Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Técnicas de microscopía para interpretar la colonización fúngica en tejidos de plantas micoheterótrofas y germinación simbiótica de semillas
En este artículo
Resumen
Este protocolo tiene como objetivo proporcionar procedimientos detallados para recolectar, fijar y mantener muestras de plantas micoheterótrofas, aplicando diferentes técnicas de microscopía como microscopía electrónica de barrido y transmisión, microscopía de luz, confocal y fluorescencia para estudiar la colonización fúngica en tejidos vegetales y semillas germinadas con hongos micorrícicos.
Resumen
La botánica estructural es una perspectiva indispensable para comprender completamente la ecología, la fisiología, el desarrollo y la evolución de las plantas. Al investigar plantas micoheterótrofas (es decir, plantas que obtienen carbono de hongos), aspectos notables de sus adaptaciones estructurales, los patrones de colonización tisular por hongos y la morfoanatomía de los órganos subterráneos pueden iluminar sus estrategias de desarrollo y sus relaciones con las hifas, la fuente de nutrientes. Otro papel importante de los hongos simbióticos está relacionado con la germinación de las semillas de orquídeas; todas las especies de Orchidaceae son micoheterótrofas durante la germinación y la etapa de plántula (micoheterotrofia inicial), incluso las que fotosintetizan en etapas adultas. Debido a la falta de reservas nutricionales en las semillas de orquídeas, los simbiontes fúngicos son esenciales para proporcionar sustratos y permitir la germinación. El análisis de las etapas de germinación desde perspectivas estructurales también puede responder preguntas importantes sobre la interacción de los hongos con las semillas. Se pueden aplicar diferentes técnicas de imagen para revelar hongos endófitos en tejidos vegetales, como se propone en este artículo. Las secciones a mano alzada y delgadas de los órganos de la planta se pueden teñir y luego observar mediante microscopía óptica. Un fluorocromo conjugado con aglutinina de germen de trigo se puede aplicar a los hongos y co-incubar con Calcofluor White para resaltar las paredes celulares de las plantas en microscopía confocal. Además, se detallan las metodologías de microscopía electrónica de barrido y transmisión para orquídeas micoheterótrofas, y se exploran las posibilidades de aplicar dichos protocolos en plantas relacionadas. La germinación simbiótica de semillas de orquídeas (es decir, en presencia de hongos micorrícicos) se describe en detalle en el protocolo, junto con las posibilidades de preparar las estructuras obtenidas de diferentes etapas de germinación para análisis con microscopía óptica, confocal y electrónica.
Introducción
La investigación estructural en botánica, que abarca la morfología y anatomía de las plantas, es básica para comprender todo el organismo1,2, y proporciona perspectivas indispensables para integrar y contribuir al conocimiento sobre la ecología, fisiología, desarrollo y evolución de las plantas3. Los métodos en morfología y anatomía vegetal actualmente comprenden protocolos, equipos y conocimientos desarrollados recientemente, así como hace más de un siglo2. La ejecución continua y la adaptación de métodos clásicos (por ejemplo, microscopía óptica) junto con técnicas más recientes (por ejemplo, microscopía confocal, microtomografía de rayos X) tienen la misma base esencial: conocimientos teóricos que permiten el desarrollo de una metodología.
La herramienta principal en anatomía y morfología vegetal es la imagen. A pesar de la idea errónea de que tales análisis son simples observaciones, dando espacio a interpretaciones subjetivas2, el análisis y la comprensión de las imágenes en esta área requieren el conocimiento de los métodos aplicados (el equipo, el tipo de análisis, los procedimientos metodológicos), los componentes celulares, la histoquímica y el cuerpo de la planta (organización y función del tejido, ontogenia, adaptaciones morfológicas). La interpretación de las imágenes obtenidas a través de una variedad de métodos puede conducir a correlacionar forma y función, descifrar la composición química de una estructura, corroborar la descripción de taxones, comprender las infecciones por fitopatógenos y otras evaluaciones similares.
Al investigar plantas micoheterótrofas (MH) (es decir, plantas no fotosintéticas que obtienen carbono de hongos micorrícicos4,5), aspectos notables de sus adaptaciones estructurales, los patrones de colonización tisular por hongos y la morfoanatomía de los órganos subterráneos pueden iluminar sus estrategias de desarrollo y relaciones con las hifas, que son la fuente de nutrientes. Los órganos subterráneos de las plantas MH suelen mostrar importantes adaptaciones relacionadas a su asociación con hongos del suelo, por lo que es esencial realizar estas investigaciones anatómicas y morfológicas6. Los órganos aéreos de las especies MH no deben ser ignorados, ya que los endófitos también pueden estar presentes en estos tejidos, incluso si no son hongos micorrícicos (observaciones personales, aún no publicadas).
Además de la esencialidad bien establecida de la asociación de hongos micorrícicos con especies MH durante todo su ciclo de vida7, todas las especies de orquídeas, incluso las autótrofas, tienen una etapa micoheterotrófica obligada inicial en ambientes naturales. Ocurre porque el embrión de las orquídeas es indiferenciado y carece de endospermo o cotiledones, siendo así incapaz de desarrollarse y establecerse en ambientes naturales sin el apoyo nutricional de socios fúngicos 4,8. Considerando eso, los protocolos de germinación simbiótica pueden ser aplicados no sólo a especies MH sino también a orquídeas fotosintéticas, con el objetivo de investigar la especificidad orquídea-hongo en germinación y desarrollo protocormo, una metodología ampliamente aplicada en iniciativas para la conservación de especies amenazadas 9,10,11.
En este ensamblaje de métodos, describimos pasos importantes involucrados en la recolección, fijación y almacenamiento de muestras de plantas MH para estudios anatómicos (sección 1), análisis de superficie y selección de muestras (sección 2), métodos de seccionamiento (a mano alzada: sección 3, microtomía: sección 4, criomicrotomía: sección 5), tinción y montaje (sección 6), fluorescencia y microscopía confocal de endófitos fúngicos (sección 7), microscopía electrónica de barrido (sección 8), y microscopía electrónica de transmisión (sección 9). Además, describimos un método de germinación simbiótica para semillas de orquídeas (MH y autótrofa, sección 10), ya que los métodos de imagen mencionados anteriormente se pueden aplicar con éxito para analizar la colonización fúngica de semillas, protocormos y plántulas en el proceso de germinación.

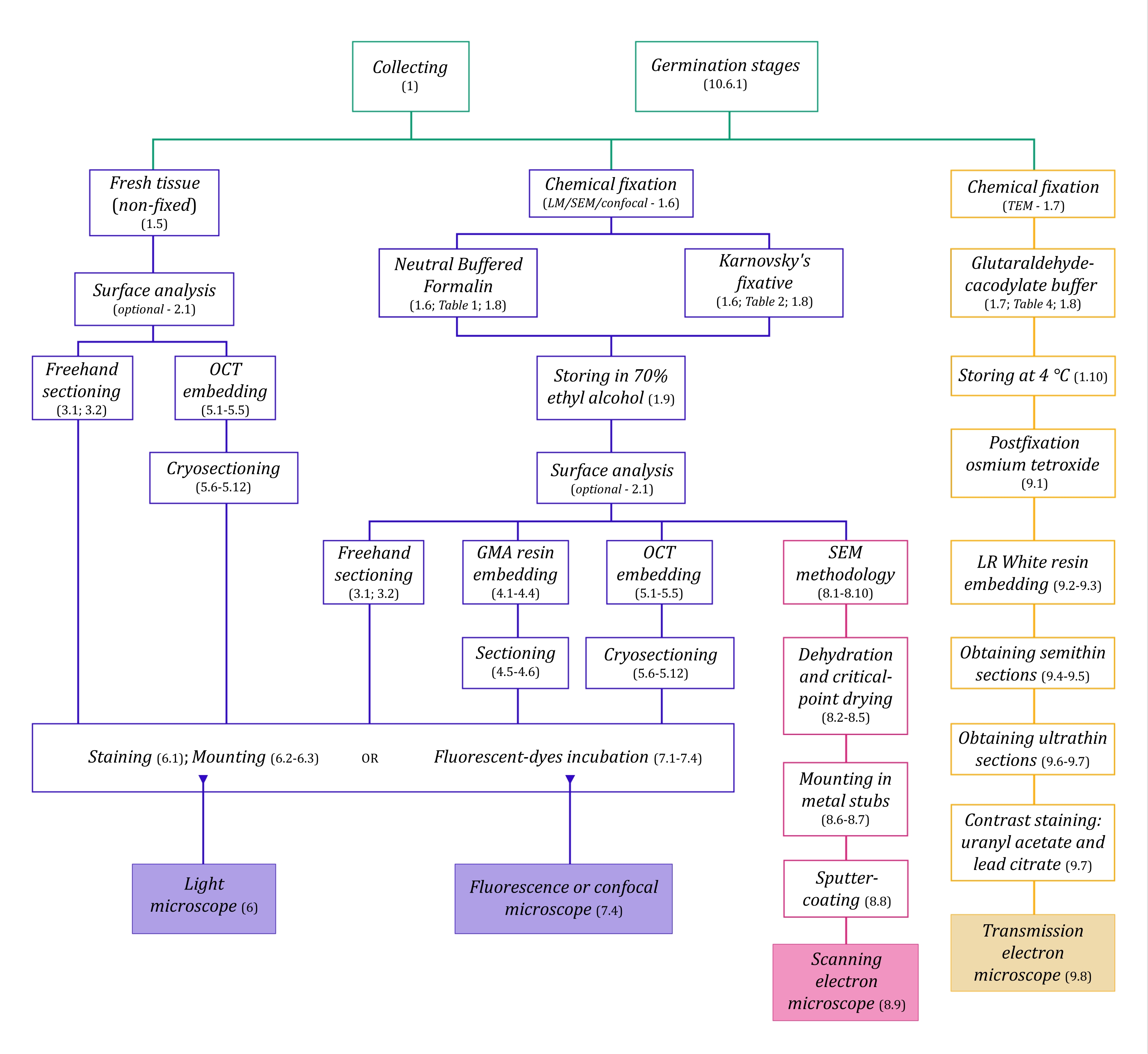
Figura 1: Resumen esquemático de los métodos de imagen. Los esquemas proporcionan indicaciones de los pasos del protocolo en los que se detallan. Abreviaturas: GMA = metacrilato de glicol, OCT = compuesto de temperatura de corte óptima, SEM = microscopía electrónica de barrido. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Las técnicas de microscopía descritas aquí en detalle (Figura 1) están precedidas por los siguientes pasos esenciales: recolectar, fijar, deshidratar, incrustar y seccionar muestras. Como los pasos son variables (Figura 1) dependiendo de la(s) técnica(s) elegida(s), es importante pensar en el futuro, considerando los fijadores que se prepararán y transportarán al sitio de recolección, cómo se deben preparar las muestras antes de la fijación, los procesos de deshidratación que se utilizarán (sección 1) y las diferentes posibilidades de incrustación y métodos de seccionamiento (secciones 4, 5, y 9). La Figura 1 resume secuencialmente todos los pasos requeridos para cada técnica de microscopía que se describen a continuación.
Protocolo
1. Recolectar, fijar y mantener muestras
NOTA: Las plantas totalmente MH generalmente se pueden encontrar en el sotobosque del bosque oscuro 12,13, principalmente en áreas húmedas y abundantes en basura, mientras que las plantas parcialmente MH se pueden encontrar en bosques más abiertos12,13. Las plantas MH generalmente tienen órganos subterráneos bien desarrollados en una variedad de formas y tamaños.
- Al recolectar especies MH, explore el suelo alrededor de la base de la planta, teniendo cuidado de no dañar los órganos subterráneos y evite tirar de las plantas del suelo para evitar desconectar los órganos aéreos de los subterráneos.
- Excave cuidadosamente alrededor de las estructuras aéreas con una paleta de jardinería mientras explora los órganos subterráneos como raíces, tallos, rizomas y órganos de almacenamiento, sin dañar estas estructuras.
- Retire las partículas de tierra para preservar las estructuras frágiles y lave delicadamente estos órganos con agua del grifo para enjuagar las partículas restantes del suelo antes de fijar las muestras.
- Las plantas MH asociadas con la hojarasca exigen atención adicional; Recoja cuidadosamente los órganos conectados al material en descomposición a través de sus hifas, evite extraer estos órganos de las estructuras conectadas y recójalos con cuidado, ya que estas partes son muy delicadas. Conserve las estructuras con tales conexiones y recoja la basura también para su análisis.
- Si opta por analizar material fresco utilizando técnicas de imagen, mantenga las muestras en bolsas de plástico cerradas con la humedad adecuada, evaporando suficiente agua e hidratando la planta, evitando que el exceso de agua esté en contacto con las muestras. Transpórelos al laboratorio inmediatamente y analice las muestras el mismo día en que fueron recolectadas, prestando atención si las muestras aún se conservan al analizarlas.
- Lleve los fijadores al sitio de recolección en recipientes bien sellados. Fijar las muestras rápidamente después de la recolección para microscopía óptica (LM) y microscopía electrónica de barrido (SEM) en cualquiera de los siguientes fijadores: formalina tamponada neutra al 10% (NBF14, Tabla 1) o solución de Karnovsky (modificada15, Tabla 2). La solución de Karnovsky se puede preparar con tampón fosfato0,2 M 15, cuya receta se describe en la Tabla 3.
- Para el análisis por microscopía electrónica de transmisión (TEM), dividir las muestras con un espesor de 4-3 mm dentro de una gota de tampón de cacodilato glutaraldehído-sódico (modificado16, Tabla 4) en secciones más pequeñas de 1-2 mm de espesor. Deseche los bordes cortados fuera de la gota. Transferir inmediatamente las secciones a un tubo de recolección con un volumen de fijador más de 10 veces mayor que el volumen de las muestras, ya que es un fijador aditivo (es decir, sus moléculas se agregan químicamente a las proteínas fijadas16).
PRECAUCIÓN: Los tres fijadores descritos son altamente tóxicos. Evite la inhalación, especialmente durante su uso en el campo. Prepare todos los fijadores en una campana extractora con guantes. No mezcle cacodilato y ácidos, ya que se puede formar gas arsénico16. - Si las muestras fijas flotan en el fijador, esto indica la presencia de gas en los tejidos vegetales. El aire y otros gases impiden que el fijador penetre en toda la muestra2. Eliminar el gas de los tejidos remuestreándolos en partes más pequeñas y usando una bomba de vacío (presión de -300 a -400 inHg) hasta que todas las muestras se hundan en el fondo de la solución17. Tenga cuidado ya que la presión excesiva ejercida por la bomba puede dañar las muestras.
- Después de al menos 48 h en la solución de Karnovsky o NBF al 10%, lavar las muestras en 0,2 M PB (Tabla 3) y deshidratarlas con una serie de etanol al 10%, 30%, 50% y finalmente al 70%. Para muestras delicadas, deshidratar durante 30 minutos en cada concentración; Para muestras más grandes, deshidratar durante 1 h o más.
NOTA: Una solución de etanol al 70% es el medio ideal para almacenar muestras. Las muestras en etanol al 70% se pueden almacenar a temperatura ambiente durante años. No almacene material vegetal durante largos períodos en los fijadores, ya que la eliminación de los agentes fijadores es un paso esencial después de la fijación2. - Conservar las muestras en glutaraldehído-cacodilato a 4 °C antes de proceder a la posfijación (paso 9.1).
| 10% de formalina tamponada neutra (NBF)14 | |
| Paso 1 | añadir 10 ml de solución de formaldehído al 37-40% en 80 ml de agua destilada |
| Paso 2 | añadir 0,4 g de fosfato de sodio monobásico monohidrato (NaH2PO4· H2O) a la solución |
| Paso 3 | añadir 0,65 g de fosfato sódico dibásico, anhidro (Na2HPO4) |
| Paso 4 | engordar el volumen a 100 mL |
Tabla 1: Receta de formalina tamponada neutra al 10% 14.
| Solución de Karnovsky (modificada15) | |
| Paso 1 | en 20 ml de agua destilada a 60-70 °C |
| Paso 2 | Añadir 0,8 g de paraformaldehído (para obtener 4% p/v), agitando |
| Paso 3 | añadir 1-4 gotas de NaOH al 40% y remover hasta que la solución se vuelva clara |
| Paso 4 | enfriarlo y añadir 30 mL de tampón fosfato 0.2 M pH 7.2 (Tabla 3) |
| Paso 5 | diluir glutaraldehído al 25% en 0,1 M PB (pH 7,2) para obtener glutaraldehído al 1% (volumen final: ~60 mL) |
| Paso 6 | añadir glutaraldehído al 1% (paso 5) a la solución obtenida en el paso 4 hasta completar hasta 100 ml de fijador |
Tabla 2: Receta de la solución de Karnovsky (modificada15).
| Tampón fosfato (PB) 0,2 M pH 7,2 | |
| Paso 1 | añadir 14,196 g de fosfato sódico dibásico, anhidro (Na2HPO4) a 400 mL de agua destilada |
| Paso 2 | añadir 13,8 g de fosfato sódico monobásico monohidrato (NaH2PO4· H2O) |
| Paso 3 | Revuelva hasta que la solución esté clara |
| Paso 4 | ajustar el volumen final a 500 ml con agua destilada |
| Paso 5 | ajustar el pH a 7.2 |
| Paso 6 | para un PB de 0,1 M, diluir 1:1 |
Tabla 3: Receta de tampón de fosfato 0.2 M.
| Tampón de cacodilato de sodio glutaraldehído al 3% 0,2 M (modificado 16) | |
| Paso 1 | Tampón de cacodilato 0,2 M: añadir 4,28 g de trihidrato de cacodilato de sodio en 100 ml de agua destilada |
| Paso 2 | ajustar el pH a 7.2 |
| Paso 3 | añadir 12 ml de glutaraldehído al 25% en 25 ml de la solución en la etapa 2 (tampón de cacodilato 0,2 M pH 7,2) |
| Paso 4 | enrasar el volumen a 100 ml con agua destilada |
Tabla 4: Receta de tampón de cacodilato de glutaraldehído sódico al 3% 0.2 M (modificado16).
2. Análisis superficial de órganos en material fijo y no fijo
- Para analizar las hifas superficiales en órganos, especialmente subterráneos, y aquellos en contacto con hojarasca, observe material fijo o fresco en un microscopio de disección (estereomicroscopio) con un aumento de 7.5x o más, dependiendo de las muestras analizadas.
- Visualice las muestras sumergidas en el fijador, etanol al 70% (si se almacena en él) o agua del grifo en el caso de material fresco. Evite la luz directa del microscopio de disección, ya que puede secar y dañar las muestras.
- Búsqueda de áreas de interés en las muestras, guiada por las hifas superficiales y rizomorfos. Seleccione muestras que contengan áreas con rizomorfos superficiales, ya que se pueden seccionar para visualizar pelotones y bobinas de hifas dentro de las células corticales en raíces y tallos.
- Después de la selección, siga los pasos 1.6 y 1.9 si las muestras aún no están fijas. Si lo desea, fotografíe muestras frescas con un microscopio óptico sin fijación, como se describe en la sección 3.
- Utilice la cámara acoplada al microscopio estereoscópico para recoger imágenes de superficies de órganos, rizomorfos y otras estructuras observadas. En tales casos, organice un color de fondo adecuado para contrastar bien con el material y, si es posible, elija un material de fondo con una superficie menos rugosa (por ejemplo, papel).
3. Secciones a mano alzada de órganos vegetales
NOTA: Las secciones a mano alzada de los órganos de las plantas pueden ser un desafío, especialmente para estructuras pequeñas y delgadas. Sin embargo, estas secciones de tejidos con endófitos fúngicos pueden, en algunos casos, evidenciar mejor las hifas y otras características en comparación con las secciones delgadas.
- Seccionar muestras frescas o fijas con una cuchilla afilada, cortarlas lo más delgadas posible y colocarlas inmediatamente en una pequeña placa de Petri con agua (si está fresca) o etanol al 70% (si está fijada). Use un pincel pequeño para manipular las secciones sin dañarlas.
- Para facilitar la seccionación de materiales más difíciles (es decir, órganos pequeños, delgados y flexibles), rodee la muestra en una estructura, por ejemplo, poliestireno o pecíolo de Cecropia . Talle el soporte para acomodar la muestra y haga una sección delgada de la muestra y el soporte por completo.
- Manchar y montar las muestras como se describe en la sección 6.
4. Incrustación de muestras de plantas en resina y seccionamiento
- Deshidratar aún más las muestras almacenadas en etanol al 70% en 80%, 96% y 2x en etanol al 100%, durante 30 min a 2 h dependiendo del tamaño y la composición de las muestras.
- Use un kit de resina de metacrilato de glicol (GMA) de acuerdo con las instrucciones del fabricante. Véase Gerrits y Horobin (1996)18 para más consideraciones. Siga los pasos de infiltración e incrustación en consecuencia.
PRECAUCIÓN: La resina GMA es tóxica, puede causar reacción alérgica e irritación de la piel, los ojos y las mucosas. Use los reactivos en una campana extractora y guantes. - Utilice bandejas de moldeo de polietileno para incrustar, seleccionadas por tamaño de muestra (por ejemplo, 13 mm x 19 mm x 5 mm para muestras más grandes, 6 mm x 8 mm x 5 mm para muestras más pequeñas). Preste atención a la orientación deseada de la muestra dentro del molde y use una aguja para ayudar a orientarse.
- Dejar para la polimerización, preferiblemente a temperatura ambiente, hasta que se solidifique por completo. El proceso de endurecimiento suele durar unas horas, aunque se recomienda desmoldar los bloques al día siguiente. Después de la polimerización, separe cuidadosamente los bloques de resina de los moldes y proceda a unir los bloques lo antes posible para evitar la curvatura del bloque.
- Lija la cara del bloque de resina que se unirá, creando una superficie plana. Pegue el bloque de resina a un cuboide de madera (se recomienda 2 cm x 2 cm x 3 cm) con un adhesivo de cianoacrilato líquido de viscosidad media (consulte la Tabla de materiales). Asegúrese de que la resina esté completamente unida para evitar comprometer la sección.
- Realice la sección en un microtomo rotativo como se describe a continuación.
PRECAUCIÓN: Las hojas de los cuchillos de microtomo son muy afiladas y pueden causar accidentes. Asegúrese de manejarlos siguiendo todas las medidas de seguridad. Antes de acercarse al cuchillo (por ejemplo, para cambiar el bloque, para humedecer la resina), bloquee el volante grueso y coloque la cubierta de seguridad de la cuchilla. Guarde las cuchillas desechables en los casos apropiados. Tenga mucho cuidado al reemplazar las cuchillas.
NOTA: Se pueden usar diferentes tipos de cuchillos (por ejemplo, desechables o fijos; vidrio o acero) para seccionar resina GMA18. La calidad de las secciones depende de qué tan afilado sea el cuchillo. Asegúrese de que el cuchillo esté bien sujeto y no pueda moverse. Es posible que sea necesario cambiar regularmente los cuchillos desechables para lograr una mejor sección.- Fije el cuboide de madera firmemente al soporte del bloque. Ajuste la orientación de la sección con los tornillos de orientación y asegúrese de un ángulo adecuado de la cuchilla utilizando la inclinación de la cuchilla. Seleccione el grosor de las secciones; use un ajuste más grueso para el recorte y un ajuste más delgado para las secciones seleccionadas, ya que las secciones GMA se adhieren más adecuadamente a la diapositiva de vidrio cuando son más delgadas; El grosor sugerido para los tejidos vegetales es de 5-8 μm.
- Antes de comenzar, enfríe la habitación, si es necesario, ya que las temperaturas más altas empeoran la calidad de las secciones. Prepare lo siguiente para la sección: un vaso de precipitados con agua destilada, una placa caliente, pinceles (al menos dos), pinzas de punta fina, una pipeta Pasteur, portaobjetos de vidrio, papel de filtro (o papel de seda) y un lápiz (para identificar las muestras que se están seccionando).
- Ajuste la placa caliente a 50 °C y coloque el vaso de precipitados sobre ella. Preste atención a las diferencias en el calentamiento dependiendo del área de la placa caliente (por lo general, el área media se calienta más que los bordes; caliente el agua en el medio, preferiblemente).
- Elija un portaobjetos de vidrio, identifíquelo con un lápiz y pipete el agua destilada tibia por toda la superficie del portaobjetos. Si es necesario, use una solución (por ejemplo, detergente y agua, o etanol al 70%) para romper la tensión entre el agua y el vidrio, de modo que todo el portaobjetos esté igualmente cubierto. Algunos tipos de portaobjetos deben limpiarse previamente con etanol al 70% para obtener una adherencia adecuada a la sección.
- Comience avanzando gradualmente la cara del bloque de resina hacia la hoja del cuchillo. No intente seccionar sin avanzar primero el bloque, o de lo contrario el equipo y el bloque pueden dañarse. Si es necesario, recorte el bloque con un grosor superior (10 μm o más).
- Al acercarse a una sección adecuada, haga un movimiento firme en un solo sentido con el volante, de modo que la sección se realice de inmediato. Mantenga un control sobre la humedad de la resina. Durante la sección, humedezca regularmente la cara del bloque que se está cortando con un pincel humedecido en agua destilada si hay un problema con las secciones de rizado. Retire el exceso de agua con un pañuelo de papel.
- Con un par de pinzas de punta fina, coloque la sección obtenida en el agua sobre el tobogán. Al entrar en contacto con el agua, la resina GMA se estira. Si es necesario, use un pincel para desplegar y estirar suavemente las secciones. Use otro pincel para mantener constantemente la cuchilla libre de residuos de resina. No cambie entre los pinceles para evitar mojar la cuchilla.
- Después de poner en cola todas las secciones deseadas en la diapositiva, seque la cara inferior de la diapositiva y colóquela sobre la placa caliente. Retire el exceso de agua de la parte superior del portaobjetos frotando suavemente con un papel de filtro (opcional). Las secciones se adherirán cuando el agua se evapore del tobogán. No deje las guías demasiado largas para evitar que las secciones se dañen por el calor excesivo.
- Guarde los portaobjetos en una caja portaobjetos, lejos del polvo y el sol, y utilícelos para teñir y otros procedimientos. Las diapositivas se pueden almacenar durante varios años.
5. Congelación de muestras de plantas y seccionamiento con un criostato
NOTA: La consideración esencial en la criosección de tejido biológico es reducir el daño debido a la formación de cristales de hielo al congelar muestras. La crioprotección generalmente se realiza mediante la infusión de soluciones químicamente inertes como glicerol o sacarosa19,20.
- Un día antes de la sección de muestra, realice los siguientes pasos.
- Diluir 100 mL de 0.2 M PB (Tabla 3) en 100 mL de agua destilada para obtener 200 mL de 0.1 M PB. Prepare soluciones de sacarosa al 10%, 20% y 30% en PB 0.1 M (por ejemplo, para una solución al 10%, agregue 2 g de sacarosa en 20 ml de tampón).
- Para muestras frescas, lávelas en 0,1 M PB durante 30 min. Para las muestras en un fijador, lávelas en el mismo tampón utilizado para preparar el fijador durante 30 minutos. Para muestras en etanol al 70%, hidratarlas en etanol al 50% y al 30% y lavar en 0,1 M PB durante 1 h en cada solución.
- Incubar las muestras durante 2 h en sacarosa al 10%, 2 h en sacarosa al 20% y 2 h en sacarosa al 30%, a temperatura ambiente. Después, incubar durante la noche en sacarosa al 30% a 4 °C (o al menos durante 3 h; el tiempo máximo es de 48 h).
- El día del seccionamiento, preparar 40% y 50% de sacarosa en 0.1 M PB; No prepare soluciones de sacarosa con más de 12 h de antelación. Incubar durante 2 h en cada concentración de sacarosa, a 4 °C.
- Para la incrustación, en moldes pequeños, hacer una capa de compuesto OCT (medio de temperatura de corte óptima, utilizado para incrustar y congelar las muestras) y mantenerlo a -20 °C para congelar. Los moldes pueden ser moldes histológicos regulares, aunque para facilitar el desmoldeo de los bloques, se pueden hacer papeles o papel de aluminio utilizando un pequeño cuboide como marco y cinta adhesiva.
- Después de que la capa inferior del compuesto OCT se congele en los moldes, trabaje dentro de una cámara de criostato (aprox. -27 ° C). Colocar las muestras incubadas en sacarosa al 50% en los moldes, en la orientación en la que serán seccionadas. La cara superior de un bloque cuboide suele ser una mejor cara de sección. Marque en el molde donde se colocan las muestras, para que el bloque se pueda recortar fácilmente y se mantenga la orientación correcta.
- Rodee las muestras en compuesto OCT y reviente cualquier burbuja de aire que toque las muestras. Congelar a -20 °C. Con los bloques completamente congelados, colóquelos dentro de la cámara de criostato (aprox. -27 °C). Desmolde cada uno solo antes de usarlo y preste atención a las marcas que indican la ubicación de la muestra dentro del bloque.
- Como el compuesto OCT se corta fácilmente con una cuchilla, recorte adecuadamente los bloques antes de colocarlos en los mandriles. Coloque un poco de compuesto OCT en el mandril de criostato y coloque el bloque de modo que la cara superior esté seccionada. Las caras con áreas más pequeñas proporcionan mejores secciones. Espere hasta que el bloque esté bien unido al mandril y pruébelo antes de comenzar a seccionar.
- Coloque el mandril firmemente en el portamandriles. Ajuste la orientación de la sección con los tornillos de orientación. Ajuste el ángulo del cuchillo usando la inclinación del cuchillo. Seleccione el grosor de las secciones. Las muestras se pueden seccionar secciones de resina más gruesas que las habituales. Las secciones en un rango entre 5-20 μm se obtienen con éxito, siendo las secciones más gruesas más fáciles de hacer (menos rizado y menos daño de las estructuras).
- Avance la cara del bloque congelado hacia la hoja del cuchillo. No intente seccionar sin hacerlo, o de lo contrario el bloque puede desprenderse del mandril y dañarse. Si es necesario, recorte el bloque con un grosor superior (10 μm o más).
- Al acercarse a una sección adecuada, coloque la placa estabilizadora (es decir, una placa transparente que retenga la sección) sobre el cuchillo y haga un movimiento firme en un solo sentido con el volante, de modo que la sección se haga de inmediato. Los problemas de rizado pueden ser causados si la placa estabilizadora necesita ser ajustada (generalmente es ajustable en referencia a la cuchilla) o si hay residuos en la cuchilla. Limpie la cuchilla constantemente con un pincel para eliminar los residuos.
- Utilice portaobjetos especiales para que las secciones se adhieran fácilmente, como portaobjetos silanizados (comerciales o preparados con 2% de aminoalquilsilano en acetona 21), o portaobjetos preparados con 500 μg/ml de poli-L-lisina en agua destilada 21 o 0,2% de gelatina (ver detalles21). Mantenga los portaobjetos a temperatura ambiente.
- Para adherir la sección a una corredera, levante la placa estabilizadora y haga que la corredera toque rápidamente la sección. Como el portaobjetos está a temperatura ambiente, la sección se derrite inmediatamente y se adhiere al portaobjetos. Preste atención para girar la cara tratada de la diapositiva hacia la sección, que puede permanecer por encima del cuchillo o en la cara interna de la placa estabilizadora. Para evitar el curvamiento de las secciones, realice este paso rápidamente tan pronto como se levante la placa y tenga cuidado de no contorsionar la sección.
- Deje el portaobjetos fuera de la cámara de criostato (a temperatura ambiente) si se le van a añadir nuevas secciones. Después de adherir todas las secciones deseadas al portaobjetos, manténgalo dentro de la cámara de criostato o en el congelador (-20 °C o menos). No exponga los portaobjetos a la humedad. Guárdelas en una caja de diapositivas y recuerde identificar las diapositivas con un lápiz.
NOTA: Las diapositivas y bloques de OCT se pueden almacenar a -20 °C, aunque no durante demasiado tiempo. Para lograr mejores resultados, use las diapositivas y los bloques dentro de unos días.
6. Tinción de secciones de plantas y endófitos para microscopía óptica
NOTA: Se pueden usar muchos tipos de tinciones para secciones de plantas. Es difícil teñir diferencialmente los hongos endófitos y los tejidos vegetales. Aunque no es un procedimiento de tinción, en la sección 7 se presenta un método para marcar estructuras de hongos (fluorescencia con un conjugado de aglutinina de germen de trigo). Las secciones a mano alzada (explicadas en la sección 3), las secciones de resina (sección 4) y las criosecciones (sección 5) se pueden teñir, aunque las tinciones a base de fenol y alcohol son un desafío para estas muestras, ya que la resina GMA y la OCT pierden adherencia al portaobjetos en estos casos.
- Utilice uno o combine los siguientes métodos habituales de tinción para muestras de plantas.
- Azul de toluidina O22,23, un método ampliamente aplicado para la tinción general de secciones de plantas. Preparar una solución de 0,05% de azul de toluidina O en 0,1 M de fosfato (pH 6,8) o tampón de citrato 0,09 M (pH 4,5-4,8), dependiendo de la especie y tipos de tejido. Incubar secciones de resina GMA durante 2-10 minutos usando un frasco de tinción de portaobjetos o colocando algunas gotas sobre las secciones si se manchan algunas diapositivas. Lavar cuidadosamente con agua destilada o tampón después de la incubación y montar los portaobjetos con agua o secarlos en una placa caliente para producir portaobjetos permanentes como se describe en el paso 6.3.
- El reactivoLugol 2 indica la presencia de almidón. Prepare una solución de yodo al 5% (I2) y yoduro de potasio (KI) al 10% en agua destilada. Manchar las secciones durante 2 min, agregando unas gotas por encima del portaobjetos, y luego lavar con agua destilada. Esta prueba histoquímica generalmente se aplica a portaobjetos temporales.
- Sudán III, IV y negroB 24,25 tiñen para diferentes lípidos. Prepare una solución de Sudán al 0,3% (III, IV o B negra) en etanol al 70%, caliéntela hasta que hierva y deje enfriar. Use el sobrenadante, fílelo e incube secciones durante 15-30 minutos en una placa de Petri cerrada. Lave las secciones cuidadosamente con etanol al 70% y agua destilada. Monte los toboganes con agua (generalmente se aplica solo a toboganes temporales).
NOTA: Como Sudán es un tinte a base de alcohol, es más adecuado para secciones a mano alzada. Realice la tinción de la sección de resina GMA con cuidado, ya que generalmente se desprenden de la diapositiva.
- Para toboganes temporales, monte las secciones en agua o glicerina y observe posteriormente. Selle el cubreobjetos con esmalte de uñas para conservarlos un poco más.
- Para portaobjetos permanentes, monte las secciones con resinas sintéticas (por ejemplo, medio de montaje rápido, consulte Tabla de materiales). Gotea unas gotas del medio de montaje (puede desbordar el cubreobjetos), coloca el cubreobjetos con cuidado para evitar burbujas y usa pinzas para la ropa para presionar la corredera contra el cubreobjetos hasta que esté completamente seca. Elimine el exceso de medio de montaje seco con una cuchilla de afeitar.
7. Aplicación de un fluorocromo conjugado a la aglutinina de germen de trigo en microscopía fluorescente y confocal
NOTA: Este método se puede aplicar a secciones a mano alzada (explicadas en la sección 3), secciones de resina (sección 4) y criosecciones (sección 5). Las criosecciones pueden ser adecuadas para fines de microscopía confocal, ya que se pueden proporcionar muestras más gruesas en comparación con las secciones de resina, pero no tan gruesas como las de mano alzada. Un fluorocromo conjugado con la aglutinina de germen de trigo (WGA, ver Tabla de materiales) se aplica a imágenes fúngicas en microscopía de fluorescencia26. Un microscopio confocal no es esencial, aunque puede proporcionar imágenes tridimensionales claras de las estructuras de las plantas27.
- Preparar una solución de 0,2 mg/ml de conjugado WGA-fluorocromo en 0,1 M PB28 (pH 7,2, comprobar paso 5.1.1 y Tabla 3). Preparar una solución de Calcofluor White al 1% en 0,1 M PB (pH 7,2). Prepare pequeñas cantidades de estas soluciones, ya que las secciones se incuban directamente con ellas.
- Incubar las secciones en los portaobjetos de vidrio durante 30 minutos en la solución conjugada WGA-fluorocromo29, utilizando suficiente volumen para cubrir las secciones, luego lavar en 0,1 M PB.
- Incubar las secciones en la solución de calcofluor, utilizando suficiente volumen como medio de montaje. La solución se puede mantener durante el período de observación.
- Coloque cubreobjetos en los portaobjetos y observe en un microscopio confocal o un microscopio de luz fluorescente utilizando los siguientes filtros: TC/GFP (excitación: 470-440, emisión: 525-550, para WGA-fluorocromo en la Tabla de materiales - la pared celular fúngica fluoresce verde bajo este filtro29) y DAPI (excitación: 358, emisión: 463, para Calcofluor White)30.
NOTA: Las imágenes tridimensionales se pueden obtener utilizando la función de la serie Z en el microscopio confocal27.
8. Microscopía electrónica de barrido de órganos vegetales
- Después de fijar las muestras, realizar la deshidratación y almacenar etanol al 70% (sección 1), una posibilidad es cortar muestras para exponer cualquier superficie deseada para el análisis SEM, si es necesario (por ejemplo, tejidos internos, estructura del ovario). Use una hoja de afeitar afilada y nueva y haga cortes con un movimiento unidireccional, evitando una apariencia dañada de estas áreas en SEM. Si es necesario, use un microscopio estereoscópico para seleccionar las muestras y considere el área de talones metálicos para determinar el tamaño de las muestras.
- Muestras adicionales de deshidratación para SEM en una serie etanólica: 80%, 96% y 2x en alcohol etílico absoluto (≥99,8%). Mantener muestras pequeñas y delicadas durante 30 minutos en cada concentración y muestras más grandes y densas durante 1 h.
- Doble los sobres pequeños con papel de seda para organizar las muestras para los próximos pasos, las muestras más grandes se pueden manejar sin un sobre. Identifique los sobres con un lápiz escribiendo una letra o un número, y mantenga un registro de todas las muestras en cada uno. Conservar las muestras en etanol absoluto, aunque no durante mucho tiempo, y proceder al paso 8.4 lo antes posible.
- Proceda al secado del punto crítico (CP). Opere un secador CP de acuerdo con los procedimientos operativos estándar. Coloque las muestras en etanol absoluto (fluido intermedio) en una cámara de presión. En el punto crítico deCO2 (31 °C, 7,3 x 106 Pa) el fluido intermedio se disuelve en el fluido de transición (dióxido de carbono líquido) y las muestras se secan31.
- Después del secado CP, almacenar las muestras lo antes posible en un recipiente de desecación, por ejemplo, un matraz sellado que contenga gel de sílice. La humedad atmosférica puede destruir las muestras si se reabsorben31.
- Use talones de metal para montar las muestras. Antes de montar, ponte guantes para manipular los talones, sumérgelos en acetona durante 5 min para eliminar cualquier grasa, y déjalos secar. Utilice una cinta adhesiva de carbono conductiva de doble cara para fijar las muestras en el trozo, y un microscopio estereoscópico para ayudar a colocar las muestras, teniendo en cuenta que la vista desde arriba es la única perspectiva posible en las imágenes SEM.
- Manipule las muestras con pinzas de punta fina, teniendo cuidado ya que la parte de la muestra que tocan las pinzas generalmente está dañada, así que intente tocar las partes colocadas lejos de las áreas de interés (por ejemplo, áreas en contacto con la cinta). Mantenga los talones con muestras en una placa de Petri sellada con gel de sílice. Continúe con el paso 8.8 lo antes posible.
- Utilice una capa de pulverización catódica para depositar una capa de metal, generalmente oro o platino, en la superficie de las muestras en una atmósfera de baja presión de un gas inerte, frecuentemente argón31. Siga los procedimientos operativos estándar cuando utilice una recubridora de pulverización catódica. El espesor del recubrimiento depende de la topografía de las muestras, generalmente entre 15-40 nm32.
- Mantenga los talones recubiertos en una placa de Petri sellada con gel de sílice, y siempre que el gel de sílice retenga la humedad, las muestras se pueden almacenar de esta manera durante semanas. Utilice un microscopio electrónico de barrido para analizar las muestras. Las muestras en vacío son golpeadas por un haz de electrones, y la emisión de señales de tal interacción se interpreta como imágenes31. Para más detalles sobre el funcionamiento de un microscopio electrónico de barrido, lea Jeffree y Read (1991)31 y Bozzola y Russell (1999)32.
- Para reutilizar los talones, tire de la cinta adhesiva y frótelos con lana de alambre. Lavar en agua del grifo, sumergir en etanol absoluto y secar adecuadamente, evitando la oxidación del metal constituyente.
9. Microscopía electrónica de transmisión
- Prefijar muestras con tampón glutaraldehído-cacodilato, como se explica en los pasos 1.7 y 1.10. Después de 12-24 h de prefijación, lavar las muestras 3 veces en tampón de cacodilato 0,2 M (pH 7,25) durante 10 min. Realizar postfijación con tetróxido de osmio al 1% (OsO4) en tampón de cacodilato 0,2 M, durante 12 h en la oscuridad, a temperatura ambiente. Lavar 3 veces con agua destilada durante 5 min.
PRECAUCIÓN: El cacodilato y el tetróxido de osmio son altamente tóxicos y no deben inhalarse. Utilícelos en campanas extractoras, siguiendo las respectivas fichas de datos de seguridad. - Deshidratar las muestras con etanol al 30%, 50%, 70% y 96%, 2x en cada concentración, durante 10 min. Luego, deshidratar 3 veces en alcohol absoluto, durante 15 minutos cada vez.
- Infiltrar las muestras en resinas acrílicas hidrófilas (ver Tabla de materiales), una vez con resina 1:1 + etanol absoluto y 3x con resina pura durante 8-12 h cada una. Realizar la polimerización en cápsulas de gelatina a 60 °C hasta que se solidifique completamente (12 h máximo17).
- Evaluar de cerca la orientación de las muestras dentro del bloque de resina; Cortar la parte superior del bloque, con una cuchilla de afeitar, haciendo una forma piramidal que concentra la muestra en el área de seccionamiento. Obtener secciones semifinas (250-500 nm)33 en un ultramicrotomo con un cuchillo de diamante y colocar sobre portaobjetos de vidrio en unas pocas gotas de agua.
- Mantener los portaobjetos en una placa caliente a 60 °C. Manchar las secciones con azul de toluidina O como en el paso 6.1.1 y dejar secar completamente la mancha. Lavar cuidadosamente con agua del grifo. Evalúe la sección obtenida dibujando cuatro cuadrantes y seleccionando el cuadrante más adecuado para el análisis.
- Recorte el bloque para que la forma piramidal concentre el cuadrante elegido en la cara superior del bloque. Producir secciones ultrafinas (50-100 nm)33,34. El espesor se evalúa de acuerdo con el color de interferencia de las secciones: las secciones con aproximadamente 70 nm aparecen plateado-oro, con aproximadamente 100 nm aparecen oro y con aproximadamente 200 nm aparecenazules 34.
- Recoja las secciones ultrafinas del agua utilizando rejillas de cobre y proceda al método de tinción de contraste con acetato de uranilo y citrato de plomo, como se describe a continuación.
- Prepare una solución de citrato de plomo (Tabla 5) y congele la solución final en tubos de microcentrífuga con 1 ml de solución en cada uno, solo descongelando justo antes de usar.
- Preparar una solución de acetato de uranilo: disolver 0,625 g de acetato de uranilo [UO 2(CH3COO)2] en 25 ml de agua destilada recientemente hervida y enfriada. Conservar en un matraz oscuro en el congelador.
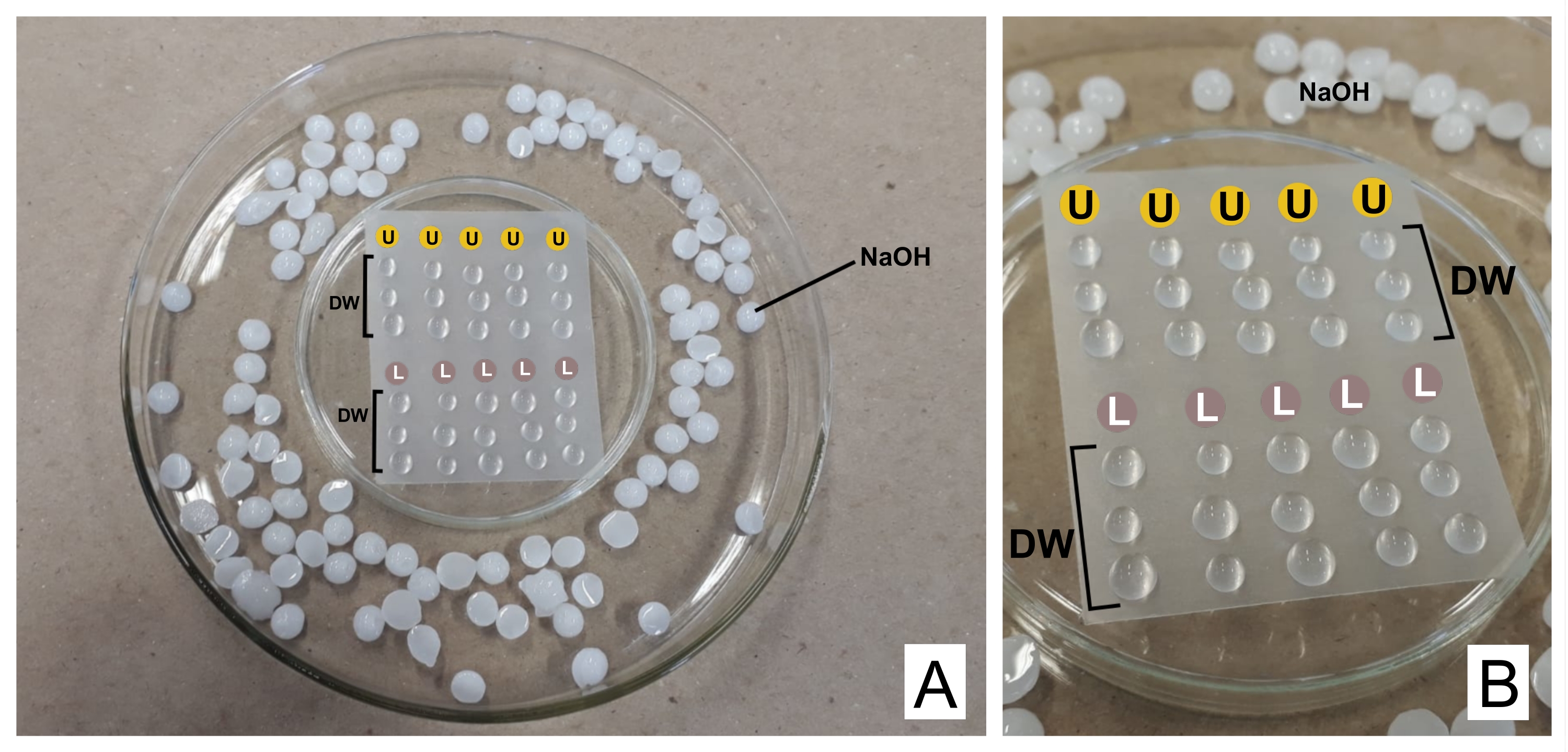
PRECAUCIÓN: El nitrato de plomo es tóxico si se ingiere. 1 N NaOH es altamente corrosivo. El acetato de uranilo es radiactivo y tóxico. No debe ingerirse, inhalarse ni entrar en contacto con la piel. - Al teñir, coloque ambos reactivos preparados en jeringas separadas de 3 ml con unidades de filtro (poro de 0,22 μm, consulte la Tabla de materiales). Prepare una placa de Petri volteada boca abajo con una película termoplástica de sellado sobre ella (ver Tabla de materiales) y dentro de una placa más ancha, con pellets de NaOH en los bordes como trampa para CO232 (ver Figura 2).
- Deseche la primera gota, y sobre la película coloque una gota de acetato de uranilo y tres gotas de agua destilada por cada rejilla manchada. Haga lo mismo con el citrato de plomo y agregue tres gotas más de agua destilada.
- Incubar la rejilla (con el lado opaco hacia abajo, donde están las secciones) en uranilo durante 30 min (tiempo variable). Lave 3 veces en las gotas de agua destilada, secando la rejilla cada vez suavemente con papel de filtro en el lado brillante. Repita con la gota de citrato de plomo (30 min) y lávelo.
- Después de al menos 4 h, analice las rejillas en un microscopio electrónico de transmisión. En este microscopio, un haz de electrones pasa a través de las secciones en vacío y la imagen se proyecta en una pantalla fluorescente. Para más detalles sobre el funcionamiento de un microscopio electrónico de transmisión, lea Bozzola y Russell (1999)32.
| solución de citrato de plomo (para tinción con contraste TEM) | |
| Paso 1 | Rodea un vaso de precipitados con papel de aluminio |
| Paso 2 | disolver 0,266 g de nitrato de plomo [Pb(NO3)2] en 6 ml de agua destilada recién hervida y enfriada |
| Paso 3 | agitar durante 2 min |
| Paso 4 | añadir 0,352 g de citrato trisódico [Na3(C6H5O7).2H2O] (la solución debe adquirir un aspecto lechoso) |
| Paso 5 | agitar durante 15 minutos, sellar el vaso de precipitados con papel de estaño y transferir la solución a un vaso de precipitados de 10 ml |
| Paso 6 | añadir 1,6 ml de NaOH 1N y 2,4 ml de agua destilada (la solución debe ser translúcida) |
| Paso 7 | si es necesario, ajuste el pH cerca de 12 |
Tabla 5: Receta de solución de citrato de plomo.

Figura 2: Esquema de tinción de contraste con soluciones de citrato de plomo y acetato de uranilo . (A) Prepare las placas de Petri, una volteada al revés (en el centro) con película termoplástica para que las gotas se puedan colocar por encima de ella, dentro de una más ancha. Los pellets de NaOH son lugares alrededor del plato central. (B) Las gotas de acetato de uranilo se colocan en los círculos con la letra U, y las gotas de citrato de plomo en los círculos marcados L. DW indican gotas de agua destilada. Las cuadrículas se tiñen secuencialmente en la columna, por lo que se pueden teñir cinco cuadrículas simultáneamente como se representa. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
10. Germinación simbiótica de semillas de orquídeas
- Asegúrese de que las soluciones y todos los materiales utilizados en la germinación simbiótica de las semillas sean estériles para evitar la contaminación. Comience esterilizándolos en autoclave durante 20 minutos a 121 °C. Los pasos de germinación simbiótica se resumen en la Figura 3.
- Desinfecte superficialmente las frutas y semillas sumergiéndolas en una solución de hipoclorito de sodio que contenga 2% de cloro activo durante 10-15 min para frutas y 7-10 min para semillas, considerando la rigidez y el grosor de la cubierta de semilla9. Las semillas delgadas y frágiles se pueden sumergir en una solución diluida de hipoclorito de sodio 1:1. Luego, lavar 3 veces en agua destilada esterilizada en autoclave para eliminar la solución de hipoclorito.
- Recuperar las semillas filtrando en tejido serigráfico y utilizar las semillas para proceder a las pruebas de germinación (preferiblemente). Si es necesario, guárdelos en sobres de papel de filtro dentro de matraces de vidrio con gel de sílice, a 4 °C, cierre herméticamente los matraces y ciérrelos con film transparente. Transfiera algunas gotas de agua del último lavado al agar dextrosa de papa (PDA, 39 g / L), para evaluar la efectividad del proceso de lavado.
- Antes de sembrar semillas en el medio de cultivo, evaluar su viabilidad a través de la prueba de tetrazolio (opcional) como se describe a continuación35.
- Incubar aproximadamente 10 mg de semillas en un tubo de microcentrífuga con 1 ml de sacarosa al 10% en agua destilada, durante 24 h a temperatura ambiente (aprox. 25 °C), a la luz.
- Retire la solución de sacarosa con una micropipeta y agregue 1 ml de solución de tetrazolio al 1% (cloruro de trifeniltetrazolio) en agua destilada. Incubar a 40 °C en un termobloque durante 24 h en la oscuridad.
- Retire la solución de tetrazolio con una micropipeta y lave las semillas con agua destilada 2 veces o hasta que se retire la solución. Retire todos los líquidos. Si es necesario, las semillas pueden almacenarse en el congelador hasta una semana (como en el paso 10.3) antes de ser analizadas.
- Resuspender las semillas en agua destilada y analizarlas bajo un microscopio óptico. Las semillas viables adquieren un color rojo claro a oscuro, mientras que las semillas no viables conservan su color natural.
- Realizar el siguiente protocolo adaptado9 para la germinación simbiótica de semillas de orquídeas.
- Incubar las semillas sobre discos de papel de filtro esterilizados en autoclave (1-2 cm de diámetro) colocados en placas de Petri con medio de cultivo de agar avena (OMA) (2,5 g/L de copos de avena y 7 g/L de agar, pH 6).
- En el centro de la placa de Petri, inocular un fragmento de medio de cultivo (aprox. 1 cm2) que contenga micelio del hongo aislado elegido para el procedimiento de germinación. Selle las placas de Petri con film transparente e incubarlas en la oscuridad a unos 25 °C o temperatura ambiente, ya que es más adecuado para el crecimiento de hongos.
- Preparar algunos platos con semillas y sin inoculación fúngica, como control negativo para la prueba de germinación.
- Analice los resultados de germinación semanalmente, recopilando datos cuantitativos y cualitativos y fotografiando protocormos y plántulas. La observación de semillas y protocormos debe realizarse utilizando un microscopio estereoscópico para una evaluación más precisa de la germinación. Utilice una fuente de luz que venga de abajo, ya que permite un mayor contraste, lo que permite discriminar el micelio fúngico de los protocormos más fácilmente.
- Recoger muestras en diferentes etapas de desarrollo y corregirlas para análisis anatómicos (sección 1). Aplicar todos los análisis de imágenes descritos anteriormente para investigar endófitos fúngicos en semillas, protocormos y plántulas durante la germinación (microscopía óptica - secciones 4, 5 y 6; confocal y fluorescencia - sección 7; SEM y TEM - secciones 8 y 9).
- Generar resultados cuantitativos siguiendo la clasificación de etapas según la Tabla 6. Las etapas describen el desarrollo habitual de las semillas de las orquídeas micoheterótrofas. Recopilar datos semanalmente y tabla con las fechas iniciales de cada etapa observada.
- Además, recopile datos cuantitativos que estimen el porcentaje y la tasa de germinación. Cuenta al menos 100 semillas o define los campos de conteo35. Demarque tres o más campos de conteo por placa de Petri, que consisten en regiones fijas con un área estandarizada, y evalúe semanalmente. Calcule los datos recopilados de acuerdo con la ecuación del índice de crecimiento (IG):

donde N 0 es el número de semillas contadas en la etapa0 , N 1 se refiere a la etapa1 , y sigue hasta la etapa 6 (registrada como N6)36.

Figura 3: Resumen esquemático de la metodología de germinación simbiótica de semillas. Los esquemas proporcionan indicaciones de pasos detallados en el protocolo. Abreviaturas: OMA = agar avena, PDA = agar dextrosa de patata. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Etapa de germinación | Descripción |
| 0 | Sin germinación |
| 1 | Hinchazón del embrión |
| 2 | Ruptura de Testa |
| 3 | Se desarrollan pelos absorbentes |
| 4 | Se desarrolla la proyección del vástago |
| 5 | Desarrollo de escamas protectoras (brácteas) |
| 6 | Se desarrollan las primeras raíces |
Tabla 6: Descripción de las etapas de desarrollo del protocormo aplicadas a los análisis periódicos de las pruebas de germinación. Modificado a partir de etapas descritas en Otero et al.36.
Resultados
Siguiendo las etapas esenciales de fijación del tejido vegetal se obtienen estructuras celulares lo más similares posible al estado vivo, considerando la morfología, el volumen y la organización espacial de los componentes y tejidos celulares16. Observe tales rasgos en las muestras después de la fijación química (Figura 4). La figura 4C-F representa muestras fijadas adecuadamente bajo microscopía ...
Discusión
Los análisis de imagen en anatomía y morfología vegetal tienen un potencial importante para cumplir objetivos y ayudar a comprender las relaciones entre las plantas micoheterótrofas y sus endófitos fúngicos indispensables, como lo demuestran los estudios de órganos subterráneos6,40, análisis estructurales de germinación simbiótica de semillas39 y estructuras aéreas y reproductivas 41 . La botánica estru...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores agradecen el financiamiento de FAEPEX y FAPESP (2015/26479-6). MPP agradece a Capes por su beca de maestría (proceso 88887.600591/2021-00) y CNPq. JLSM agradece al CNPq por las becas de productividad (303664/2020-7). Los autores también agradecen el acceso a equipos y asistencia proporcionados por LME (Laboratorio de Microscopía Electrónica - IB/Unicamp), INFABiC (Instituto Nacional de Ciencia y Tecnología sobre Fotónica Aplicada a la Biología Celular - Unicamp) y LaBiVasc (Laboratorio de Biología Vascular - DBEF/IB/Unicamp); LAMEB (UFSC) y Eliana de Medeiros Oliveira (UFSC) por contribuciones al protocolo de crioprotección; LME para contribuciones al protocolo TEM.
Materiales
| Name | Company | Catalog Number | Comments |
| Acetone | Sigma-Aldrich | 179124 | (for SEM stubs mounting) |
| Agar-agar (AA) | Sigma-Aldrich | A1296 | (for seeds germination tests) |
| Calcofluor White Stain | Sigma-Aldrich | 18909 | fluorescent dye (detects cellulose) |
| Citrate Buffer Solution, 0.09M pH 4.8 | Sigma-Aldrich | C2488 | (for toluidine blue O staining) |
| Conductive Double-Sided Carbon Tape | Fisher Scientific | 50-285-81 | (for SEM) |
| Confocal Microscope | Zeiss | (any model) | |
| Copper Grids | Sigma-Aldrich | G4776 | (for TEM) |
| Critical-point dryer | Balzers | (any model) | |
| Cryostat | Leica Biosystems | (any model) | |
| Dissecting microscope | Leica Biosystems | (= stereomicroscope, any model) | |
| Entellan | Sigma-Aldrich | 107960 | rapid mounting medium for microscopy |
| Ethyl alcohol, pure (≥99.5%) | Sigma-Aldrich | 459836 | (= ethanol, for dehydration processes) |
| Formaldehyde solution, 37% | Sigma-Aldrich | 252549 | (for NBF solution preparation) |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128 | histological tissue fixative |
| Gelatin capsules for TEM | Fisher Scientific | 50-248-71 | (for resin polymerisation in TEM) |
| Gelatin solution, 2% in H2O | Sigma-Aldrich | G1393 | (dilute for slides preparation - OCT adherence) |
| Glutaraldehyde solution, 25% | Sigma-Aldrich | G6257 | (for Karnovsky’s solution preparation) |
| HistoResin | Leica Biosystems | 14702231731 | glycol methacrylate (GMA) embedding kit |
| Iodine | Sigma-Aldrich | 207772 | (for Lugol solution preparation) |
| Lead(II) nitrate | Sigma-Aldrich | 228621 | Pb(NO3)2 (for TEM contrast staining) |
| Light Microscope | Olympus | (any model) | |
| LR White acrylic resin | Sigma-Aldrich | L9774 | hydrophilic acrylic resin for TEM |
| Lugol solution | Sigma-Aldrich | 62650 | (for staining) |
| Metal stubs for specimen mounts | Rave Scientific | (for SEM, different models) | |
| Microtome | Leica Biosystems | manual rotary microtome or other model | |
| Oatmeal agar (OMA) | Millipore | O3506 | (for seeds germination tests) |
| OCT Compound, Tissue-Tek | Sakura Finetek USA | 4583 | embedding medium for frozen tissues |
| Osmium tetroxide | Sigma-Aldrich | 201030 | OsO4 (for TEM postfixation) |
| Parafilm M | Sigma-Aldrich | P7793 | sealing thermoplastic film |
| Paraformaldehyde | Sigma-Aldrich | 158127 | (for Karnovsky’s solution preparation) |
| Poly-L-lysine solution, 0.1% in H2O | Sigma-Aldrich | P8920 | (for slides preparation - OCT adherence) |
| Poly-Prep Slides | Sigma-Aldrich | P0425 | poly-L-lysine coated glass slides |
| Polyethylene Molding Cup Trays | Polysciences | 17177A-3 | (6x8x5 mm, for embbeding samples in GMA resin) |
| Polyethylene Molding Cup Trays | Polysciences | 17177C-3 | (13x19x5 mm, for embbeding samples in GMA resin) |
| Potassium iodide | Sigma-Aldrich | 221945 | (for Lugol solution preparation) |
| Potato Dextrose Agar (PDA) | Millipore | 70139 | (for seeds germination tests) |
| Scanning Electron Microscope | Jeol | (any model) | |
| Silane [(3-Aminopropyl)triethoxysilane] | Sigma-Aldrich | A3648 | (for slides preparation - OCT adherence) |
| Silane-Prep Slides | Sigma-Aldrich | S4651 | glass slides coated with silane |
| Silica gel orange, granular | Supelco | 10087 | (for dessicating processes) |
| Sodium cacodylate trihydrate | Sigma-Aldrich | C0250 | (for glutaraldehyde-sodium cacodylate buffer) |
| Sodium hydroxide | Sigma-Aldrich | S5881 | NaOH (for Karnovsky’s solution preparation and TEM contrast staining) |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044 | NaClO (for seeds surface disinfection) |
| Sodium phosphate dibasic, anhydrous | Sigma-Aldrich | 71640 | Na2HPO4 (for NBF solution and PB preparation) |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638 | NaH2PO4·H2O (for NBF and PB) |
| Sputter coater | Balzers | (any model) | |
| Sucrose | Sigma-Aldrich | S0389 | C12H22O11 (for cryoprotection and germination test) |
| Sudan III | Sigma-Aldrich | S4131 | (for staining) |
| Sudan IV | Sigma-Aldrich | 198102 | (for staining) |
| Sudan Black B | Sigma-Aldrich | 199664 | (for staining) |
| Syringe | (3 mL, any brand, for TEM contrast staining) | ||
| Syringe Filter Unit, Millex-GV 0.22 µm | Millipore | SLGV033R | PVDF, 33 mm, gamma sterilized (for TEM contrast staining) |
| Tek Bond Super Glue 793 | Tek Bond Saint-Gobain | 78072720018 | liquid cyanoacrylate adhesive, medium viscosity |
| Toluidine Blue O | Sigma-Aldrich | T3260 | (for staining) |
| Transmission Electron Microscope | Jeol | (any model) | |
| Triphenyltetrazolium chloride | Sigma-Aldrich | T8877 | (for the tetrazolium test in seeds germination) |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804 | Na3(C6H5O7)·2H2O (for TEM contrast staining) |
| Ultramicrotome | Leica Biosystems | (any model) | |
| Uranyl acetate | Fisher Scientific | 18-607-645 | UO2(CH3COO)2 (for TEM contrast staining) |
| Vacuum pump | (any model) | ||
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | TermoFisher Scientific | W11261 | fluorescent dye-conjugated lectin (detects sialic acid and N-acetylglucosaminyl residues) |
Referencias
- Evert, R. F. . Esau’s Plant Anatomy: Meristems, Cells, and Tissues of the Plant Body: Their Structure, Function, and Development. , (2006).
- Yeung, E. C. T., Stasolla, C., Sumner, M. J., Huang, B. Q. . Plant Microtechniques and Protocols. , (2015).
- Sokoloff, D. D., Jura-Morawiec, J., Zoric, L., Fay, M. F. Plant anatomy: at the heart of modern botany. Botanical Journal of the Linnean Society. 195 (3), 249-253 (2021).
- Leake, J. R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytologist. 127 (2), 171-216 (1994).
- Bidartondo, M. I. The evolutionary ecology of myco-heterotrophy. New Phytologist. 167 (2), 335-352 (2005).
- Imhof, S., Massicotte, H. B., Melville, L. H., Peterson, R. L. Subterranean morphology and mycorrhizal structures. Mycoheterotrophy. , 157-214 (2013).
- Rasmussen, H. N., Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos. 118 (3), 334-345 (2009).
- Rasmussen, H. N., Dixon, K. W., Jersáková, J., Těšitelová, T. Germination and seedling establishment in orchids: a complex of requirements. Annals of Botany. 116 (3), 391-402 (2015).
- Zettler, L. W. Terrestrial orchid conservation by symbiotic seed germination: techniques and perspectives. Selbyana. 18 (2), 188-194 (1997).
- Stewart, S. L., Kane, M. E. Symbiotic seed germination and evidence for in vitro mycobiont specificity in Spiranthes brevilabris (Orchidaceae) and its implications for species-level conservation. In Vitro Cellular & Developmental Biology - Plant. 43 (3), 178-186 (2007).
- Zhao, D. -. K., et al. Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Frontiers in Plant Science. 12, 701152 (2021).
- Selosse, M. A., Roy, M. Green plants that feed on fungi: facts and questions about mixotrophy. Trends in Plant Science. 14 (2), 64-70 (2009).
- Merckx, V. S. F. T., Mennes, C. B., Peay, K. G., Geml, J. Evolution and diversification. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 215-244 (2013).
- Boon, M. E., Drijver, J. Routine Cytological Staining Techniques: Theoretical Background and Practice. Macmillan International Higher Education. , (1986).
- Karnovsky, M. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. Journal of Cell Biology. 27 (2), 137-138 (1964).
- Hayat, M. . Fixation for Electron Microscopy. , (1981).
- Roland, J. C., Vian, B. General preparation and staining of thin sections. Electron Microscopy of Plant Cells. 1, 675 (1991).
- Gerrits, P. O., Horobin, R. W. Glycol methacrylate embedding for light microscopy: basic principles and trouble-shooting. Journal of Histotechnology. 19 (4), 297-311 (1996).
- Zhang, Z., Niu, L., Chen, X., Xu, X., Ru, Z. Improvement of plant cryosection. Frontiers in Biology. 7 (4), 374-377 (2012).
- BeneŠ, K. On the media improving freeze-sectioning of plant material. Biologia Plantarum. 15 (1), 50-56 (1973).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. Cold Spring Harbor Protocols. 2008 (5), (2008).
- Sakai, W. S. Simple method for differential staining of paraffin embedded plant material using toluidine blue O. Stain Technology. 48 (5), 247-249 (1973).
- O’Brien, T., Feder, N., McCully, M. E. Polychromatic staining of plant cell walls by toluidine blue O. Protoplasma. 59 (2), 368-373 (1964).
- Ventrella, M. C., Almeida, A. L., Nery, L. A., Coelho, V. P. d. e. M. Métodos Histoquímicos Aplicados às Sementes. Universidade Federal de Viçosa. , (2013).
- Pearse, A. G. E. . Histochemistry, Theoretical and Applied. , (1960).
- Andrade-Linares, D. R., Franken, P. Fungal endophytes in plant roots: taxonomy, colonization patterns, and functions. Symbiotic Endophytes. , 311-334 (2013).
- Wymer, C. L., Beven, A. F., Boudonck, K., Lloyd, C. W. Confocal microscopy of plant cells. Confocal Microscopy Methods and Protocols. , 103-130 (1999).
- Marques, J. P. R., Soares, M. K. M. Manual de Técnicas Aplicadas à Histopatologia Vegetal. FEALQ. , (2021).
- Navarro, B. L., Marques, J. P. R., Appezzato-da-Glória, B., Spósito, M. B. Histopathology of Phakopsora euvitis on Vitis vinifera. European Journal of Plant Pathology. 154 (4), 1185-1193 (2019).
- Marques, J. P. R., et al. Sugarcane cell wall-associated defense responses to infection by Sporisorium scitamineum. Frontiers in Plant Science. 9, 698 (2018).
- Jeffree, C. E., Read, N. D. Ambient-and low-temperature scanning electron microscopy. Electron Microscopy of Plant Cells. , 313-413 (1991).
- Bozzola, J. J., Russell, L. D. . Electron Microscopy: Principles and Techniques for Biologists. , (1999).
- Murray, S. Basic transmission and scanning electron microscopy. Introduction to electron Microscopy for Biologists. , 3-18 (2008).
- . Glossary of TEM terms Available from: https://www.jeol.co.jp/en/words/emterms/ (2021)
- Seaton, P. T., et al. Orchid seed and pollen: a toolkit for long-term storage, viability assessment and conservation. Orchid Propagation: From Laboratories to Greenhouses—Methods and Protocols. , 71-98 (2018).
- Otero, J. T., Ackerman, J. D., Bayman, P. Differences in mycorrhizal preferences between two tropical orchids. Molecular Ecology. 13 (8), 2393-2404 (2004).
- Koch, R. A., et al. Marasmioid rhizomorphs in bird nests: Species diversity, functional specificity, and new species from the tropics. Mycologia. 112 (6), 1086-1103 (2020).
- Webster, J., Weber, R. . Introduction to Fungi. , (2007).
- Sisti, L. S., et al. The role of non-mycorrhizal fungi in germination of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. Frontiers in Plant Science. 10, 1589 (2019).
- Martos, F., et al. Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytologist. 184 (3), 668-681 (2009).
- Alves, M. F., et al. Reproductive development and genetic structure of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. BMC Plant Biology. 21 (1), 332 (2021).
- Merckx, V. S. F. T. Mycoheterotrophy: an introduction. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 1-17 (2013).
- Hall, J. L., Hawes, C. . Electron Microscopy of Plant Cells. , (1991).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados