
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Serielle Block-Face-Rasterelektronenmikroskopie (SBEM) zur Untersuchung dendritischer Stacheln
In diesem Artikel
Zusammenfassung
Die serielle Block-Face-Rasterelektronenmikroskopie (SBEM) wird angewendet, um dendritische Stacheln im murinen Hippocampus abbilden und analysieren zu können.
Zusammenfassung
Die dreidimensionale Elektronenmikroskopie (3D EM) bietet die Möglichkeit, morphologische Parameter dendritischer Stacheln mit nanoskaliger Auflösung zu analysieren. Darüber hinaus können einige Merkmale der dendritischen Wirbelsäule, wie das Volumen der Wirbelsäule und die postsynaptische Dichte (PSD) (die den postsynaptischen Teil der Synapse darstellt), das Vorhandensein eines präsynaptischen Terminals und ein glattes endoplasmatisches Retikulum oder eine atypische Form der PSD (z. B. multiinnervierte Stacheln), nur mit 3D-EM beobachtet werden. Durch den Einsatz der seriellen Blockflächen-Rasterelektronenmikroskopie (SBEM) ist es möglich, 3D-EM-Daten einfacher und reproduzierbarer zu erhalten als bei herkömmlichen seriellen Schnitten. Hier zeigen wir, wie man Hippocampusproben der Maus für die SBEM-Analyse vorbereitet und wie dieses Protokoll mit immunfluoreszenzuntersuchungen dendritischer Stacheln kombiniert werden kann. Die milde Fixierungsperfusion ermöglicht es uns, Immunfluoreszenzstudien mit Lichtmikroskopie an einer Hälfte des Gehirns durchzuführen, während die andere Hälfte für SBEM vorbereitet wurde. Dieser Ansatz reduziert die Anzahl der Tiere, die für die Studie verwendet werden sollen.
Einleitung
Die meisten erregenden Synapsen im zentralen Nervensystem befinden sich auf dendritischen Stacheln - kleinen Vorsprüngen einer neuronalen Membran. Diese Vorsprünge bilden begrenzte biochemische Kompartimente, die die intrazelluläre Signaltransduktion steuern. Die strukturelle Plastizität von dendritischen Stacheln und Synapsen steht in engem Zusammenhang mit den funktionellen Veränderungen der synaptischen Wirksamkeit, die so wichtigen Prozessen wie Lernen und Gedächtniszugrunde liegen 1,2. Es ist wichtig zu beachten, dass die Elektronenmikroskopie (EM) die einzige Technik ist, mit der festgestellt werden kann, ob eine dendritische Wirbelsäule einen präsynaptischen Eingang hat. Die EM-Auflösung ist auch erforderlich, um ultrastrukturelle Details wie die Form einer postsynaptischen Dichte (PSD), die einen postsynaptischen Teil einer Synapse darstellt, oder die Abmessungen einer dendritischen Wirbelsäule sowie die Größe und Form eines axonalen Boutons zu untersuchen. Darüber hinaus ist es mit EM möglich, Synapsen und ihre Umgebung zu visualisieren.
Dank der Fortschritte in der Bildgebung und Computertechnologie ist es möglich, ganze neuronale Schaltkreise zu rekonstruieren. Volumenelektronenmikroskopische Techniken wie die serielle Transmissionselektronenmikroskopie (ssTEM), die serielle Blockflächen-Rasterelektronenmikroskopie (SBEM) und die fokussierte Ionenstrahl-Rasterelektronenmikroskopie (FIB-REM) werden häufig für neuronale Schaltkreisrekonstruktionen verwendet3.
In unseren Studien wird die SBEM-Methode erfolgreich eingesetzt, um die strukturelle Plastizität von dendritischen Stacheln und PSDs in Proben des Maus-Hippocampus und organotypischer Hirnschnitte zu untersuchen 4,5. Das SBEM basiert auf der Installation eines Miniatur-Ultramikrotoms in der Rasterelektronenmikroskopkammer6,7,8,9. Die Oberseite des Probenblocks wird abgebildet, und dann wird die Probe in einer bestimmten Tiefe durch das Ultramikrotom geschnitten, wodurch eine neue Blockfläche frei wird, die erneut abgebildet wird und dann der Prozess wiederholt wird8. Dadurch bleibt nur das Bild einer Blockfläche übrig, während die geschnittene Scheibe als Schutt verloren geht. Aus diesem Grund wird SBEM als destruktive Technik bezeichnet, was bedeutet, dass es nicht möglich ist, denselben Ort erneut abbilden zu können. Der Vorteil der destruktiven On-Block-Methoden besteht jedoch darin, dass sie nicht unter Warping-Problemen und Abschnittsverlusten leiden, die die Datenqualität und die Datenanalyse erheblich beeinträchtigen können3. Darüber hinaus bietet SBEM die Möglichkeit, ein relativ großes Sichtfeld ( > 0,5 mm × 0,5 mm) mit hoher Auflösung3 abbilden.
Um SBEM einsetzen zu können, müssen Proben aufgrund des rückgestreuten Elektronendetektors, der für die Bildaufnahme verwendet wird, nach einem dedizierten, kontrastreichen Protokoll vorbereitet werden. Wir zeigen hier, wie die Probenvorbereitung nach dem Protokoll durchgeführt wird, das auf einem von Deerinck10 (National Center for Microscopy and Imaging Research (NCMIR) -Methode) entwickelten Verfahren basiert, wobei reduzierte Osmium-Thiocarbohydrazid-Osmium (rOTO) -Flecken verwendetwerden,die in den 1980er Jahren entwickelt wurden8,11. Darüber hinaus führen wir einen zweistufigen Fixierungsansatz mit leichter Fixierungsperfusion ein, der es ermöglicht, dasselbe Gehirn sowohl für Immunfluoreszenzstudien mit Lichtmikroskopie als auch sbEM zu verwenden.
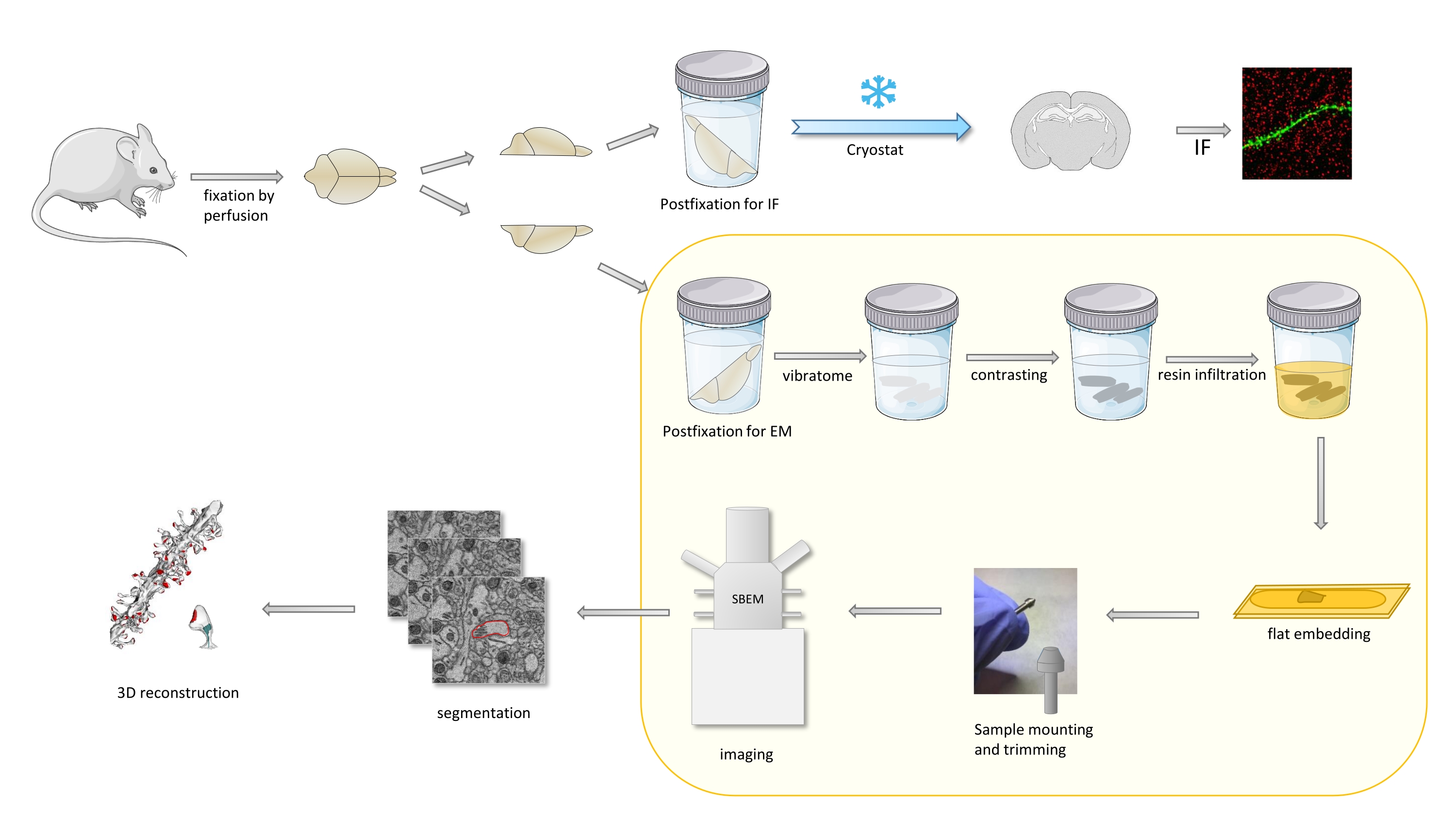
Im Protokoll wird ein Mausgehirn hauptsächlich mit einem milden Fixiermittel fixiert und dann in Zwei hälften geschnitten, und eine Hemisphäre wird postfixiert und für Immunfluoreszenz (IF) vorbereitet, während die andere für EM-Studien vorbereitet wird (Abbildung 1).

Abbildung 1. Schematische Darstellung des Workflows für die dendritischen Stacheln Vorbereitung für die Analyse mit SBEM. Mäuse wurden geopfert und mit einem milden primären Fixiermittel durchtränkt. Das Gehirn wurde in Zwei hälften geschnitten, und eine Hemisphäre wurde mit Immunfluoreszenz (IF) -dediziertem Fixiermittel postfixiert, kryogeschützt, mit einem Kryostaten geschnitten und für IF-Studien verarbeitet, während die andere Hemisphäre mit EM-Fixiermitteln postfixiert, mit dem Vibratom geschnitten und für EM-Studien vorbereitet wurde. Gehirnschnitte für SBEM-Studien wurden kontrastiert, flach in Harz eingebettet, dann wurde eine CA1-Region des Hippocampus am Pin montiert und mit SBEM abgebildet (Abbildung 1). Der Teil des Protokolls, der in einem gelben Kasten hervorgehoben ist, wurde im Video gezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protokoll
Die Forschung wurde in Übereinstimmung mit den Richtlinien des Nencki-Instituts und der Genehmigung des Lokalen Ethikkomitees durchgeführt. Die Studien wurden in Übereinstimmung mit der Richtlinie des Rates der Europäischen Gemeinschaften vom 24. November 1986 (86/609/EWG), dem polnischen Tierschutzgesetz, durchgeführt und von der ersten lokalen Ethikkommission in Warschau genehmigt. Es wurden alle Anstrengungen unternommen, um die Anzahl der verwendeten Tiere und ihr Leiden zu minimieren.
ACHTUNG: Alle unten beschriebenen Verfahren müssen in einem Laborabzug durchgeführt werden. Aufgrund der Gefährlichkeit der verwendeten Reagenzien. Persönliche Sicherheitsmaßnahmen wie Handschuhe, Laborkittel, Schutzbrille und eine Gesichtsmaske sind erforderlich.
1. Herstellung des Fixiermittels für die Perfusion (2 % gew/vol Paraformaldehyd (PFA) und 0,5 % vol/vol Glutaraldehyd (GA) in 0,1 M Phosphatpuffer (PB), pH 7,4)
HINWEIS: Bereiten Sie die Fixierlösung am selben Tag vor, an dem sie verwendet wird, und lagern Sie sie nicht länger als 3 Stunden. Bei Zeitmangel am Vortag 2% PFA in 0,1 M PB zubereiten, bei 4 °C lagern und kurz vor der Perfusion frisches GA hinzufügen.
- Nehmen Sie 400 ml steriles doppelt destilliertes Wasser (ddH2O) und erhitzen Sie es mit einer Rührplatte auf 60 °C. Dann fügen Sie 20 g PFA hinzu. Fügen Sie Tropfen von 1 M NaOH hinzu, bis PFA vollständig gelöst ist, und lassen Sie die Mischung abkühlen.
- Fügen Sie 500 ml 0,2 M PB (pH 7,4) hinzu.
- Filtern Sie die Lösung, um eventuelle Ablagerungen zu entfernen und kühlen Sie sie auf 4 °C ab.
- Kurz vor der Perfusion 20 ml 25% GA in die Lösung geben und dann das Volumen mit ddH2O auf 1 L auffüllen.
2. Herstellung von Postperfusionsfixiermitteln für SBEM (2% wt/vol PFA und 2,5% vol/vol GA in 0,1 M PB, pH 7,4)
- Nehmen Sie 50 ml des Fixiermittels zur Perfusion (2% PFA und 0,5% GA in 0,1 M PB).
- Fügen Sie 5 ml 25% GA hinzu.
3. Herstellung von Postperfusionsfixiermittel für die IF-Färbung (4% PFA in phosphatgepufferter Kochsalzlösung (PBS))
- Eine Tablette mit 1x PBS (pH 7,4) in einem gereinigten und entionisierten Wasser(H2O) gemäß den Anweisungen des Herstellers auflösen.
- Verwenden Sie eine Rührplatte, um die Lösung auf 60 °C zu erhitzen und fügen Sie 40 g PFA hinzu.
- Fügen Sie Tropfen von 1 M NaOH hinzu, bis das PFA vollständig gelöst ist, und lassen Sie die Mischung abkühlen.
- Stellen Sie den pH-Wert der Lösung auf 7,5 mit 1 M HCl ein und beladen Sie dann das Volumen mit H2O auf 1 L.
- Filtern Sie die Lösung, um Ablagerungen zu entfernen.
4. Transkardiale Perfusion von Tieren
HINWEIS: Alle PFA- und GA-Abfälle müssen gemäß den lokalen Vorschriften gesammelt und zur Entsorgung gelagert werden. Anästhesie und Perfusion sollten den lokalen Vorschriften folgen. In dem beschriebenen Protokoll wurden erwachsene 3 Monate alte und 20±1 Monate alte weibliche Thy1-GFP(M)-Mäuse (Thy1-GFP +/-)12 exprimierendes grünes Fluoreszenzprotein (GFP) in einer spärlich verteilten Population glutamaterger Neuronen verwendet, aber auch jedes andere kann verwendet werden. Die Tiere wurden als Heterozygoten mit dem C57BL/6J-Hintergrund im Tierhaus des Nencki Institute of Experimental Biology gezüchtet.
- Vor der Perfusion eine Maus durch Verabreichung einer Ketamin/Xylazin-Mischung (bis zu 90 mg/kg Körpergewicht Ketamin und 10 mg/kg Körpergewicht Xylazin) mittels intraperitonealer Injektion (27-Gauge-Nadel) betäuben.
- Beurteilen Sie, ob die Tiefe der Anästhesie ausreichend ist, indem Sie den Reflex auf Schmerzreize (Kneifen) und den Hornhautreflex (Schielen) überprüfen.
- Nach 20 Minuten führen Sie eine intraperitoneale Injektion (27-Gauge-Nadel) von Natriumpentobarbital (50 mg/kg Körpergewicht) durch.
- Perfusionsen einer Maus nach einem perfusionschirurgischen Protokoll, das von Gage et al.13 beschrieben wird (siehe Punkt 4; Abbildung 5-6). Beginnen Sie mit einer Perfusionspumpe mit einem 0,1 M Phosphatpuffer, pH 7,4 (30 ml) für 3 Minuten und fahren Sie mit 2% PFA und 0,5% GA in 0,1 M PB, pH 7,4 für 6 Minuten (80 ml) fort.
- Sezieren Sie das Gehirn vorsichtig vom Schädel und teilen Sie es in zwei Hälften (siehe Abbildung 9-10 in Gage et al., 201213). Legen Sie ein Stück in eine Durchstechflasche, die Fixiermittel für SBEM enthält, und das zweite in die Durchstechflasche mit 4% PFA / PBS für die IF-Färbung.
- Halten Sie die Hemisphären über Nacht bei 4 °C im Fixiermittel.
5. Gehirnschnitte Vorbereitung für die Elektronenmikroskopie
- Wählen Sie die Vibrationseinstellungen (Klingenverfahrgeschwindigkeit: 0,075 mm/s, Schnittfrequenz: 80 Hz).
- Legen Sie die Scheibenkammer in die Halterung, befestigen Sie sie am Vibratom und umgeben Sie sie mit Eis. Legen Sie dann eine Rasierklinge in den Vibratom-Klingenhalter.
- Verwenden Sie einen Löffel oder ein ähnliches Objekt und legen Sie das gekühlte Gehirn (dorsale Oberfläche nach oben) auf eine harte Schnittfläche (z. B. einen Glasdeckel der Petrischale). Um eine koronale Scheibe des Hippocampus vorzubereiten, machen Sie einen senkrechten Schnitt zwischen der Gehirnhemisphäre und dem Kleinhirn mit einer Rasierklinge oder einem Skalpell und entfernen Sie so das Kleinhirn. Der Riechkolben kann auch entfernt werden.
- Tragen Sie Cyanacrylatkleber auf die trockene Plattform des Vibratoms auf.
- Nehmen Sie das Gehirn mit einer Zette auf und trocknen Sie es vorsichtig auf Filterpapier.
- Kleben Sie die Halbkugel mit der Rostralspitze nach oben auf die Plattform in der Nähe der Schneidklinge. Befestigen Sie die Plattform an der Halterung und füllen Sie sie sofort mit eiskalten 0,1 M PB, pH 7,4. Wenn der senkrechte Schnitt richtig gemacht wird, steht die Hemisphäre gerade nach oben und bietet einen Winkel von 90 °, der für einen symmetrischen koronalen Schnitt mit dem Hippocampus erforderlich ist. Stellen Sie sicher, dass das Gehirn mit PB bedeckt ist.
- Positionieren Sie die Vibratomklinge vor der Hemisphäre und senken Sie sie auf die koronale Seite der Hemisphäre. Senken Sie die Klinge in kaudaler Richtung weiter auf 400 μm und beginnen Sie mit dem Schneiden. Schneiden Sie weiter, bis die ersten beiden Scheiben vollständig vom Gewebeblock getrennt sind.
- Ziehen Sie die Klinge ein und senken Sie weitere 100 μm, dann schneiden Sie sie erneut.
- Wenn der Hippocampus sichtbar wird (verwenden Sie den Maus-Hirnatlas Paxinos und Franklin, 200414), sammeln Sie die Scheiben mit dem kleinen Pinsel oder der verbreiterten Pasteur-Pipette aus Kunststoff.
- Die Scheiben in eine 12-Well-Platte geben, die mit kaltem 0,1 M PB, pH 7,4 gefüllt ist.
- Sezieren Sie den Hippocampus in einer mit 0,1 M PB, pH 7,4 gefüllten Glas-Petrischale mit einer Rasierklinge oder einem Skalpell und legen Sie ihn in Glasfläschchen mit dem gleichen Phosphatpuffer.
HINWEIS: Zur Langzeitlagerung der Scheiben 0,1 M PB mit 0,05% Natriumazid(NaN3)ergänzen.
6. Hirnprobenvorbereitung zur Immunfärbung
- Nach der Fixierung über Nacht in 4% PFA/PBS-Lösung in einem Abzug das Hirngewebe in die Kryokonservierungslösung (30% Saccharose in PBS mit 0,05%NaN3)geben und bei 4 °C für 2 Tage (bis zum Versenken) aufbewahren.
- Bereiten Sie eine Frostschutzlösung (15% Saccharose / 30% Ethylenglykol / 0,05% NaN3/ PBS) vor.
- Stellen Sie die Schranktemperatur des Kryostaten auf -19 °C ein und stellen Sie sicher, dass die Temperatur erreicht ist, bevor Sie fortfahren. Achten Sie beim Schnitten darauf, dass die Schranktemperatur zwischen -18 °C und -20 °C bleibt.
- Verwenden Sie einen Löffel oder ein ähnliches Objekt und legen Sie das gekühlte Gehirn (dorsale Oberfläche nach oben) auf eine harte Schnittfläche (z. B. einen Glasdeckel der Petrischale). Um eine koronale Scheibe des Hippocampus vorzubereiten, machen Sie einen senkrechten Schnitt zwischen der Gehirnhemisphäre und dem Kleinhirn mit einer Rasierklinge oder einem Skalpell und entfernen Sie so das Kleinhirn.
- Wählen Sie eine vorgekühlte Probenscheibe, bedecken Sie sie mit einem Medium zum Einfrieren auf einem Befreiendegal und befestigen Sie die Hemisphäre mit einer Rostralspitze nach oben an der Scheibe und warten Sie, bis die Probe vollständig eingefroren ist. Legen Sie die Probenscheibe in einen Probenkopf ein.
- Setzen Sie eine Klinge in einen Klingenhalter in der Kryokammer ein und schneiden Sie 40 μm dicke Scheiben.
- Die Scheiben mit einem kleinen Pinsel auf die mit kalter Frostschutzlösung gefüllte 96-Well-Platte geben und die Abschnitte vorsichtig abrollen (nach jeder Runde schneiden eine Scheibe sammeln, um zu verhindern, dass sie in der Scheibenkammer verloren gehen).
HINWEIS: Gehirne oder Abschnitte können lange Zeit in einer Frostschutzlösung bei -20 °C gelagert werden.
7. Immunfärbung von Hirnscheiben
HINWEIS: Alle Färbeschritte wurden in einer 24-Well-Platte auf einem Plattform-Shaker durchgeführt.
- Waschen Sie die Scheiben dreimal mit PBS, jedes Mal für 6 Minuten.
- Inkubieren Sie die Scheiben in 300 μL Blocklösung (5% normales Eselserum (NDS)/0,3% Triton X-100) für 1 Stunde mit sanftem Schütteln auf einem Rotator.
- Die Scheiben werden mit dem primären Antikörper gegen PSD-95, verdünnt 1:500 in 5% NDS/0,3% Triton X-100/PBS (300 μL pro Vertiefung), über Nacht bei 4 °C inkubiert. Die Endkonzentration des primären Antikörpers beträgt 2 μg/ml.
- Waschen Sie die Scheiben dreimal mit 0,3% Triton X-100/PBS bei Raumtemperatur (RT), jeweils 6 Minuten lang.
- Inkubieren Sie die Scheiben mit dem sekundären Antikörper, der 1:500 in 300 μL 0,3% Triton X-100/PBS verdünnt ist, für 90 Minuten. Die Endkonzentration des sekundären Antikörpers beträgt 4 μg /ml.
- Waschen Sie die Scheiben dreimal mit PBS, jedes Mal für 6 Minuten.
- Montieren Sie die Scheiben mit einem Montagemedium auf Dias und schließen Sie sie dann mit einem Abdeckschieber.
- Untersuchen Sie die Proben mit einem konfokalen Mikroskop (63 × Ölobjektiv, NA 1,4, Pixelgröße 0,13 μm × 0,13 μm).
HINWEIS: Für die Langzeitlagerung: Bewahren Sie die Proben bei 4 °C auf, schützen Sie sie vor Licht.
8. SBEM-Probenvorbereitung
ACHTUNG: Aufgrund der Gefährlichkeit der verwendeten Reagenzien müssen alle unten beschriebenen Verfahren in einem Laborabzug durchgeführt werden. Bevor Sie diese Chemikalien verwenden, lesen Sie sorgfältig die von den Herstellern bereitgestellten Sicherheitsdatenblätter und fragen Sie den Sicherheitsbeauftragten nach den lokalen Regeln, um eine sichere Handhabung und Abfallentsorgung zu gewährleisten.
- Beispielkontrast
HINWEIS: Scheibenwaschungen und inkubation bei Raumtemperatur sollten mit leichtem Schütteln (z. B. auf einem Plattformstreuer) durchgeführt werden. Autoklaven entgastes Wasser wurde verwendet.- Waschen Sie die Scheiben mit kaltem 0,1 M PB, pH 7,4 fünfmal, jedes Mal für 3 Minuten.
- Bereiten Sie eine 1:1-Mischung aus 4% wässrigem Osmiumtetroxid und 3% Kaliumferrocyanid (1:1 vol) vor. Das Endprodukt wird braun. Tauchen Sie die Proben in diese Mischung, legen Sie sie auf Eis und ab diesem Stadium ist es wichtig, sie vor Licht zu schützen. Inkubieren Sie sie mit sanftem Schütteln für 1 Stunde.
- In der Zwischenzeit eine Thiocarbohydrazid (TCH) -Lösung vorbereiten. 10 mL doppelt destilliertesH2O (ddH2O) und 0,1 g TCH mischen und 1 Stunde in einen bei 60 °C eingestellten Ofen geben. Es ist wichtig, die Lösung von Zeit zu Zeit zu schwenken (z. B. alle 10 Minuten). Wenn Sie fertig sind, kühlen Sie es auf Raumtemperatur ab.
- Waschen Sie die Scheiben fünfmal mit ddH2O, jedes Mal für 3 Minuten.
- Filtern Sie TCH-Lösung mit einem 0,22 μm Spritzenfilter und tauchen Sie die Probe in gefilterte Lösung. Die Scheiben werden schwarz. Inkubieren Sie sie für 20 Minuten bei Raumtemperatur.
- Waschen Sie die Proben fünfmal mit ddH2O, jedes Mal für 3 Minuten.
- Inkubieren Sie die Proben mit einer 2% igen wässrigen Lösung von OsO4 für 30 Minuten bei Raumtemperatur.
- Waschen Sie die Proben fünfmal mit ddH2O, jedes Mal für 3 Minuten.
- Die Proben in gefiltertes 1% wässriges Uranylacetat geben und über Nacht bei 4 °C inkubieren. Verwenden Sie einen 0,22 μm Spritzenfilter für die Filtration.
- Bereiten Sie L-Asparaginsäurelösung vor, indem Sie 0,4 g L-Asparaginsäure und 80 ml ddH2O mischen, den pH-Wert auf 3,8 einstellen, um das Auflösen zu erleichtern, und dann mit Wasser auf 100 ml aufladen.
- Beginnen Sie am nächsten Tag mit Waltons Lead Aspartate15 Vorbereitung. 0,066 g Bleinitrat mit 10 ml L-Asparaginsäurelösung (Nummer 8.1.10) vorgewärmt auf 60 °C mischen und den pH-Wert auf 5,5 (gemessen bei 60 °C) mit 1 M NaOH einstellen. Die Durchstechflasche mit Bleiaspartat verschließen und bei 60 °C 30 Minuten in einem Wasserbad stehen lassen. Die Lösung sollte klar sein. Wenn es bewölkt wird, muss es verworfen und ein neues vorbereitet werden.
- In der Zwischenzeit die Proben fünfmal, jeweils 3 Minuten lang, mit entgastem ddH2Owaschen. Anschließend 30 Minuten im Ofen bei 60 °C eingestellt aufbewahren.
- Tauchen Sie die Proben in die frisch zubereitete Bleiaspartatlösung und ubatieren Sie sie 20 Minuten lang im Bei 60 °C eingestellten Ofen.
- Waschen Sie die Proben fünfmal mit entgastem ddH2O, jeweils für 3 Minuten.
- Dehydratisierung und Harzeinbettung
- Bereiten Sie Epoxidharz vor. Wiegen Sie die Zutaten (33,3 g Komponente A/M, 33,3 g Komponente B und 1 g Komponente D) und mischen Sie das Harz mindestens 30 Minuten lang gut (z. B. auf einem Rotationsschüttler in einem 15-ml-Röhrchen schütteln), bevor Sie 16 Tropfen Beschleuniger DMP 30 hinzufügen. Weitere 10 Minuten umrühren.
HINWEIS: Wiegen Sie die Zutaten unter dem Abzug. Diese Menge an Komponenten ergibt etwa 60 ml Harz, was für 15 Fläschchen mit Proben ausreicht. Sie können weniger vorbereiten oder den Rest des Harzes in einer Spritze bei 4 °C lagern und am nächsten Tag verwenden. Denken Sie daran, die Spitze der Spritze zu versiegeln. - Durchstechflaschen mit abgestuften Ethanolverdünnungen (30%, 50%, 70%, 90%, 100%, 100% Ethanol in Wasser und 100% auf einem Molekularsieb getrocknet) vorbereiten.
- Mischen Sie das Harz mit 100% Ethanol im Verhältnis 1:1, um 50% Harz zu erhalten. Mischen Sie es gut.
- Die Proben werden für 5 Minuten in jeder Ethanolverdünnung ab 30% getrocknet und endend mit 100% wasserfreiem Ethanol, das auf Molekularsieb getrocknet wird, getrocknet. Denken Sie daran, dass die Proben niemals vollständig trocknen dürfen.
- Infiltrieren Sie die Proben zuerst in 50% Harz für 30 Minuten, dann in 100% Harz für eine Stunde und dann wieder in 100% Harz über Nacht. Führen Sie alle Infiltrationsschritte mit konstantem langsamem Schütteln durch.
- Am nächsten Tag legen Sie die Proben für eine Stunde in frisches 100% Harz und betten sie dann zwischen Fluorpolymer-Einbettungsplatten ein. Entfetten Sie Stücke der Einbettungsplatte mit Ethanol und betten Sie die Proben flach zwischen zwei Schichten ein, wobei zwei Glasobjektträger als Unterstützung verwendet werden. Versuchen Sie, Luftblasen in Harz auf oder in der Nähe der Probe zu vermeiden.
- Die Proben im Ofen bei 70 °C mindestens 48 Stunden aushärten.
- Bereiten Sie Epoxidharz vor. Wiegen Sie die Zutaten (33,3 g Komponente A/M, 33,3 g Komponente B und 1 g Komponente D) und mischen Sie das Harz mindestens 30 Minuten lang gut (z. B. auf einem Rotationsschüttler in einem 15-ml-Röhrchen schütteln), bevor Sie 16 Tropfen Beschleuniger DMP 30 hinzufügen. Weitere 10 Minuten umrühren.
- Trimmen und Montieren
- Trennen Sie die Einbettungsblätter und schneiden Sie ein Stück der eingebetteten Probe (ca. 1 mm x 1 mm) mit einer Rasierklinge. Übertragen Sie es auf einen Parafilm. Dadurch wird die Gefahr minimiert, dass die Probe aufgrund von Elektrostatik verloren geht.
- Nehmen Sie einen Aluminiumstift, der mit Ethanol entfettet wurde. Mischen Sie ein leitfähiges Epoxidharz gut und verwenden Sie eine kleine Menge davon, um die Probe am Stift zu befestigen. Härten Sie leitfähiges Epoxidharz bei 70 °C für 10 Minuten aus.
- Schneiden Sie jede Seite des Probenblocks mit dem Diamantmesser ab und polieren Sie dann die Vorderseite des Blocks, bis das Gewebe freigelegt ist.
HINWEIS: Bestätigen Sie in diesem Schritt das Vorhandensein einer Region von Interesse, indem Sie einige Abschnitte sammeln und toluidinblaueFärbung 16 durchführen oder unter dem Elektronenmikroskop überprüfen. - Reduzieren Sie die Größe der Probe so weit wie möglich. Anschließend die Probe mit leitfähiger Farbe auf den Stift einmahlen und aushärten (24 Stunden bei Raumtemperatur oder im Ofen bei 65 °C für 40 Minuten).
- Um Ladeartefakte zu minimieren, beschichten Sie die Proben mit einer dünnen Schicht Gold oder Gold / Palladium.
9. SBEM-Bildgebung
- Legen Sie den Stift mit einer Probe in die Kammer des seriellen Rasterelektronenmikroskops. Richten Sie die Probe am Messer aus. Schließen Sie die Kammer und stellen Sie die Parameter ein.
- Sammeln Sie einen Stapel bilder mit der gewünschten Vergrößerung, Pixelgröße, Slice-Dicke, beschleunigter Spannung (EHT), Blende, Druck usw.
HINWEIS: Die Parameter hängen von der Probe und dem Ziel des Experiments ab. Beispielhafte erste Bildgebungseinstellungen sind in der Materialtabelle enthalten.
10.3D Rekonstruktionen
HINWEIS: Für die unten genannten Schritte verwenden wir Open-Access-Software wie z.B. FijiJ17 (ImageJ Version 1.49b), Microscopy Image Browser (MIB)18 und Reconstruct19, aber es können verschiedene andere Software verwendet werden.
- Konvertieren Sie digitale Mikrographendateien (dm) in das TIFF-Format. Importieren Sie zuerst die Bildsequenz (in FijiJ: File > Import > Image sequence > Choose 8 Bit Format) und speichern Sie sie dann als TIFF (in FijiJ: File > Save As > Image Sequence > Choose Tiff).
- Passen Sie Helligkeit und Kontrast des Bildstapels an (in FijiJ: Bild > passen Sie > Helligkeit / Kontrast an) und denoise es bei Bedarf (verwenden Sie das DenoisEM-Plugin für FijJ20: Plugins > DenoiseEM > Denoise).
- Richten Sie den Stack aus (in FijiJ: Plugins > StagReg oder MIB: Dataset > Alignment Tools).
- Segment dendritische Stacheln und Synapsen (In der MIB- oder Reconstruct-Software stehen umfassende Tutorials zur Verfügung (Siehe Materialtabelle für Details).
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Mit der oben beschriebenen Methode können kontrastreiche Bilder des Gehirngewebes der Maus mit guter Auflösung erhalten werden. Ein großes Sichtfeld, das die SBEM-Technik bietet, ermöglicht die präzise Auswahl der region von Interesse. Das große Bild der CA1-Region des Hippocampus wurde aufgenommen, um die Länge des Stratum radiatum (SR) zu messen (Abbildung 2A) und die Bildgebung genau in der Mitte einzustellen (Abbildung 2B). Als nächstes wurd...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Es gibt viele Variationen der primären NCMIR-Methode, die von Deerinck im Jahr 2010 beschrieben wurde10. Die Grundprinzipien bleiben gleich, aber je nach Art des untersuchten Materials werden geringfügige Änderungen umgesetzt. Es wurde bereits beschrieben, dass verschiedene Harze verwendet werden können, um Proben für SBEM einzubetten, und zum Beispiel im Falle von Pflanzen ist Spurr's aufgrund seiner niedrigen Viskosität, die eine bessere Infiltration durch die Zellwände ermöglicht, das H...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
SBEM-Bildgebung, Lichtmikroskopie-Bildgebung und elektronenmikroskopische Probenvorbereitung wurden mit den Geräten des Laboratory of Imaging Tissue and Function durchgeführt, das als bildgebende Kerneinrichtung am Nencki Institute of Experimental Biology dient.
Zur Vorbereitung von Abbildung 1 wurde das Bild einer Maus (Souris_02) und eine Durchstechflasche aus dem https://smart.servier.com/ verwendet.
Diese Arbeit wurde durch das Grant Opus des Nationalen Wissenschaftszentrums (Polen) (UMO-2018/31/B/NZ4/01603) unterstützt, das kr verliehen wurde.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade,  98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands | 50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands | 10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
Referenzen
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693(2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , Pt 2 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329(2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564(2012).
- Paxinos, G., Franklin, K. B. J. The mouse brain in stereotaxic coordinates. , Acad. Press. San Diego, Calif. (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340(2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771(2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084(2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. Biological specimen preparation for transmission electron microscopy. , Princeton University Press. Princeton. (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten