
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Microscopia eletrônica de varredura de blocos de bloco serial (SBEM) para o estudo de espinhas dendríticas
Neste Artigo
Resumo
A Microscopia eletrônica de varredura de blocos de blocos serial (SBEM) é aplicada à imagem e análise de colunas dendríticas no hipocampo murino.
Resumo
A microscopia eletrônica tridimensional (3D EM) oferece a possibilidade de analisar parâmetros morfológicos de colunas dendríticas com resolução de nanoescala. Além disso, algumas características das espinhas dendríticas, como volume da coluna vertebral e densidade pós-sináptica (PSD) (representando parte pós-sináptica da sinapse), presença de terminal pré-sináptico e retículo endoplasmático liso ou forma atípica de PSD (por exemplo, colunas multi-inervadas), podem ser observadas apenas com EM 3D. Ao empregar microscopia eletrônica de varredura de blocos serial (SBEM), é possível obter dados EM 3D mais fáceis e de forma mais reprodutível do que ao realizar seções seriais tradicionais. Aqui mostramos como preparar amostras hipocampais de camundongos para análise do SBEM e como este protocolo pode ser combinado com o estudo de imunofluorescência de espinhas dendríticas. A fixação leve da perfusão nos permite realizar estudos de imunofluorescência com microscopia leve em metade do cérebro, enquanto a outra metade foi preparada para o SBEM. Essa abordagem reduz o número de animais a serem utilizados para o estudo.
Introdução
A maioria das sinapses excitatórias no sistema nervoso central estão localizadas em espinhas dendríticas - pequenas saliências de uma membrana neuronal. Essas saliências formam compartimentos bioquímicos confinados que controlam a transdução de sinal intracelular. A plasticidade estrutural das espinhas e sinapses dendríticas está intimamente relacionada com as mudanças funcionais na eficácia sináptica que fundamentam processos tão importantes como a aprendizagem e a memória1,2. É importante notar que a microscopia eletrônica (EM) é a única técnica que permite determinar se uma coluna dendrítica tem uma entrada pré-sináptica. A resolução EM também é necessária para estudar detalhes ultraestruturais, como a forma de uma densidade postiáspática (PSD), representando uma parte postsiáspática de uma sinapse, ou dimensões de uma coluna dendrítica, bem como o tamanho e a forma de um bouton axonal. Além disso, com o EM é possível visualizar sinapses e seus arredores.
Graças aos avanços nas tecnologias de imagem e computação é possível reconstruir circuitos neurais inteiros. Técnicas de microscopia eletrônica de volume, como microscopia eletrônica de transmissão de seção serial (ssTEM), microscopia eletrônica de varredura de rosto serial (SBEM) e microscopia eletrônica de varredura de feixe de íons (FIB-SEM) são comumente utilizadas para reconstruções de circuito neuronal3.
Em nossos estudos, o método SBEM é empregado com sucesso para investigar a plasticidade estrutural de espinhos dendráticos e PSDs em amostras do hipocampo do camundongo e das fatias cerebrais organotípicas 4,5. O SBEM é baseado na instalação de um ultramicromo em miniatura dentro da câmara de microscópio eletrônico de varredura6,7,8,9. A parte superior do bloco de amostra é imageda e, em seguida, a amostra é cortada a uma profundidade especificada pelo ultramicrotome, revelando um novo bloco-face, que é novamente imagedo e, em seguida, o processo é repetido8. Como resultado, apenas a imagem de um rosto de bloco é deixada enquanto a fatia que foi cortada é perdida como detritos. É por isso que o SBEM é chamado de técnica destrutiva, o que significa que não é possível imaginar o mesmo lugar novamente. No entanto, a vantagem dos métodos destrutivos no bloco é que eles não sofrem de problemas de deformação e perda de seção que podem afetar significativamente a qualidade dos dados e a análise de dados3. Além disso, a SBEM dá a possibilidade de imagem de um campo de visão relativamente grande (> 0,5 mm × 0,5 mm) em alta resolução3.
Para empregar o SBEM, as amostras devem ser preparadas de acordo com um protocolo dedicado e altamente contrastante devido ao detector de elétrons backscattered usado para a aquisição de imagens. Mostramos aqui como realizar a preparação da amostra de acordo com o protocolo baseado em um procedimento desenvolvido pelo Deerinck10 (Centro Nacional de Pesquisa em Microscopia e Imagem (NCMIR), utilizando manchas reduzidas de osmium-thiocarbohydrazida-osmium (rOTO) desenvolvidas na década de 19808,11. Além disso, introduzimos uma abordagem de fixação em duas etapas, com perfusão de fixação leve que permite usar o mesmo cérebro tanto para estudos de imunofluorescência com microscopia leve e SBEM.
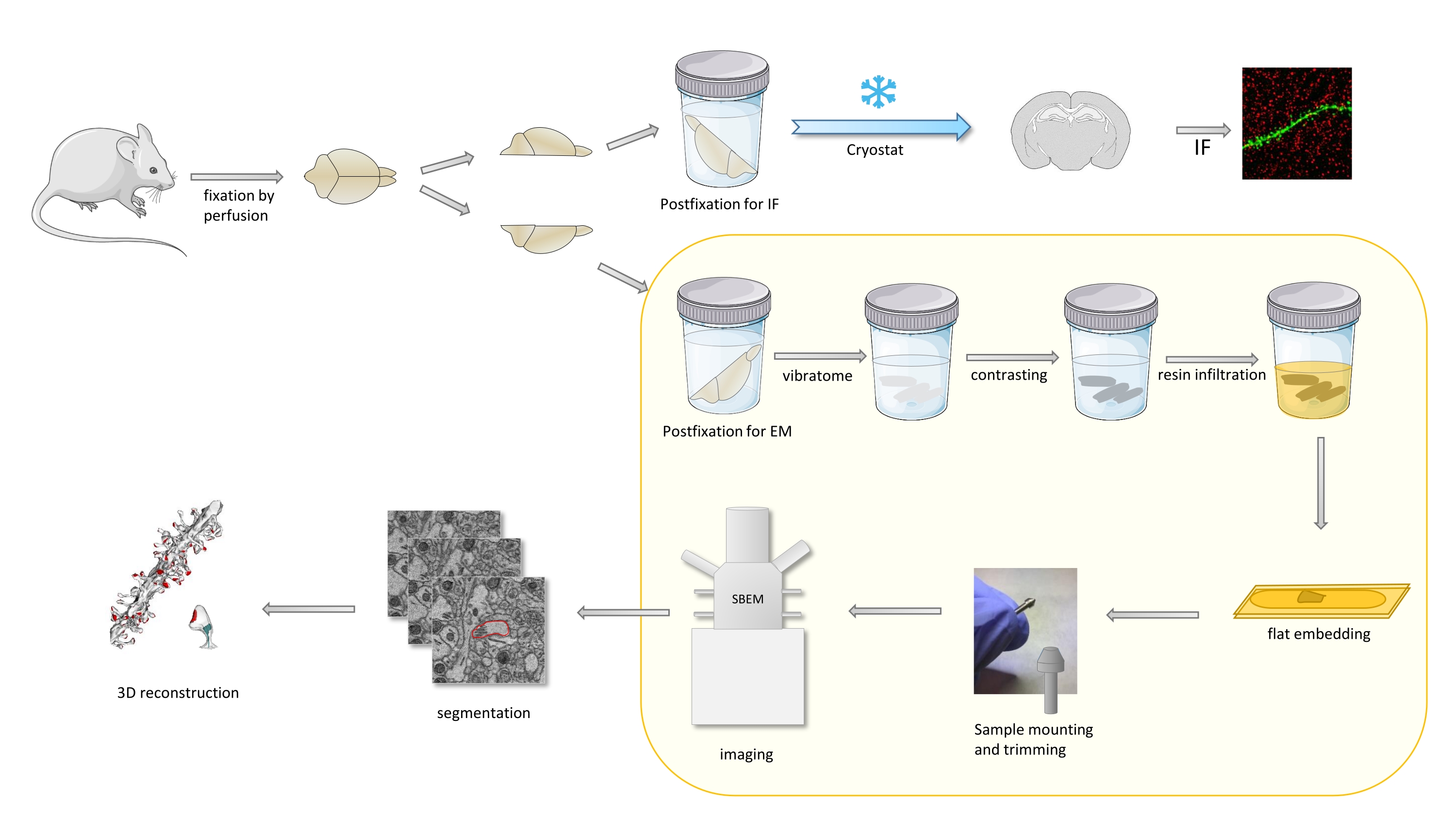
No protocolo, um cérebro de camundongo é fixado principalmente com um fixador leve, e depois cortado em metades, e um hemisfério é postfixado e preparado para imunofluorescência (IF), enquanto o outro para estudos EM(Figura 1).

Figura 1. Representação esquemática do fluxo de trabalho para a preparação das lombadas dendríticas para a análise com a SBEM. Os ratos foram sacrificados e perfundidos com um leve fixador primário. O cérebro foi cortado em metades, e um hemisfério foi postfixado com imunofluorescência (IF) dedicada fixação, crioprotegida, fatiada usando um criostat e processada para estudos if, enquanto o outro hemisfério foi postfixado com FIXAtivo EM, fatiado com o vibratome e preparado para estudos EM. As fatias cerebrais para estudos da SBEM foram contrastadas, planas embutidas em resina, em seguida, uma região ca1 do hipocampo foi montada ao pino, e imageada com SBEM (Figura 1). A parte do protocolo destacada em uma caixa amarela foi destaque no vídeo. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocolo
A pesquisa foi realizada em conformidade com as diretrizes do Instituto Nencki e a permissão do Comitê De ética Local. Os estudos foram realizados de acordo com a Diretiva do Conselho europeu de Comunidades de 24 de Novembro de 1986 (86/609/CEE), Lei de Proteção Animal da Polônia e aprovado pelo primeiro Comitê de Ética Local em Varsóvia. Todos os esforços foram feitos para minimizar o número de animais utilizados e seu sofrimento.
ATENÇÃO: Todos os procedimentos descritos abaixo devem ser realizados em um capô de fumaça de laboratório. Devido à natureza perigosa dos reagentes utilizados. São necessárias medidas de segurança pessoal, como luvas, jaleco, óculos de segurança e máscara facial.
1. Preparação do fixador para perfusão (2% wt/vol paraformaldeído (PFA) e 0,5% vol/vol glutaraldeído (GA) em 0,1 M tampão fosfato (PB), pH 7,4)
NOTA: Prepare a solução fixativa no mesmo dia em que será usada e não a armazene por mais tempo do que por 3 horas. Em caso de falta de tempo, prepare 2% de PFA em 0,1 M PB no dia anterior, armazene-o a 4 °C e adicione ga fresco pouco antes da perfusão.
- Pegue 400 mL de água dupla destilada estéril (ddH2O) e aqueça-a a 60 °C usando uma placa quente agitada. Em seguida, adicione 20 g de PFA. Adicione gotas de 1 M NaOH até que o PFA esteja totalmente dissolvido e deixe a mistura esfriar.
- Adicione 500 mL de 0,2 M PB (pH 7.4).
- Filtre a solução para remover qualquer depósito e esfrie-a a 4 °C.
- Pouco antes da perfusão, adicione 20 mL de 25% de GA à solução e, em seguida, reescode o volume com ddH2O a 1 L.
2. Preparação de fixação pós-perfusão para SBEM (2% wt/vol PFA e 2,5% vol/vol GA em 0,1 M PB, pH 7.4)
- Tome 50 mL do fixador para perfusão (2% PFA e 0,5% GA em 0,1 M PB).
- Adicione 5 mL de 25% de GA.
3. Preparação de fixação pós-perfusão para coloração IF (4% PFA em soro fisiológico tamponado de fosfato (PBS))
- Dissolva um comprimido de 1x PBS (pH 7.4) em uma água purificada e deionizada (H2O) de acordo com as instruções do fabricante.
- Use uma placa quente para aquecer a solução a 60 °C e adicione 40 g de PFA.
- Adicione gotas de 1 M NaOH até que o PFA esteja totalmente dissolvido e deixe a mistura esfriar.
- Ajuste o pH da solução para 7,5 com 1 M HCl e, em seguida, ressarca o volume com H2O a 1 L.
- Filtre a solução para remover quaisquer depósitos.
4. Perfusão transcárvia de animais
NOTA: Todos os resíduos de PFA e GA devem ser coletados e armazenados para descarte de acordo com as regulamentações locais. Anestesia e perfusão devem seguir as normas locais. No protocolo descrito,± foram utilizados camundongos thy1-GFP(M) (Thy1-GFP +/-)12 expressando proteína de fluorescência verde (GFP) em uma população pouco distribuída de neurônios glutamatergicos, mas qualquer outro também pode ser usado. Os animais foram criados como heterozigotes com o fundo C57BL/6J na Casa animal do Instituto Nencki de Biologia Experimental.
- Antes de anestesiar um rato por administração de uma mistura de cetamina/xilazina (até 90 mg/kg de cetamina de peso corporal e 10 mg/kg de xilazina) através de injeção intraperitoneal (agulha de 27 bitola).
- Avalie se a profundidade da anestesia é suficiente verificando o reflexo aos estímulos de dor (beliscar) e o reflexo da córnea (squinting).
- Após 20 minutos, realize uma injeção intraperitoneal (agulha de calibre 27) de pentobarbital de sódio (50 mg/kg de peso corporal).
- Perfuse um rato de acordo com um protocolo de cirurgia de perfusão descrito por Gage et al.13 (ver ponto 4; Figura 5-6). Utilizando uma bomba de perfusão comece com um tampão fosfato de 0,1 M, pH 7.4 (30 mL) por 3 minutos e continue com 2% PFA e 0,5% GA em 0,1 M PB, pH 7,4 por 6 minutos (80 mL).
- Disseque suavemente o cérebro do crânio e divida-o ao meio (ver Figura 9-10 em Gage et al., 201213). Coloque uma peça em um frasco contendo fixação para SBEM e a segunda no frasco com 4% pfa/PBS para coloração IF.
- Mantenha os hemisférios na fixação a 4 °C durante a noite.
5. Cérebro corta preparação para microscopia eletrônica
- Escolha as configurações do vibratome (velocidade de viagem da lâmina: 0,075 mm/s, frequência de corte: 80 Hz).
- Coloque a câmara de fatia no suporte, anexe-a ao vibratome e cerque-a com gelo. Em seguida, coloque uma lâmina de barbear no suporte da lâmina vibratome.
- Use uma colher, ou objeto semelhante e coloque o cérebro resfriado (superfície dorsal para cima) em uma superfície de corte resistente (por exemplo, uma tampa de placa de Petri de vidro). Para preparar uma fatia coronal do hipocampo faça um corte perpendicular entre o hemisfério cerebral e o cerebelo com uma lâmina de barbear ou bisturi, removendo assim o cerebelo. A lâmpada olfativa também pode ser removida.
- Aplique cola de cianoacrilato na plataforma seca do vibratome.
- Pegue o cérebro com fórceps e seque-o cuidadosamente no papel do filtro.
- Cole o hemisfério à plataforma perto da lâmina de corte com a ponta rostral para cima. Conecte a plataforma ao suporte e preencha-a imediatamente com gelo frio de 0,1 M PB, pH 7.4. Se o corte perpendicular for devidamente feito, o hemisfério ficará em pé fornecendo ângulo de 90° necessário para fazer um corte coronal simétrico contendo o hipocampo. Certifique-se de que o cérebro está coberto de PB.
- Posicione a lâmina vibratome na frente do hemisfério e abaixe-a para o lado coronal do hemisfério. Abaixe a lâmina para 400 μm ainda mais na direção caudal e comece a cortar. Continue cortando até que as duas primeiras fatias estejam completamente separadas do bloco de tecido.
- Retire a lâmina e baixe mais 100 μm, depois corte novamente.
- Quando o hipocampo se torna visível (use o cérebro do rato atlas Paxinos e Franklin, 200414) colete as fatias com o pequeno pincel ou pipeta pasteur de plástico alargada.
- Transfira as fatias para uma placa de 12 poços cheia de frio de 0,1 M PB, pH 7.4.
- Disseque o hipocampo em uma placa de vidro Petri cheia de 0,1 M PB, pH 7.4 usando uma lâmina de barbear ou bisturi, e coloque-o em frascos de vidro com o mesmo tampão de fosfato.
NOTA: Para armazenamento a longo prazo das fatias suplemento 0,1 M PB com azida de sódio de 0,05% (NaN3).
6. Preparação da amostra cerebral para imunostaining
- Após fixação durante a noite em 4% solução PFA/PBS em um capô de fumaça, coloque o tecido cerebral na solução de criopreservação (30% de sacarose em PBS com 0,05% NaN3) e mantenha-o a 4 °C por 2 dias (até afundar).
- Prepare uma solução anticongelante (15% de sacarose/30% de etileno glicol/0,05% NaN3/PBS).
- Coloque a temperatura do gabinete do criostat em -19 °C e certifique-se de que a temperatura seja atingida antes de prosseguir. Durante a secção, certifique-se de que a temperatura do gabinete permaneça entre -18 °C e -20 °C.
- Use uma colher, ou objeto semelhante e coloque o cérebro resfriado (superfície dorsal para cima) em uma superfície de corte resistente (por exemplo, uma tampa de placa de Petri de vidro). Para preparar uma fatia coronal do hipocampo faça um corte perpendicular entre o hemisfério cerebral e o cerebelo com uma lâmina de barbear ou bisturi, removendo assim o cerebelo.
- Selecione um disco de espécime pré-resfriado, cubra-o com um meio para congelar em uma prateleira de liberação e usando fórceps fixe o hemisfério no disco com uma ponta rostral para cima e espere até que o espécime esteja completamente congelado. Insira o disco da amostra em uma cabeça de espécime.
- Instale uma lâmina em um suporte de lâmina dentro da câmara criogênica e corte fatias de 40 μm de espessura.
- Transfira fatias com um pequeno pincel para a placa de 96 poços cheia de solução anticongelante a frio e desenrole suavemente as seções (colete uma fatia após cada rodada de corte para evitar que elas se percam na câmara de corte).
NOTA: Cérebros ou seções podem ser armazenados em uma solução anticongelante a -20 °C por um longo tempo.
7. Imunostaining de fatias cerebrais
NOTA: Todas as etapas de coloração foram realizadas em uma placa de 24 poços em um agitador de plataforma.
- Lave as fatias com PBS três vezes, cada vez por 6 minutos.
- Incubar fatias em 300 μL de solução de bloqueio (5% de soro normal de burro (NDS)/0,3% Triton X-100) por 1 hora com agitação suave em um rotador.
- Incubar as fatias com o anticorpo primário contra PSD-95 diluído 1:500 em 5% NDS/0,3% Triton X-100/PBS (300 μL por poço) durante a noite a 4 °C. A concentração final do anticorpo primário é de 2 μg/mL.
- Lave as fatias com 0,3% Triton X-100/PBS à temperatura ambiente (RT) três vezes, cada vez por 6 minutos.
- Incubar fatias com o anticorpo secundário diluído 1:500 em 300 μL de 0,3% Triton X-100/PBS por 90 minutos. A concentração final do anticorpo secundário é de 4 μg /mL.
- Lave as fatias com PBS três vezes, cada vez por 6 minutos.
- Monte as fatias em slides usando um meio de montagem e, em seguida, feche-as com um slide de cobertura.
- Examine as amostras usando um microscópio confocal (63 × objetivo do óleo, NA 1.4, tamanho do pixel 0,13 μm × 0,13 μm).
NOTA: Para armazenamento a longo prazo: mantenha as amostras a 4 °C, proteja-as contra a luz.
8. Preparação da amostra SBEM
ATENÇÃO: Devido à natureza perigosa dos reagentes utilizados todos os procedimentos descritos abaixo devem ser realizados em um capô de fumaça de laboratório. Antes de usar esses produtos químicos leia atentamente as Folhas de Dados de Segurança do Material fornecidas pelos fabricantes e pergunte ao oficial de segurança sobre as regras locais para garantir o manuseio seguro e o descarte de resíduos.
- Contraste de amostra
NOTA: As lavagens de fatias e a incubação da temperatura ambiente devem ser realizadas com leve agitação (por exemplo, em um agitador de plataforma). Foi utilizada água desgassada autoclave.- Lave as fatias com frio de 0,1 M PB, pH 7,4 cinco vezes, cada vez por 3 minutos.
- Prepare uma mistura de 1:1 de 4% de tetroxida de ósmio aquoso e ferrocianida de 3% de potássio (1:1 por vol). O produto final ficará marrom. Mergulhe as amostras nesta mistura, coloque-as no gelo, e a partir desta fase é importante protegê-las da luz. Incubar-os com agitação suave por 1 hora.
- Enquanto isso, prepare uma solução de tiocarbohydrazida (TCH). Misture 10 mL de H2O destilado duplo (ddH2O) e 0,1 g de TCH e coloque-o em um forno a 60 °C por 1 hora. É importante rodar a solução de tempos em tempos (por exemplo, a cada 10 minutos). Quando estiver pronto, esfrie-o à temperatura ambiente.
- Lave as fatias com ddH2O cinco vezes, cada vez por 3 minutos.
- Filtrar a solução TCH usando um filtro de seringa de 0,22 μm e imergir a amostra em solução filtrada. As fatias ficarão pretas. Incuba-os por 20 minutos em temperatura ambiente.
- Lave as amostras com ddH2O cinco vezes, cada vez por 3 minutos.
- Incubar as amostras com uma solução aquosa de 2% de OsO4 por 30 minutos em temperatura ambiente.
- Lave as amostras com ddH2O cinco vezes, cada vez por 3 minutos.
- Coloque as amostras no acetato de urânyl 1% aquoso filtrado e incuba-as a 4 °C durante a noite. Use filtro de seringa de 0,22 μm para filtragem.
- Prepare a solução de ácido L-aspartic misturando 0,4 g de ácido L-aspartic e 80 mL de ddH2O, ajuste o pH para 3,8 para facilitar a dissolução e, em seguida, cubra com água a 100 mL.
- No dia seguinte, comece com a preparação de Walton aspartate15. Misture 0,066 g de nitrato de chumbo com 10 mL de solução de ácido L-aspartic (ponto 8.1.10) pré-aquecido a 60 °C e ajuste o pH para 5,5 (medido a 60 °C) com 1 M NaOH. Feche o frasco com aspartato de chumbo e deixe-o a 60 °C por 30 minutos em um banho de água. A solução deve ser clara. Se ficar nublado, deve ser descartado e um novo precisa ser preparado.
- Enquanto isso, lave as amostras com ddH2O desgaseada cinco vezes, cada vez por 3 minutos. Em seguida, mantenha-os no forno a 60 °C por 30 minutos.
- Mergulhe as amostras na solução de aspartato de chumbo recém-preparada e incuba-as no forno a 60 °C por 20 minutos.
- Lave as amostras com ddH2O desgaseada cinco vezes, cada vez por 3 minutos.
- Desidratação e incorporação de resina
- Prepare resina epóxi. Pesar os ingredientes (33,3 g do componente A/M, 33,3 g do componente B e 1 g do componente D) e misturar bem a resina (por exemplo, agite-a em um agitador rotativo em um tubo de 15 mL) por pelo menos 30 minutos antes de adicionar 16 gotas de acelerador DMP 30. Mexa novamente por mais 10 minutos.
NOTA: Pesar os ingredientes sob o capô da fumaça. Esta quantidade de componentes dá aproximadamente 60 mL de resina o que é suficiente para 15 frascos com amostras. Você pode preparar menos ou armazenar o resto da resina em uma seringa a 4 °C e utilizá-la no dia seguinte. Lembre-se de selar a ponta da seringa. - Prepare frascos com diluições classificadas de etanol (30%, 50%, 70%, 90%, 100%, 100% etanol na água e 100% seco em uma peneira molecular).
- Misture a resina com 100% de etanol em proporção 1:1 para obter 50% de resina. Misture bem.
- Desidrate as amostras por 5 minutos em cada diluição do etanol a partir de 30% e terminando com etanol 100% anidro seco em peneira molecular. Lembre-se, as amostras nunca devem secar completamente.
- Infiltrar as amostras primeiro em 50% de resina por 30 minutos, em seguida em 100% de resina por uma hora, e depois novamente em 100% de resina durante a noite. Realize todos os passos de infiltração com constante agitação lenta.
- No dia seguinte, coloque as amostras em resina fresca de 100% por uma hora e, em seguida, incorpore-as entre folhas de incorporação de fluoropolímeros. Desengrasse peças de folha de incorporação com etanol, e plana incorpore as amostras entre duas camadas dela, usando dois slides de vidro como suporte. Tente evitar bolhas de ar em resina ou perto da amostra.
- Cure as amostras no forno a 70 °C por pelo menos 48 horas.
- Prepare resina epóxi. Pesar os ingredientes (33,3 g do componente A/M, 33,3 g do componente B e 1 g do componente D) e misturar bem a resina (por exemplo, agite-a em um agitador rotativo em um tubo de 15 mL) por pelo menos 30 minutos antes de adicionar 16 gotas de acelerador DMP 30. Mexa novamente por mais 10 minutos.
- Aparamento e montagem
- Separe as folhas de incorporação e corte um pedaço da amostra incorporada (aproximadamente 1 mm x 1 mm) com uma lâmina de barbear. Transfira para um parafilm. Isso minimizará o perigo de perder a amostra devido à eletrostática.
- Pegue um pino de alumínio que foi desengordurado com etanol. Misture bem um epóxi condutivo e use um pouco de quantidade para montar a amostra no pino. Curar epóxi condutivo a 70 °C por 10 minutos.
- Corte cada lado do bloco de amostra com a faca de diamante e, em seguida, polir a face do bloco até que o tecido seja exposto.
NOTA: Nesta etapa, confirme a presença de uma região de interesse coletando algumas seções e realizando coloração azul toluidina16 ou verificação sob o microscópio eletrônico. - Reduza o tamanho da amostra o máximo possível. Em seguida, coloque a amostra no pino com tinta condutiva e cure-a (por 24 horas em temperatura ambiente ou no forno a 65 °C por 40 minutos).
- Para minimizar os artefatos de carregamento, cubra as amostras com uma fina camada de ouro ou ouro/paládio.
9. Imagem SBEM
- Coloque o pino com uma amostra na câmara do microscópio eletrônico de varredura serial face de bloco. Alinhe a amostra à faca. Feche a câmara e defina os parâmetros.
- Colete uma pilha de imagens na ampliação desejada, tamanho do pixel, espessura da fatia, tensão acelerada (EHT), abertura, pressão, etc.
NOTA: Os parâmetros dependem da amostra e do objetivo do experimento. Configurações de imagem iniciais exemplares estão incluídas na tabela de materiais.
Reconstruções .3D 10
NOTA: Para as etapas mencionadas abaixo, usamos software de acesso aberto, por exemplo, FijiJ17 (Versão ImageJ 1.49b), Microscopy Image Browser (MIB)18 e Reconstruct19, mas vários outros softwares podem ser empregados.
- Converta arquivos de micrografo digital (dm) em formato TIFF. Primeiro importe a sequência de imagem (em FijiJ: Arquivo > importe > sequência de imagem > Escolha o formato de 8 bits) e depois salve como TIFF (em FijiJ: Arquivo > Salvar como > sequência de imagem > Escolha Tiff).
- Ajuste o brilho e o contraste da pilha de imagens (em FijiJ: Imagem > Ajuste > Brilho/Contraste) e, se necessário, desemnize-o (use plugin DenoisEM para FijJ20 : Plugins > DenoiseEM > Denoise).
- Alinhe a pilha (em FijiJ: Plugins > StagReg ou MIB: Dataset > Ferramentas de Alinhamento).
- As espinhas e sinapses dendríticas do segmento (em software MIB ou Reconstruct, tutoriais abrangentes estão disponíveis (Veja Tabela de Materiais para mais detalhes).
Access restricted. Please log in or start a trial to view this content.
Resultados
Usando o método descrito acima de alto contraste, podem ser obtidas imagens de boa resolução do tecido cerebral do camundongo. Um grande campo de visão proporcionado pela técnica da SBEM facilita a seleção precisa da região de interesse. A grande imagem da região ca1 do hipocampo foi tomada para medir o comprimento do radiador de estrato (SR) (Figura 2A) e para definir a imagem precisamente no centro (Figura 2B). Em seguida, a pilha de imagens ...
Access restricted. Please log in or start a trial to view this content.
Discussão
Existem muitas variações do método NCMIR primário descrito por Deerinck em 201010. Os princípios básicos permanecem os mesmos, mas, dependendo do tipo de material estudado, pequenas mudanças são implementadas. Foi descrito anteriormente que diferentes resinas podem ser usadas para incorporar espécimes para SBEM e, por exemplo, no caso de plantas, Spurr é a resina de escolha devido à sua baixa viscosidade que permite melhor infiltração através das paredescelulares 22...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Foram realizadas imagens da SBEM, imagens de microscopia leve e preparação de amostras de microscopia eletrônica com o uso do equipamento do Laboratório de Tecido e Função de Imagem, que serve como uma instalação de núcleo de imagem no Instituto Nencki de Biologia Experimental.
Para a preparação da Figura 1, foi utilizada a imagem de um rato (Souris_02), e um frasco da https://smart.servier.com/.
Este trabalho foi apoiado pelo Centro Nacional de Ciência (Polônia) Grant Opus (UMO-2018/31/B/NZ4/01603) concedido à KR.
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade,  98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands | 50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands | 10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
Referências
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693(2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , Pt 2 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329(2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564(2012).
- Paxinos, G., Franklin, K. B. J. The mouse brain in stereotaxic coordinates. , Acad. Press. San Diego, Calif. (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340(2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771(2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084(2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. Biological specimen preparation for transmission electron microscopy. , Princeton University Press. Princeton. (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados