être jeté et un nouveau doit être préparé.
- En attendant, laver les échantillons avec du ddHdégazé 2O cinq fois, à chaque fois pendant 3 minutes. Conservez-les ensuite au four à 60 °C pendant 30 minutes.
- Immergez les échantillons dans la solution d’aspartate de plomb fraîchement préparée et incubez-les dans le four réglé à 60 °C pendant 20 minutes.
- Laver les échantillons avec du ddHdégazé 2O cinq fois, à chaque fois pendant 3 minutes.
- Déshydratation et incorporation de résine
- Préparez la résine époxy. Peser les ingrédients (33,3 g du composant A/M, 33,3 g du composant B et 1 g du composant D) et bien mélanger la résine (p. ex., la secouer sur un agitateur rotatif dans un tube de 15 mL) pendant au moins 30 minutes avant d’ajouter 16 gouttes d’accélérateur DMP 30. Remuer à nouveau pendant encore 10 minutes.
REMARQUE: Peser les ingrédients sous la hotte. Cette quantité de composants donne environ 60 mL de résine ce qui est suffisant pour 15 flacons avec des échantillons. Vous pouvez préparer moins ou stocker le reste de la résine dans une seringue à 4 °C et l’utiliser le lendemain. N’oubliez pas de sceller la pointe de la seringue. - Préparer des flacons avec des dilutions graduées d’éthanol (30%, 50%, 70%, 90%, 100%, 100% d’éthanol dans l’eau et 100% séché sur un tamis moléculaire).
- Mélanger la résine avec 100% d’éthanol dans une proportion de 1:1 pour obtenir 50% de résine. Mélangez bien.
- Déshydrater les échantillons pendant 5 minutes dans chaque dilution d’éthanol à partir de 30% et se terminant par de l’éthanol anhydre à 100% séché sur tamis moléculaire. N’oubliez pas que les échantillons ne doivent jamais sécher complètement.
- Infiltrez d’abord les échantillons dans 50% de résine pendant 30 minutes, puis dans 100% de résine pendant une heure, puis à nouveau dans 100% de résine pendant la nuit. Effectuez toutes les étapes d’infiltration avec des secousses lentes constantes.
- Le lendemain, placez les échantillons dans de la résine fraîche à 100% pendant une heure, puis incorporez-les entre des feuilles d’incorporation de fluoropolymères. Dégraissez des morceaux de feuille d’encastrement avec de l’éthanol et incorporez à plat les échantillons entre deux couches de celui-ci, en utilisant deux lames de verre comme support. Essayez d’éviter les bulles d’air dans la résine sur ou à proximité de l’échantillon.
- Durcir les échantillons au four à 70 °C pendant au moins 48 heures.
- Préparez la résine époxy. Peser les ingrédients (33,3 g du composant A/M, 33,3 g du composant B et 1 g du composant D) et bien mélanger la résine (p. ex., la secouer sur un agitateur rotatif dans un tube de 15 mL) pendant au moins 30 minutes avant d’ajouter 16 gouttes d’accélérateur DMP 30. Remuer à nouveau pendant encore 10 minutes.
- Rognage et montage
- Séparez les feuilles d’encastrement et découpez un morceau de l’échantillon incorporé (environ 1 mm x 1 mm) avec une lame de rasoir. Transférez-le sur un parafilm. Cela minimisera le risque de perdre l’échantillon en raison de l’électrostatique.
- Prenez une broche en aluminium qui a été dégraissée avec de l’éthanol. Mélangez bien un époxy conducteur et utilisez-en une petite quantité pour monter l’échantillon sur la broche. Durcir l’époxy conducteur à 70 °C pendant 10 minutes.
- Coupez chaque côté du bloc d’échantillon avec le couteau à diamant, puis polissez la face du bloc jusqu’à ce que le tissu soit exposé.
REMARQUE: À cette étape, confirmez la présence d’une région d’intérêt en collectant certaines sections et en effectuant une coloration au bleu toluidine16 ou en vérifiant au microscope électronique. - Réduisez autant que possible la taille de l’échantillon. Ensuite, broyez l’échantillon à l’épingle avec de la peinture conductrice et durcissez-le (pendant 24 heures à température ambiante ou au four à 65 °C pendant 40 minutes).
- Pour minimiser les artefacts de charge, recouvrez les échantillons d’une fine couche d’or ou d’or/palladium.
9. Imagerie SBEM
- Placez la broche avec un échantillon dans la chambre du microscope électronique à balayage à face de bloc en série. Alignez l’échantillon sur le couteau. Fermez la chambre et définissez les paramètres.
- Collectez une pile d’images au grossissement souhaité, à la taille des pixels, à l’épaisseur de la tranche, à la tension accélérée (EHT), à l’ouverture, à la pression, etc.
REMARQUE : Les paramètres dépendent de l’échantillon et de l’objectif de l’expérience. Des paramètres d’imagerie initiaux exemplaires sont inclus dans le tableau des matériaux.
10.3D reconstructions
REMARQUE: Pour les étapes mentionnées ci-dessous, nous utilisons des logiciels en libre accès, par exemple FijiJ17 (ImageJ version 1.49b), Microscopy Image Browser (MIB)18 et Reconstruct19, mais divers autres logiciels peuvent être utilisés.
- Convertissez des fichiers de micrographie numérique (dm) au format TIFF. Importez d’abord la séquence d’images (dans FijiJ : Fichier > Importer > Séquence d’images > Choisissez le format 8 bits), puis enregistrez-la au format TIFF (en FidjiJ : Fichier > Enregistrer sous > Séquence d’images > Choisissez Tiff).
- Ajustez la luminosité et le contraste de la pile d’images (en FidjiJ: Image > Ajuster > Luminosité / Contraste) et, si nécessaire, débruissez-le (utilisez le plugin DenoisEM pour FijJ20: Plugins > DenoiseEM > Denoise).
- Alignez la pile (dans FijiJ: Plugins > StagReg ou MIB: Dataset > Alignment Tools).
- Segment des épines dendritiques et des synapses (Dans les logiciels MIB ou Reconstruct, des tutoriels complets sont disponibles (voir la table des matériaux pour plus de détails).
Résultats
En utilisant la méthode décrite ci-dessus à contraste élevé, des images de bonne résolution du tissu cérébral de la souris peuvent être obtenues. Un large champ de vision fourni par la technique SBEM facilite la sélection précise de la région d’intérêt. La grande image de la région CA1 de l’hippocampe a été prise pour mesurer la longueur de la couche radiale (SR)(Figure 2A)et pour fixer l’imagerie précisément au centre (Figure 2B
Discussion
Il existe de nombreuses variantes de la méthode NCMIR primaire décrite par Deerinck en 201010. Les principes de base restent les mêmes mais, selon le type de matériel étudié, de légers changements sont mis en œuvre. Il a été décrit précédemment que différentes résines peuvent être utilisées pour intégrer des spécimens pour SBEM et par exemple dans le cas des plantes, Spurr est la résine de choix en raison de sa faible viscosité qui permet une meilleure infiltration à travers ...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
L’imagerie SBEM, l’imagerie par microscopie optique et la préparation d’échantillons de microscopie électronique ont été réalisées à l’utilisation de l’équipement du Laboratoire d’imagerie des tissus et des fonctions qui sert d’installation centrale d’imagerie à l’Institut Nencki de biologie expérimentale.
Pour la préparation de la figure 1, l’image d’une souris (Souris_02) et un flacon de la https://smart.servier.com/ ont été utilisés.
Ce travail a été soutenu par la subvention Opus (UMO-2018/31/B/NZ4/01603) du Centre national des sciences (Pologne) attribuée à KR.
matériels
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade,  98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands | 50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands | 10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
Références
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693 (2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. . NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. J. . The mouse brain in stereotaxic coordinates. , (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771 (2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084 (2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. . Biological specimen preparation for transmission electron microscopy. , (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.