
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Techniques de microscopie pour interpréter la colonisation fongique dans les tissus végétaux mycohétérotrophes et germination symbiotique des graines
Dans cet article
Résumé
Ce protocole vise à fournir des procédures détaillées pour la collecte, la fixation et la conservation d’échantillons de plantes mycohétérotrophes, en appliquant différentes techniques de microscopie telles que la microscopie électronique à balayage et à transmission, la microscopie optique, confocale et à fluorescence pour étudier la colonisation fongique dans les tissus et les graines des plantes germées avec des champignons mycorhiziens.
Résumé
La botanique structurale est une perspective indispensable pour bien comprendre l’écologie, la physiologie, le développement et l’évolution des plantes. Lors de la recherche sur les plantes mycohétérotrophes (c’est-à-dire les plantes qui obtiennent du carbone des champignons), des aspects remarquables de leurs adaptations structurelles, les modèles de colonisation des tissus par les champignons et la morphoanatomie des organes souterrains peuvent éclairer leurs stratégies de développement et leurs relations avec les hyphes, la source de nutriments. Un autre rôle important des champignons symbiotiques est lié à la germination des graines d’orchidées; toutes les espèces d’Orchidaceae sont mycohétérotrophes pendant la germination et le stade des plantules (mycohétérotrophie initiale), même celles qui photosynthétisent à l’âge adulte. En raison du manque de réserves nutritionnelles dans les graines d’orchidées, les symbiotes fongiques sont essentiels pour fournir des substrats et permettre la germination. L’analyse des stades de germination par perspective structurelle peut également répondre à des questions importantes concernant l’interaction des champignons avec les graines. Différentes techniques d’imagerie peuvent être appliquées pour dévoiler des champignons endophytes dans les tissus végétaux, comme le proposent cet article. Des sections à main levée et minces d’organes végétaux peuvent être colorées puis observées à l’aide de la microscopie optique. Un fluorochrome conjugué à l’agglutinine de germe de blé peut être appliqué sur les champignons et co-incubé avec Calcofluor White pour mettre en évidence les parois cellulaires végétales en microscopie confocale. En outre, les méthodologies de microscopie électronique à balayage et à transmission sont détaillées pour les orchidées mycohétérotrophes, et les possibilités d’appliquer de tels protocoles dans des plantes apparentées sont explorées. La germination symbiotique des graines d’orchidées (c’est-à-dire en présence de champignons mycorhiziens) est décrite en détail dans le protocole, ainsi que les possibilités de préparer les structures obtenues à partir de différentes étapes de la germination pour des analyses par microscopie optique, confocale et électronique.
Introduction
La recherche structurale en botanique, couvrant la morphologie et l’anatomie des plantes, est fondamentale pour comprendre l’organisme entier1,2, et fournit des perspectives indispensables pour intégrer et contribuer à la connaissance de l’écologie, de la physiologie, du développement et de l’évolution des plantes3. Les méthodes en morphologie et anatomie des plantes comprennent actuellement des protocoles, des équipements et des connaissances développés récemment ainsi qu’il y a plus d’un siècle2. L’exécution et l’adaptation continues de méthodes classiques (par exemple, la microscopie optique) ainsi que des techniques plus récentes (par exemple, la microscopie confocale, la microtomographie à rayons X) ont la même base essentielle: des connaissances théoriques permettant le développement d’une méthodologie.
L’outil principal en anatomie et morphologie des plantes est l’image. Malgré l’idée fausse que de telles analyses sont de simples observations, laissant place aux interprétations subjectives2, l’analyse et la compréhension des images dans ce domaine nécessitent une connaissance des méthodes appliquées (équipement, type d’analyse, procédures méthodologiques), des composants cellulaires, de l’histochimie et du corps végétal (organisation et fonction tissulaire, ontogenèse, adaptations morphologiques). L’interprétation des images obtenues par diverses méthodes peut conduire à corréler la forme et la fonction, à déchiffrer la composition chimique d’une structure, à corroborer la description des taxons, à comprendre les infections par les phytopathogènes et à d’autres évaluations similaires.
Lors de la recherche sur les plantes mycohétérotrophes (MH) (c’est-à-dire les plantes non photosynthétiques qui obtiennent du carbone à partir de champignons mycorhiziens4,5), des aspects remarquables de leurs adaptations structurelles, les modèles de colonisation des tissus par les champignons et la morphoanatomie des organes souterrains peuvent éclairer leurs stratégies de développement et leurs relations avec les hyphes, qui sont la source de nutriments. Les organes souterrains des plantes MH présentent généralement des adaptations importantes liées à leur association avec les champignons du sol, il est donc essentiel d’effectuer ces investigations anatomiques et morphologiques6. Les organes aériens des espèces MH ne doivent pas être ignorés, car les endophytes peuvent également être présents dans ces tissus, même s’ils ne sont pas des champignons mycorhiziens (observations personnelles, pas encore publiées).
Outre l’essentialité bien établie de l’association des champignons mycorhiziens avec les espèces MH tout au long de leur cycle de vie7, toutes les espèces d’orchidées, même les autotrophes, ont un stade mycohétérotrophe obligatoire initial dans les milieux naturels. Elle se produit parce que l’embryon des orchidées est indifférencié et dépourvu d’endosperme ou de cotylédons, étant donc incapable de se développer et de s’établir dans des environnements naturels sans le soutien nutritionnel de partenaires fongiques 4,8. Considérant que, les protocoles de germination symbiotique peuvent être appliqués non seulement aux espèces MH, mais aussi aux orchidées photosynthétiques, visant à étudier la spécificité orchidée-champignon dans la germination et le développement du protocorme, une méthodologie largement appliquée dans les initiatives de conservation des espèces menacées 9,10,11.
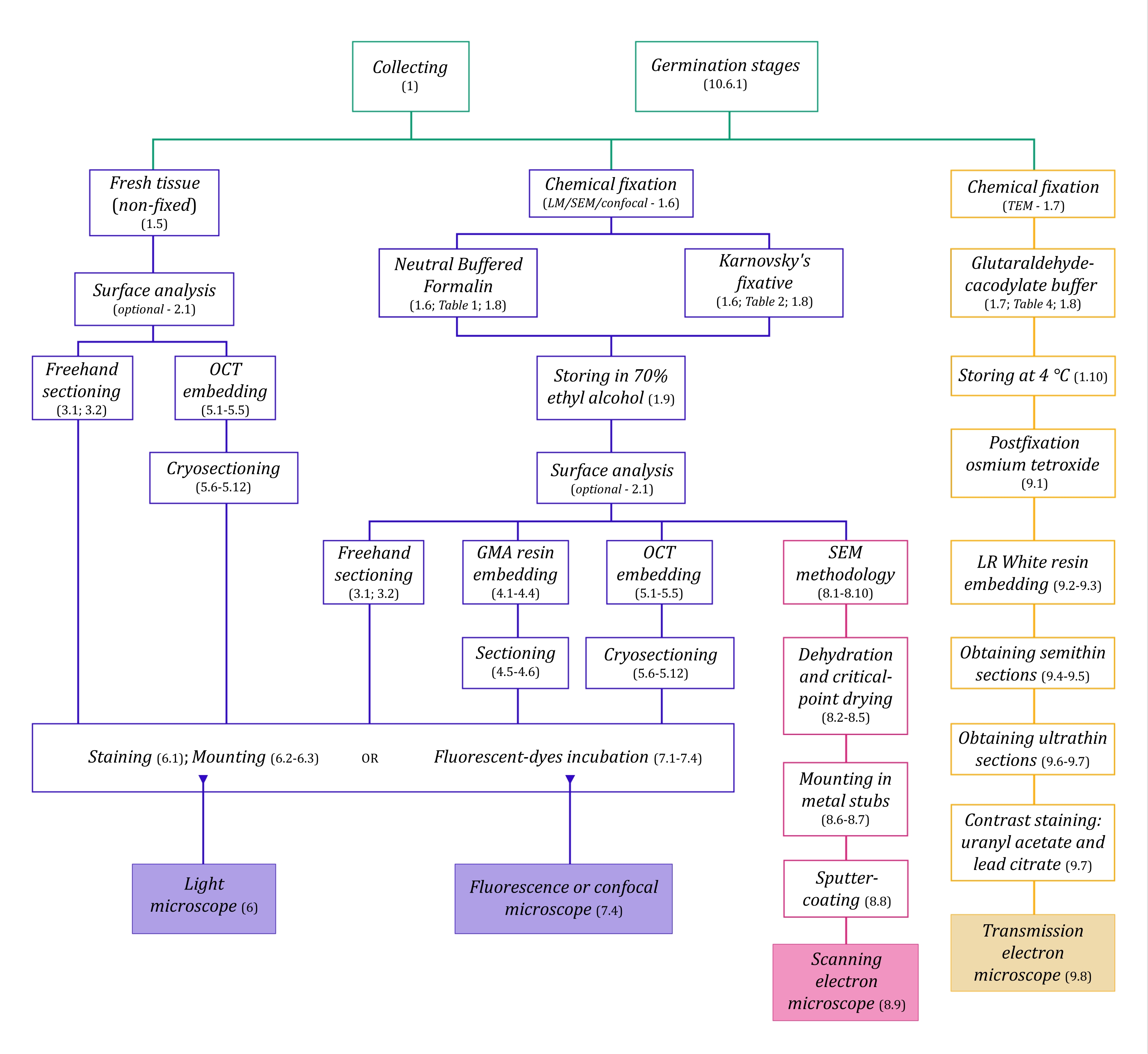
Dans cet assemblage de méthodes, nous décrivons les étapes importantes impliquées dans la collecte, la fixation et le stockage d’échantillons de plantes MH pour les études anatomiques (section 1), l’analyse de surface et la sélection des échantillons (section 2), les méthodes de sectionnement (à main levée: section 3, microtomie: section 4, cryomicrotomie: section 5), coloration et montage (section 6), la fluorescence et la microscopie confocale des endophytes fongiques (section 7), la microscopie électronique à balayage (section 8), et la microscopie électronique à transmission (section 9). De plus, nous décrivons une méthode de germination symbiotique pour les graines d’orchidées (MH et autotrophe, section 10), car les méthodes d’imagerie mentionnées précédemment peuvent être appliquées avec succès pour analyser la colonisation fongique des graines, des protocormes et des plantules dans le processus de germination.

Figure 1 : Résumé schématique des méthodes d’imagerie. Les schémas fournissent des indications sur les étapes du protocole dans lesquelles ils sont détaillés. Abréviations : GMA = méthacrylate de glycol, OCT = composé à température de coupe optimale, MEB = microscopie électronique à balayage. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Les techniques de microscopie décrites ici en détail (Figure 1) sont précédées des étapes essentielles suivantes : prélèvement, fixation, déshydratation, enrobage et sectionnement des échantillons. Comme les étapes sont variables (figure 1) en fonction de la ou des techniques choisies, il est important de penser à l’avenir, en tenant compte des fixateurs à préparer et à transporter vers le site de prélèvement, de la façon dont les échantillons doivent être préparés avant la fixation, des processus de déshydratation à utiliser (section 1), des différentes possibilités d’encastrement et des méthodes de sectionnement (sections 4, 5, et 9). La figure 1 résume séquentiellement toutes les étapes requises pour chaque technique de microscopie décrite en détail ci-dessous.
Protocole
1. Collecte, fixation et conservation des échantillons
NOTE: Les plantes entièrement MH peuvent généralement être trouvées dans le sous-étage de la forêt sombre 12,13, principalement dans les zones humides et abondantes en litière, tandis que les plantes partiellement MH peuvent être trouvées dans les forêts plus ouvertes12,13. Les plantes MH ont généralement des organes souterrains bien développés dans une variété de formes et de tailles.
- Lors de la collecte d’espèces MH, explorez le sol autour de la base végétale, en prenant soin de ne pas endommager les organes souterrains, et évitez de tirer les plantes du sol pour éviter de déconnecter les organes aériens des organes souterrains.
- Creusez soigneusement autour des structures aériennes à l’aide d’une truelle de jardinage tout en explorant les organes souterrains comme les racines, les tiges, les rhizomes et les organes de stockage, sans endommager ces structures.
- Enlevez les particules de sol pour préserver les structures fragiles et lavez délicatement ces organes à l’eau du robinet pour rincer les particules de sol restantes avant de fixer les échantillons.
- Les plantes MH associées à la litière de feuilles exigent une attention particulière; Collectez soigneusement les organes connectés au matériau en décomposition à travers leurs hyphes, évitez de retirer ces organes des structures connectées et collectez-les soigneusement car ces parties sont très délicates. Préservez les structures avec de telles connexions et collectez également la litière pour analyse.
- Si vous choisissez d’analyser des matériaux frais à l’aide de techniques d’imagerie, conservez les échantillons dans des sacs en plastique fermés avec suffisamment d’humidité, suffisamment d’eau s’évapore et hydrate la plante, empêchant ainsi un excès d’eau d’entrer en contact avec les échantillons. Transportez-les immédiatement au laboratoire et analysez les échantillons le jour même où ils ont été prélevés, tout en veillant à ce que les échantillons soient encore conservés lors de leur analyse.
- Transporter les fixateurs jusqu’au site de collecte dans des contenants bien fermés. Fixer rapidement les échantillons après le prélèvement pour la microscopie optique (LM) et la microscopie électronique à balayage (MEB) dans l’un des fixateurs suivants : formol tamponné neutre à 10 % (FBN14, tableau 1) ou solution de Karnovsky (modifié15, tableau 2). La solution de Karnovsky peut être préparée avec du tampon phosphate15 0,2 M, dont la recette est décrite dans le tableau 3.
- Pour l’analyse par microscopie électronique à transmission (MET), couper les échantillons d’une épaisseur de 4-3 mm à l’intérieur d’une goutte de tampon cacodylate de glutaraldéhyde-sodium (modifié16, tableau 4) en sections plus petites de 1-2 mm d’épaisseur. Jetez les bords coupés à l’extérieur de la goutte. Transférer immédiatement les sections dans un tube collecteur avec un volume de fixateur plus de 10 fois supérieur au volume des échantillons, car il s’agit d’un fixateur additif (c’est-à-dire que ses molécules sont ajoutées chimiquement aux protéines fixées16).
ATTENTION : Les trois fixateurs décrits sont très toxiques. Éviter l’inhalation, surtout lors de leur utilisation sur le terrain. Préparez tous les fixateurs dans une hotte à l’aide de gants. Ne mélangez pas de cacodylate et d’acides, car l’arsenic gazeux peut se former16. - Si les échantillons fixés flottent dans le fixateur, cela indique la présence de gaz dans les tissus végétaux. L’air et les autres gaz empêchent le fixateur de pénétrer dans l’ensemble de l’échantillon2. Éliminer le gaz des tissus en les prélevant à nouveau en plus petites parties et en utilisant une pompe à vide (pression de -300 à -400 inHg) jusqu’à ce que tous les échantillons coulent au fond de la solution17. Soyez prudent car une pression excessive exercée par la pompe peut endommager les échantillons.
- Après au moins 48 h dans la solution de Karnovsky ou 10% NBF, laver les échantillons dans 0,2 M PB (tableau 3) et les déshydrater avec une série de 10%, 30%, 50% et enfin 70% d’éthanol. Pour les échantillons délicats, déshydrater pendant 30 min à chaque concentration; Pour les échantillons plus gros, déshydrater pendant 1 h ou plus.
REMARQUE: Une solution d’éthanol à 70% est le milieu idéal pour stocker les échantillons. Les échantillons dans de l’éthanol à 70 % peuvent être conservés à température ambiante pendant des années. Ne stockez pas le matériel végétal pendant de longues périodes dans les fixateurs, car l’élimination des agents fixateurs est une étape essentielle après la fixation2. - Conserver les échantillons dans du glutaraldéhyde-cacodylate à 4 °C avant de procéder à la postfixation (étape 9.1).
| 10 % de formol tamponné neutre (FBN)14 | |
| Étape 1 | ajouter 10 mL de solution de formaldéhyde à 37-40 % dans 80 mL d’eau distillée |
| Étape 2 | ajouter 0,4 g de phosphate de sodium monobasique monohydraté (NaH2PO4· H2O) à la solution |
| Étape 3 | ajouter 0,65 g de phosphate de sodium dibasique, anhydre (Na2HPO4) |
| Étape 4 | porter le volume à 100 mL |
Tableau 1 : Recette de formol tamponné neutre à 10 % 14.
| Solution de Karnovski (modifiée 15) | |
| Étape 1 | dans 20 mL d’eau distillée à 60-70 °C |
| Étape 2 | Ajouter 0,8 g de paraformaldéhyde (pour obtenir 4% p/v), en agitant |
| Étape 3 | ajouter 1 à 4 gouttes de NaOH à 40% et remuer jusqu’à ce que la solution devienne claire |
| Étape 4 | refroidir et ajouter 30 mL de tampon phosphate 0,2 M pH 7,2 (tableau 3) |
| Étape 5 | diluer 25 % de glutaraldéhyde dans 0,1 M PB (pH 7,2) pour obtenir 1 % de glutaraldéhyde (volume final : ~60 mL) |
| Étape 6 | ajouter 1 % de glutaraldéhyde (étape 5) à la solution obtenue à l’étape 4 jusqu’à obtention d’une dose de fixation de 100 mL |
Tableau 2 : Recette de la solution de Karnovsky (15 modifié).
| Tampon phosphate 0,2 M (PB) pH 7,2 | |
| Étape 1 | ajouter 14,196 g de phosphate dibasique de sodium anhydre (Na2HPO4) à 400 mL d’eau distillée |
| Étape 2 | ajouter 13,8 g de phosphate de sodium monobasique monohydraté (NaH2PO4· H2O) |
| Étape 3 | remuer jusqu’à ce que la solution soit claire |
| Étape 4 | régler le volume final à 500 mL avec de l’eau distillée |
| Étape 5 | ajuster le pH à 7,2 |
| Étape 6 | pour un PB de 0,1 M, diluer 1:1 |
Tableau 3 : Recette tampon phosphate 0,2 M.
| 3% glutaraldéhyde 0,2 M tampon cacodylate de sodium (modifié 16) | |
| Étape 1 | Tampon cacodylate 0,2 M : ajouter 4,28 g de trihydrate de cacodylate de sodium dans 100 mL d’eau distillée |
| Étape 2 | ajuster le pH à 7,2 |
| Étape 3 | ajouter 12 mL de glutaraldéhyde à 25 % dans 25 mL de la solution à l’étape 2 (tampon cacodylate 0,2 M pH 7,2) |
| Étape 4 | porter le volume à 100 mL avec de l’eau distillée |
Tableau 4 : Recette tampon de glutaraldéhyde 0,2 M à 3 % de cacodylate de sodium (16 modifié).
2. Analyse de surface des organes dans le matériel fixe et non fixe
- Pour analyser les hyphes superficiels dans les organes, en particulier souterrains, et ceux en contact avec la litière de feuilles, observez le matériel fixe ou frais dans un microscope à dissection (stéréomicroscope) à un grossissement de 7,5x ou plus, selon les échantillons analysés.
- Visualisez les échantillons immergés dans le fixateur, l’éthanol à 70% (s’il y est stocké) ou l’eau du robinet dans le cas d’une matière fraîche. Empêchez la lumière directe du microscope à dissection, car elle peut sécher et endommager les échantillons.
- Rechercher des zones d’intérêt dans les échantillons, guidés par les hyphes superficiels et les rhizomorphes. Sélectionner des échantillons qui contiennent des zones avec des rhizomorphes superficiels, car ceux-ci peuvent être sectionnés pour visualiser les pelotons et les serpentins d’hyphes dans les cellules corticales des racines et des tiges.
- Après la sélection, suivez les étapes 1.6 et 1.9 si les échantillons ne sont pas encore fixés. Si vous le souhaitez, photographiez des échantillons frais à l’aide d’un microscope optique sans fixation, comme décrit à la section 3.
- Utilisez la caméra couplée au stéréomicroscope pour recueillir des images à partir de surfaces d’organes, de rhizomorphes et d’autres structures observées. Dans de tels cas, faites en sorte qu’une couleur d’arrière-plan adéquate contraste bien avec le matériau et, si possible, choisissez un matériau de fond avec une surface moins rugueuse (par exemple, du papier).
3. Sections à main levée d’organes végétaux
REMARQUE: Les sections à main levée des organes végétaux peuvent être difficiles, en particulier pour les structures petites et minces. Cependant, ces sections de tissus avec des endophytes fongiques peuvent, dans certains cas, mieux montrer les hyphes et d’autres caractéristiques par rapport aux sections minces.
- Couper les échantillons frais ou fixes à l’aide d’une lame tranchante, les couper aussi fins que possible et les placer immédiatement dans une petite boîte de Pétri avec de l’eau (si fraîche) ou de l’éthanol à 70% (si fixe). Utilisez un petit pinceau pour manipuler les sections sans les endommager.
- Pour faciliter la sectionnement de matériaux plus difficiles (c.-à-d. organes petits, minces et flexibles), entourez l’échantillon d’une structure, par exemple du polystyrène ou du pétiole de Cecropia . Sculptez le support pour accueillir l’échantillon et faites une fine section de l’échantillon et du support ensemble.
- Colorer et monter les échantillons comme décrit à la section 6.
4. Incorporation d’échantillons de plantes dans la résine et sectionnement
- Déshydrater davantage les échantillons stockés dans de l’éthanol à 70% dans 80%, 96% et 2x dans de l’éthanol à 100%, pendant 30 min à 2 h selon la taille et la composition des échantillons.
- Utilisez un kit de résine de méthacrylate de glycol (GMA) conformément aux instructions du fabricant. Voir Gerrits et Horobin (1996)18 pour d’autres considérations. Suivez les étapes d’infiltration et d’incorporation en conséquence.
ATTENTION: La résine GMA est toxique, elle peut provoquer une réaction allergique et une irritation de la peau, des yeux et des muqueuses. Utilisez les réactifs dans une hotte et utilisez des gants. - Utiliser des plateaux de moulage en polyéthylène pour l’encastrement, choisis selon la taille des échantillons (p. ex., 13 mm x 19 mm x 5 mm pour les plus gros échantillons, 6 mm x 8 mm x 5 mm pour les plus petits). Faites attention à l’orientation souhaitée de l’échantillon à l’intérieur du moule et utilisez une aiguille pour aider à l’orientation.
- Laisser pour polymérisation, de préférence à température ambiante, jusqu’à ce qu’il soit complètement solidifié. Le processus de durcissement prend généralement quelques heures, bien qu’il soit recommandé de démouler les blocs le lendemain. Après polymérisation, détachez soigneusement les blocs de résine des moules et procédez à la fixation des blocs dès que possible pour éviter la courbure des blocs.
- Poncez la face du bloc de résine qui sera attaché, créant ainsi une surface plane. Collez le bloc de résine sur un cuboïde en bois (2 cm x 2 cm x 3 cm recommandé) avec un adhésif cyanoacrylate liquide de viscosité moyenne (voir le tableau des matériaux). Assurez-vous que la résine est complètement fixée pour éviter de compromettre la section.
- Effectuer la section dans un microtome rotatif comme décrit ci-dessous.
ATTENTION : Les lames des couteaux microtome sont très tranchantes et peuvent provoquer des accidents. Assurez-vous de les manipuler en suivant toutes les mesures de sécurité. Avant de vous approcher du couteau (par exemple, pour changer le bloc, pour humidifier la résine), verrouillez le volant grossier et placez le couvercle de sécurité de la lame. Entreposer les lames jetables dans les cas appropriés. Soyez extrêmement prudent lorsque vous remplacez des lames.
NOTE: Différents types de couteaux (par exemple, jetables ou fixes; verre ou acier) peuvent être utilisés pour sectionner la résine GMA18. La qualité des sections dépend de la netteté du couteau. Assurez-vous que le couteau est bien attaché et ne peut pas bouger. Les couteaux jetables peuvent avoir besoin d’être changés régulièrement pour obtenir une meilleure section.- Fixez fermement le cuboïde en bois au support de bloc. Ajustez l’orientation du sectionnement à l’aide des vis d’orientation et assurez-vous d’un angle adéquat du couteau à l’aide de l’inclinaison du couteau. Sélectionnez l’épaisseur des sections; utiliser un réglage plus épais pour le rognage et un réglage plus mince pour les sections sélectionnées, car les sections GMA adhèrent mieux à la lame de verre lorsqu’elles sont plus minces; L’épaisseur suggérée pour les tissus végétaux est de 5 à 8 μm.
- Avant de commencer, refroidissez la pièce, si nécessaire, car des températures plus élevées aggravent la qualité des sections. Préparez les éléments suivants pour le sectionnement : un bécher avec de l’eau distillée, une plaque chauffante, des pinceaux (au moins deux), une pince à épiler à pointe fine, une pipette Pasteur, des lames de verre, du papier filtre (ou du papier de soie) et un crayon (pour identifier les échantillons à sectionner).
- Régler la plaque chauffante à 50 °C et placer le bécher dessus. Faites attention aux différences de chauffage en fonction de la zone de la plaque chauffante (généralement, la zone centrale chauffe plus que les bords; chauffez l’eau au milieu, de préférence).
- Choisissez une lame de verre, identifiez-la avec un crayon et pipette l’eau distillée chaude sur toute la surface de la lame. Si nécessaire, utilisez une solution (p. ex., détergent et eau, ou éthanol à 70 %) pour briser la tension entre l’eau et le verre, de sorte que toute la lame soit recouverte de manière égale. Certains types de lames doivent être prénettoyés avec de l’éthanol à 70 % pour obtenir une adhérence adéquate de la section.
- Commencez par avancer progressivement la face du bloc de résine vers la lame du couteau. N’essayez pas de sectionner sans avancer le bloc au préalable, sinon l’équipement et le bloc peuvent être endommagés. Si nécessaire, coupez le bloc en utilisant une épaisseur supérieure (10 μm et plus).
- Lorsque vous approchez d’une section appropriée, effectuez un mouvement ferme à sens unique avec le volant, de sorte que la section soit faite immédiatement. Gardez un œil sur l’humidité de la résine. Pendant le sectionnement, hydratez régulièrement la face du bloc à couper à l’aide d’un pinceau trempé dans de l’eau distillée s’il y a un problème avec les sections de curling. Enlevez l’excès d’eau avec un papier de soie.
- Avec une paire de pincettes à pointe fine, placez la section obtenue dans l’eau au-dessus de la glissière. Au contact de l’eau, la résine GMA s’étire. Si nécessaire, utilisez un pinceau pour déplier et étirer doucement les sections. Utilisez un autre pinceau pour garder constamment la lame exempte de débris de résine. Ne passez pas d’un pinceau à l’autre pour éviter de mouiller la lame.
- Après avoir mis en file d’attente toutes les sections souhaitées dans la glissière, séchez la face inférieure de la lame et placez-la au-dessus de la plaque chauffante. Retirez l’excès d’eau du haut de la lame en tamponnant doucement avec un papier filtre (facultatif). Les sections adhèrent lorsque l’eau s’évapore du toboggan. Ne laissez pas les lames trop longtemps pour éviter que les sections ne soient endommagées par une chaleur excessive.
- Rangez les lames dans une boîte à lames, à l’abri de la poussière et du soleil, et utilisez-les pour la coloration et d’autres procédures. Les diapositives peuvent être stockées pendant plusieurs années.
5. Congélation d’échantillons de plantes et sectionnement à l’aide d’un cryostat
NOTE: La considération essentielle dans la cryosectionnement des tissus biologiques est de réduire les dommages dus à la formation de cristaux de glace lors de la congélation des échantillons. La cryoprotection se fait généralement par infusation de solutions chimiquement inertes telles que le glycérol ou le saccharose19,20.
- Un jour avant la section de l’échantillon, effectuez les étapes suivantes.
- Diluer 100 mL de 0,2 M PB (tableau 3) dans 100 mL d’eau distillée pour obtenir 200 mL de 0,1 M PB. Préparer des solutions de saccharose à 10 %, 20 % et 30 % dans 0,1 M PB (p. ex., pour une solution à 10 %, ajouter 2 g de saccharose dans 20 mL de tampon).
- Pour les échantillons frais, les laver dans 0,1 M PB pendant 30 min. Pour les échantillons dans un fixateur, les laver dans le même tampon que celui utilisé pour préparer le fixateur pendant 30 min. Pour les échantillons dans de l’éthanol à 70%, hydratez-les dans de l’éthanol à 50% et 30% et lavez dans 0,1 M PB pendant 1 h dans chaque solution.
- Incuber les échantillons pendant 2 h dans 10% saccharose, 2 h dans 20% saccharose et 2 h dans 30% saccharose, à température ambiante. Ensuite, incuber pendant une nuit dans 30% de saccharose à 4 °C (ou au moins pendant 3 h; la durée maximale est de 48 h).
- Le jour du sectionnement, préparer 40% et 50% de saccharose dans 0,1 M PB; Ne préparez pas de solutions de saccharose plus de 12 h à l’avance. Incuber pendant 2 h dans chaque concentration de saccharose, à 4 °C.
- Pour l’enrobage, dans de petits moules, réaliser une couche de composé OCT (milieu de température de coupe optimale, utilisé pour l’encastrement et la congélation des échantillons) et la conserver à -20 °C pour la congeler. Les moules peuvent être des moules histologiques réguliers, bien que pour faciliter le démoulage des blocs, ceux en papier ou en papier d’aluminium peuvent être fabriqués en utilisant un petit cuboïde comme cadre et ruban adhésif.
- Une fois que la couche inférieure du composé OCT est congelée dans les moules, travailler à l’intérieur d’une chambre de cryostat (env. -27 °C). Placer les échantillons incubés dans 50% de saccharose dans les moules, dans l’orientation dans laquelle ils seront sectionnés. La face supérieure d’un bloc cuboïde est généralement une meilleure face de sectionnement. Marquez dans le moule où les échantillons sont placés, afin que le bloc puisse être facilement coupé et que l’orientation correcte soit maintenue.
- Entourer les échantillons dans un composé OCT et faire éclater toute bulle d’air touchant les échantillons. Congelez-les à -20 °C. Les blocs étant complètement congelés, placez-les à l’intérieur de la chambre du cryostat (env. -27 °C). Démoulez chacun seulement avant de les utiliser et faites attention aux marques qui indiquent l’emplacement de l’échantillon à l’intérieur du bloc.
- Comme le composé OCT est facilement coupé avec une lame, coupez les blocs de manière appropriée avant de les positionner sur les mandrins. Mettez un peu de composé OCT dans le mandrin du cryostat et positionnez le bloc de manière à ce que la face supérieure soit sectionnée. Les faces avec des zones plus petites offrent de meilleures sections. Attendez que le bloc soit bien attaché au mandrin et testez-le avant de commencer à sectionner.
- Placez fermement le mandrin dans le porte-mandrin. Ajustez l’orientation du sectionnement à l’aide des vis d’orientation. Ajustez l’angle du couteau à l’aide de l’inclinaison du couteau. Sélectionnez l’épaisseur des sections. Les échantillons peuvent être sectionnés plus épais que les sections de résine habituelles. Des sections comprises entre 5 et 20 μm sont obtenues avec succès, les sections plus épaisses étant plus faciles à réaliser (moins de curling et moins de dommages aux structures).
- Avancez la face du bloc gelé vers la lame du couteau. N’essayez pas de sectionner sans le faire, sinon le bloc peut être détaché du mandrin et endommagé. Si nécessaire, coupez le bloc en utilisant une épaisseur supérieure (10 μm et plus).
- Lorsque vous approchez d’une section appropriée, placez la plaque antiroulis (c’est-à-dire une plaque transparente qui retiendra la section) au-dessus du couteau et effectuez un mouvement ferme à sens unique avec le volant, de sorte que la section soit faite immédiatement. Des problèmes de curling peuvent être causés si la plaque anti-roulis doit être ajustée (elle est généralement réglable en référence à la lame) ou s’il y a des débris dans la lame. Nettoyez constamment la lame avec un pinceau pour enlever les débris.
- Utilisez des lames spéciales pour que les sections se fixent facilement, comme des lames silanisées (commerciales ou préparées avec 2% d’aminoalkylsilane dans de l’acétone 21), ou des lames préparées avec 500 μg/mL de poly-L-lysine dans de l’eau distillée 21 ou 0,2% de gélatine (voir détails21). Maintenez les lames à température ambiante.
- Pour coller la section à une glissière, soulevez la plaque antiroulis et faites rapidement toucher la section par la glissière. Comme la glissière est à température ambiante, la section fond immédiatement et adhère à la glissière. Faites attention à tourner la face traitée de la glissière vers la section, qui peut rester au-dessus du couteau ou sur la face interne de la plaque antiroulis. Pour éviter l’enroulement des sections, effectuez cette étape rapidement dès que la plaque est soulevée et veillez à ne pas contorsionner la section.
- Laissez la lame à l’extérieur de la chambre du cryostat (à température ambiante) si de nouvelles sections doivent y être ajoutées. Après avoir collé toutes les sections souhaitées à la lame, conservez-la à l’intérieur de la chambre du cryostat ou au congélateur (-20 °C ou moins). N’exposez pas les lames à l’humidité. Conservez-les dans une boîte à diapositives et n’oubliez pas d’identifier les diapositives avec un crayon.
REMARQUE: Les lames et les blocs de PTO peuvent être stockés à -20 °C, mais pas trop longtemps. Pour obtenir de meilleurs résultats, utilisez les diapositives et les blocs en quelques jours.
6. Coloration des sections de plantes et des endophytes pour la microscopie optique
REMARQUE: De nombreux types de taches peuvent être utilisés pour les sections de plantes. Il est difficile de colorer différemment les champignons endophytes et les tissus végétaux. Bien qu’il ne s’agisse pas d’une procédure de coloration, une méthode de marquage des structures de champignons est présentée à la section 7 (fluorescence avec un conjugué agglutinine de germe de blé). Les sections à main levée (expliquées à la section 3), les sections de résine (section 4) et les cryosections (section 5) peuvent être colorées, bien que les colorations à base de phénol et d’alcool soient difficiles pour ces échantillons, car la résine GMA et l’OCT perdent leur adhérence à la lame dans ces cas.
- Utilisez une ou combinez les méthodes de coloration habituelles suivantes pour les échantillons de plantes.
- Toluidine bleu O22,23, une méthode largement appliquée pour la coloration générale des sections de plantes. Préparer une solution de 0,05% de bleu de toluidine O dans 0,1 M de phosphate (pH 6,8) ou 0,09 M de tampon de citrate (pH 4,5-4,8), selon les espèces et les types de tissus. Incuber les sections de résine GMA pendant 2 à 10 minutes à l’aide d’un pot de coloration à lames ou en plaçant quelques gouttes au-dessus des sections si peu de lames sont tachées. Laver soigneusement avec de l’eau distillée ou un tampon après l’incubation et monter les lames avec de l’eau ou les sécher sur une plaque chauffante pour produire des lames permanentes comme décrit à l’étape 6.3.
- Le réactif de Lugol2 indique la présence d’amidon. Préparer une solution d’iode à 5 % (I2) et d’iodure de potassium (KI) à 10 % dans de l’eau distillée. Colorer les sections pendant 2 min, en ajoutant quelques gouttes au-dessus de la glissière, puis laver à l’eau distillée. Ce test histochimique est généralement appliqué sur des lames temporaires.
- Soudan III, IV et noir B24,25 coloration pour différents lipides. Préparer une solution à 0,3% de Soudan (III, IV ou noir B) dans de l’éthanol à 70%, la réchauffer jusqu’à ébullition et laisser refroidir. Utilisez le surnageant, filtrez-le et incuber les sections pendant 15 à 30 minutes dans une boîte de Petri fermée. Lavez soigneusement les sections avec de l’éthanol à 70% et de l’eau distillée. Montez les toboggans avec de l’eau (généralement appliqué uniquement aux toboggans temporaires).
REMARQUE: Comme le Soudan est un colorant à base d’alcool, il convient mieux aux sections à main levée. Effectuez soigneusement la coloration de la section de résine GMA car ils se détachent généralement de la lame.
- Pour les lames temporaires, montez les sections dans de l’eau ou de la glycérine et observez par la suite. Scellez le couvercle avec du vernis à ongles pour les conserver un peu plus longtemps.
- Pour les glissières permanentes, montez les sections avec des résines synthétiques (p. ex., support de montage rapide, voir Tableau des matériaux). Égoutter quelques gouttes du support de montage (il peut déborder du couvercle), placez le couvercle avec précaution pour éviter les bulles et utilisez des pinces à linge pour presser la glissière contre la lamelle de couverture jusqu’à ce qu’elle soit complètement sèche. Enlevez l’excès de support de montage séché avec une lame de rasoir.
7. Application d’un fluorochrome conjugué à l’agglutinine de germe de blé en fluorescence et en microscopie confocale
NOTE: Cette méthode peut être appliquée aux sections à main levée (expliqué dans la section 3), aux sections de résine (section 4) et aux cryosections (section 5). Les cryosections peuvent convenir à des fins de microscopie confocale, car des échantillons plus épais peuvent être fournis par rapport aux sections de résine, mais pas aussi épais que les sections à main levée. Un fluorochrome conjugué à l’agglutinine de germe de blé (WGA, voir Tableau des matériaux) est appliqué à l’imagerie fongique en microscopie à fluorescence26. Un microscope confocal n’est pas essentiel, bien qu’il puisse fournir des images tridimensionnelles claires des structures végétales27.
- Préparer une solution de 0,2 mg/mL de conjugué WGA-fluorochrome dans 0,1 M PB28 (pH 7,2, vérifier étape 5.1.1 et Tableau 3). Préparer une solution de 1% de Calcofluor White dans 0,1 M PB (pH 7,2). Préparez de petites quantités de ces solutions, car les sections sont directement incubées avec elles.
- Incuber les sections dans les lames de verre pendant 30 min dans la solution conjuguée WGA-fluorochrome29, en utilisant suffisamment de volume pour couvrir les sections, puis laver dans 0,1 M PB.
- Incuber les sections dans la solution de calcofluor, en utilisant suffisamment de volume comme support de montage. La solution peut être maintenue pendant la période d’observation.
- Placez des lames de couverture sur les lames et observez au microscope confocal ou au microscope à fluorescence à l’aide des filtres suivants: TC/GFP (excitation: 470-440, émission: 525-550, pour WGA-fluorochrome dans le tableau des matériaux - paroi cellulaire fongique fluorescente verte sous ce filtre29) et DAPI (excitation: 358, émission: 463, pour Calcofluor White)30.
NOTE: Des images tridimensionnelles peuvent être obtenues à l’aide de la fonction Z-series dans le microscope confocal27.
8. Microscopie électronique à balayage des organes végétaux
- Après avoir fixé les échantillons, effectué la déshydratation et stocké dans de l’éthanol à 70 % (section 1), une possibilité consiste à couper des échantillons pour exposer toute surface souhaitée pour l’analyse SEM, si nécessaire (p. ex. tissus internes, structure des ovaires). Utilisez une lame de rasoir tranchante et neuve et faites des coupes avec un mouvement unidirectionnel, en évitant une apparence endommagée de ces zones dans SEM. Si nécessaire, utilisez un stéréomicroscope pour sélectionner les échantillons et considérez la zone des souches métalliques pour déterminer la taille des échantillons.
- Autres échantillons déshydratés pour le MEB dans une série éthanolique : 80 %, 96 % et 2x dans de l’alcool éthylique absolu (≥99,8 %). Conserver les échantillons petits et délicats pendant 30 minutes à chaque concentration et les échantillons plus grands et plus denses pendant 1 h.
- Pliez de petites enveloppes en utilisant du papier de soie pour organiser les échantillons pour les prochaines étapes, les échantillons plus grands peuvent être manipulés sans enveloppe. Identifiez les enveloppes avec un crayon en écrivant une lettre ou un chiffre, et tenez un journal de tous les échantillons de chacune. Conservez les échantillons dans de l’éthanol absolu, mais pas longtemps, et passez à l’étape 8.4 dès que possible.
- Procéder au séchage au point critique (CP). Faire fonctionner un sécheur CP conformément aux procédures d’utilisation normalisées. Placer les échantillons dans de l’éthanol absolu (fluide intermédiaire) dans une chambre de pression. Au point critique de CO2 (31 °C, 7,3 x 106 Pa), le fluide intermédiaire se dissout dans le fluide de transition (dioxyde de carbone liquide) et les échantillons sont séchés31.
- Après séchage CP, conserver les échantillons dès que possible dans un récipient de dessiccation, par exemple une fiole scellée contenant du gel de silice. L’humidité atmosphérique peut détruire les échantillons s’ils sont réabsorbés31.
- Utilisez des bouts métalliques pour monter les échantillons. Avant de monter, mettez des gants pour manipuler les talons, plongez-les dans l’acétone pendant 5 minutes pour éliminer toute graisse et laissez-les sécher. Utilisez un ruban adhésif conducteur à double face pour fixer les échantillons sur la tige et un stéréomicroscope pour aider à positionner les échantillons, en gardant à l’esprit que la vue d’en haut est la seule perspective possible dans les images SEM.
- Manipulez les échantillons avec une pince à épiler à pointe fine, en faisant attention car la partie de l’échantillon touchée par la pince à épiler est généralement endommagée, alors essayez de toucher les parties éloignées des zones d’intérêt (par exemple, les zones en contact avec la bande). Maintenir les souches avec les échantillons dans une boîte de Petri scellée avec du gel de silice. Passez à l’étape 8.8 dès que possible.
- Utilisez une pulvérisation pour déposer une couche de métal, généralement de l’or ou du platine, à la surface des échantillons dans une atmosphère à basse pression d’un gaz inerte, souvent l’argon31. Suivez les procédures opérationnelles normalisées lorsque vous utilisez un manteau de pulvérisation. L’épaisseur du revêtement dépend de la topographie des échantillons, généralement entre 15 et 40 nm32.
- Conservez les souches enrobées dans une boîte de Petri scellée avec du gel de silice, et à condition que le gel de silice retienne l’humidité, les échantillons peuvent être stockés de cette façon pendant des semaines. Utilisez un microscope électronique à balayage pour analyser les échantillons. Les échantillons sous vide sont frappés par un faisceau d’électrons, et l’émission de signaux provenant d’une telle interaction est interprétée comme des images31. Pour plus de détails sur l’utilisation d’un microscope électronique à balayage, lire Jeffree et Read (1991)31 et Bozzola et Russell (1999)32.
- Pour réutiliser les tiges, tirez le ruban adhésif et frottez-les avec de la laine métallique. Lavez-les à l’eau du robinet, immergez-les dans de l’éthanol absolu et séchez-les adéquatement, en empêchant l’oxydation du métal constitutif.
9. Microscopie électronique à transmission
- Préfixer les échantillons avec un tampon glutaraldéhyde-cacodylate comme expliqué aux étapes 1.7 et 1.10. Après 12-24 h de préfixation, laver les échantillons 3x dans un tampon cacodylate 0,2 M (pH 7,25) pendant 10 min. Effectuer la postfixation avec du tétroxyde d’osmium à 1% (OsO4) dans un tampon cacodylate 0,2 M, pendant 12 h dans l’obscurité, à température ambiante. Laver 3x à l’eau distillée pendant 5 min.
ATTENTION : Le cacodylate et le tétroxyde d’osmium sont très toxiques et ne doivent pas être inhalés. Utilisez-les dans les hottes, en suivant les fiches de données de sécurité respectives. - Déshydrater les échantillons avec de l’éthanol à 30%, 50%, 70% et 96%, 2x à chaque concentration, pendant 10 min. Ensuite, déshydrater 3x dans de l’alcool absolu, pendant 15 min à chaque fois.
- Infiltrer les échantillons dans des résines acryliques hydrophiles (voir Tableau des matériaux), une fois avec 1:1 résine + éthanol absolu et 3x avec de la résine pure pendant 8-12 h chacun. Procéder à la polymérisation dans des capsules de gélatine à 60 °C jusqu’à solidification complète (12 h maximum17).
- Évaluer de près l’orientation des échantillons à l’intérieur du bloc de résine; Coupez la partie supérieure du bloc, avec une lame de rasoir, en formant une forme pyramidale qui concentre l’échantillon dans la zone de sectionnement. Obtenir des sections semi-minces (250-500 nm)33 dans un ultramicrotome avec un couteau diamant et placer sur des lames de verre dans quelques gouttes d’eau.
- Conserver les lames sur une plaque chauffante à 60 °C. Colorer les sections avec du bleu de toluidine O comme à l’étape 6.1.1 et laisser sécher complètement. Lavez soigneusement à l’eau du robinet. Évaluez la section obtenue en dessinant quatre quadrants et en sélectionnant le quadrant le plus approprié pour l’analyse.
- Coupez le bloc de sorte que la forme pyramidale concentre le quadrant choisi sur la face supérieure du bloc. Produire des sections ultraminces (50-100 nm)33,34. L’épaisseur est évaluée en fonction de la couleur d’interférence des sections: les sections d’environ 70 nm apparaissent argent-or, environ 100 nm apparaissent en or et avec environ 200 nm apparaissent bleues34.
- Recueillir les sections ultraminces de l’eau à l’aide de grilles en cuivre et procéder à la méthode de coloration de contraste avec de l’acétate d’uranyle et du citrate de plomb, comme décrit ci-dessous.
- Préparer une solution de citrate de plomb (tableau 5) et congeler la solution finale dans des tubes à microcentrifugation avec 1 mL de solution dans chacun, en ne décongelant que juste avant utilisation.
- Préparer une solution d’acétate d’uranyle : dissoudre 0,625 g d’acétate d’uranyle [UO 2(CH3COO)2] dans 25 mL d’eau distillée fraîchement bouillie et refroidie. Conserver dans une fiole foncée au congélateur.
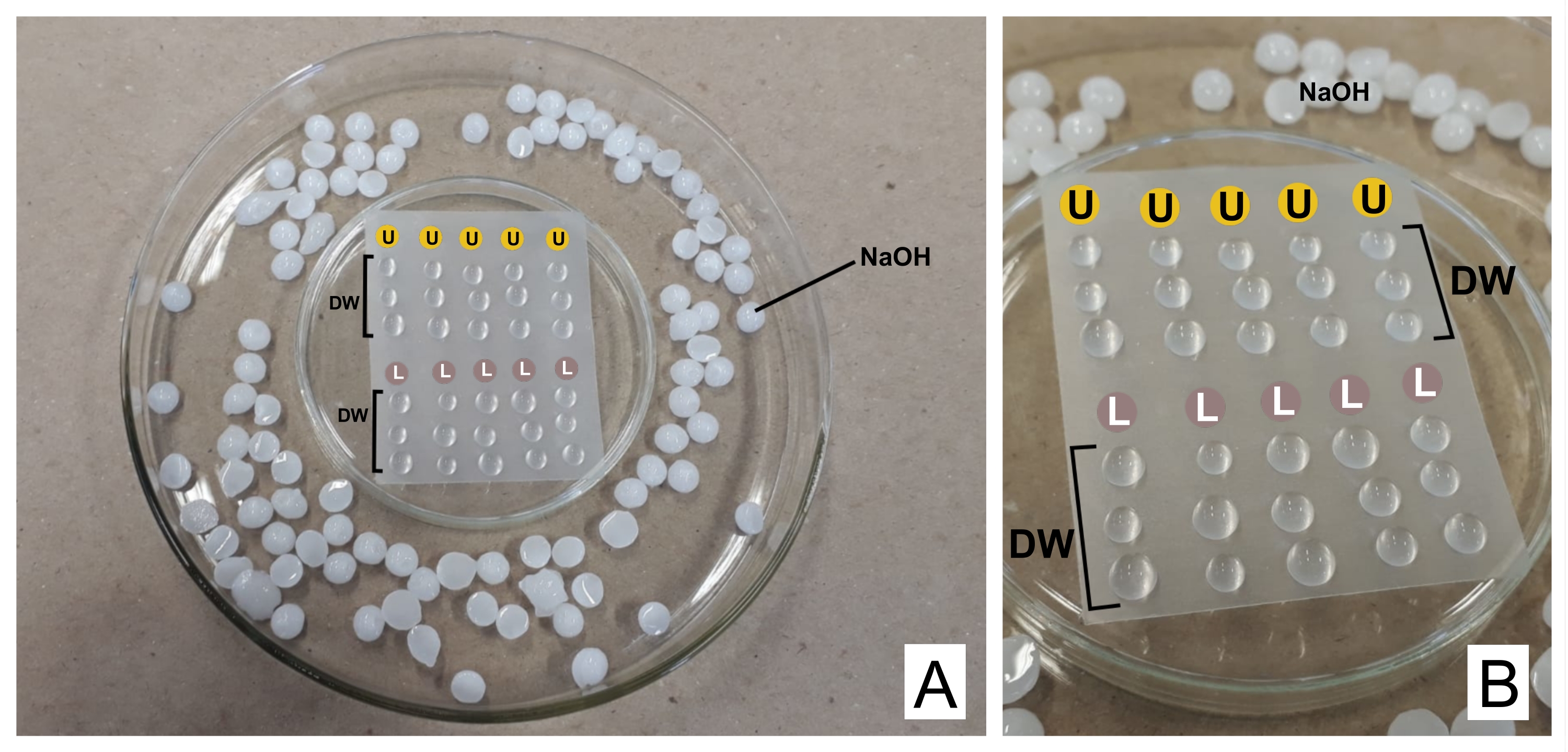
ATTENTION : Le nitrate de plomb est toxique en cas d’ingestion. 1 N NaOH est très corrosif. L’acétate d’uranyle est radioactif et toxique. Il ne doit pas être ingéré, inhalé ou entrer en contact avec la peau. - Lors de la coloration, placez les deux réactifs préparés dans des seringues séparées de 3 ml avec des unités filtrantes (pores de 0,22 μm, voir le tableau des matériaux). Préparer une boîte de Petri retournée avec un film thermoplastique d’étanchéité dessus (voir le tableau des matériaux) et à l’intérieur d’une boîte plus large, avec des granulés de NaOH sur les bords comme piège pour le CO 2 32 (voir la figure 2).
- Jeter la première goutte et, sur le film, placer une goutte d’acétate d’uranyle et trois gouttes d’eau distillée pour chaque grille tachée. Faites de même avec le citrate de plomb et ajoutez trois gouttes supplémentaires d’eau distillée.
- Incuber la grille (avec le côté opaque vers le bas, où se trouvent les sections) dans de l’uranyle pendant 30 min (temps variable). Lavez 3x dans les gouttes d’eau distillée, en séchant la grille à chaque fois doucement avec du papier filtre du côté brillant. Répétez avec la goutte de citrate de plomb (30 min) et lavez-la.
- Après au moins 4 h, analysez les grilles au microscope électronique à transmission. Dans ce microscope, un faisceau d’électrons traverse les sections sous vide et l’image est projetée sur un écran fluorescent. Pour plus de détails sur l’utilisation d’un microscope électronique à transmission, lire Bozzola et Russell (1999)32.
| solution de citrate de plomb (pour la coloration de contraste TEM) | |
| Étape 1 | entourer un bécher de papier d’aluminium |
| Étape 2 | dissoudre 0,266 g de nitrate de plomb [Pb(NO3)2] dans 6 mL d’eau distillée récemment bouillie et refroidie |
| Étape 3 | agiter pendant 2 min |
| Étape 4 | ajouter 0,352 g de citrate trisodique [Na3(C6H5O7).2H2O] (la solution doit acquérir un aspect laiteux) |
| Étape 5 | agiter pendant 15 min, sceller le bécher avec du papier d’aluminium et transférer la solution dans un bécher de 10 mL |
| Étape 6 | ajouter 1,6 mL de NaOH 1N et 2,4 mL d’eau distillée (la solution doit être translucide) |
| Étape 7 | si nécessaire, ajuster le pH proche de 12 |
Tableau 5 : Recette de solution de citrate de plomb.

Figure 2 : Schéma de coloration de contraste avec des solutions de citrate de plomb et d’acétate d’uranyle. (A) Préparez les boîtes de Petri, l’une retournée (au centre) avec un film thermoplastique afin que des gouttes puissent être placées au-dessus, à l’intérieur d’une plus large. Les granulés de NaOH sont des endroits autour du plat central. (B) Les gouttes d’acétate d’uranyle sont placées dans les cercles avec la lettre U, et les gouttes de citrate de plomb dans les cercles marquées L. DW indique des gouttes d’eau distillée. Les grilles sont colorées séquentiellement dans la colonne, de sorte que cinq grilles peuvent être colorées simultanément comme représenté. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
10. Germination symbiotique des graines d’orchidées
- S’assurer que les solutions et tous les matériaux utilisés dans la germination symbiotique des graines sont stériles pour éviter toute contamination. Commencez par les autoclaver pendant 20 min à 121 °C. Les étapes de germination symbiotique sont résumées à la figure 3.
- Désinfecter superficiellement les fruits et les graines en les immergeant dans une solution d’hypochlorite de sodium contenant 2% de chlore actif pendant 10-15 min pour les fruits et 7-10 min pour les graines, compte tenu de la rigidité et de l’épaisseur du tégument9. Les graines minces et fragiles peuvent être immergées dans une solution d’hypochlorite de sodium dilué 1:1. Ensuite, laver 3x dans de l’eau distillée autoclavée pour éliminer la solution d’hypochlorite.
- Récupérez les graines en les filtrant dans un tissu sérigraphique et utilisez les graines pour procéder aux tests de germination (de préférence). Si nécessaire, les conserver dans des enveloppes en papier filtre à l’intérieur de flacons en verre avec du gel de silice, à 4 °C, fermer hermétiquement les flacons et les sceller avec un film alimentaire. Transférer quelques gouttes d’eau du dernier lavage sur gélose au dextrose de pomme de terre (PDA, 39 g/L) pour évaluer l’efficacité du processus de lavage.
- Avant de semer des graines dans le milieu de culture, évaluer leur viabilité au moyen du test au tétrazolium (facultatif) décrit ci-dessous35.
- Incuber environ 10 mg de graines dans un tube microcentrifuge contenant 1 mL de saccharose à 10 % dans de l’eau distillée, pendant 24 h à température ambiante (environ 25 °C), à la lumière.
- Retirer la solution de saccharose à l’aide d’une micropipette et ajouter 1 mL de solution de tétrazolium à 1 % (chlorure de triphényltétrazolium) dans de l’eau distillée. Incuber à 40 °C dans un thermobloc pendant 24 h dans l’obscurité.
- Retirer la solution de tétrazolium à l’aide d’une micropipette et laver les graines à l’eau distillée 2x ou jusqu’à élimination de la solution. Retirez tous les liquides. Si nécessaire, les graines peuvent être conservées au congélateur jusqu’à une semaine (comme à l’étape 10.3) avant d’être analysées.
- Remettez les graines en suspension dans de l’eau distillée et analysez-les au microscope optique. Les graines viables acquièrent une couleur rouge clair à foncé, tandis que les graines non viables conservent leur couleur naturelle.
- Effectuerle protocole 9 adapté suivant pour la germination symbiotique des graines d’orchidées.
- Incuber les graines sur des disques de papier filtre autoclavés (1-2 cm de diamètre) placés dans des boîtes de Petri avec un milieu de culture en gélose à l’avoine (OMA) (2,5 g/L de flocons d’avoine et 7 g/L d’agar, pH 6).
- Au centre de la boîte de Pétri, inoculer un fragment de milieu de culture (environ 1 cm2) contenant du mycélium du champignon isolé choisi pour la procédure de germination. Scellez les boîtes de Petri avec un film alimentaire et incuberez-les dans l’obscurité à environ 25 ° C ou à température ambiante, car il est plus adéquat pour la croissance fongique.
- Préparez des plats avec des graines et sans inoculation fongique, comme témoin négatif pour le test de germination.
- Analyser les résultats de germination chaque semaine, en recueillant des données quantitatives et qualitatives et en photographiant les protocormes et les semis. L’observation des graines et des protocormes doit être effectuée à l’aide d’un stéréomicroscope pour une évaluation plus précise de la germination. Utilisez une source de lumière venant d’en bas, car elle permet un plus grand contraste, ce qui permet de distinguer plus facilement le mycélium fongique des protocormes.
- Prélever des échantillons à différents stades de développement et les corriger pour les analyses anatomiques (section 1). Appliquer toutes les analyses d’images décrites précédemment pour étudier les endophytes fongiques dans les graines, les protocormes et les plantules pendant la germination (microscopie optique - sections 4, 5 et 6; confocale et fluorescence - section 7; SEM et TEM - articles 8 et 9).
- Générer des résultats quantitatifs suivant la classification des étapes selon le tableau 6. Les étapes décrivent le développement habituel des graines d’orchidées mycohétérotrophes. Recueillir des données chaque semaine et les tableaux avec les dates initiales de chaque étape observée.
- De plus, recueillir des données quantitatives estimant le pourcentage et le taux de germination. Comptez au moins 100 graines ou définissez des champs de comptage35. Délimiter trois champs de comptage ou plus par boîte de Pétri, composés de régions fixes avec une superficie normalisée, et évaluer chaque semaine. Calculez les données collectées selon l’équation de l’indice de croissance (IG) :

où N 0 est le nombre de semences comptées au stade0 , N 1 se réfère au stade1 , et il suit jusqu’au stade 6 (enregistré comme N6)36.

Figure 3 : Résumé schématique de la méthodologie de germination symbiotique des semences. Les schémas fournissent des indications sur les étapes détaillées du protocole. Abréviations : OMA = gélose à l’avoine, PDA = gélose au dextrose de pomme de terre. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Stade de germination | Description |
| 0 | Pas de germination |
| 1 | Gonflement de l’embryon |
| 2 | Testa rupture |
| 3 | Les poils absorbants se développent |
| 4 | La projection des tiges se développe |
| 5 | Développement d’écailles protectrices (bractées) |
| 6 | Les premières racines se développent |
Tableau 6 : Description des stades de développement du protocorme appliqués aux analyses périodiques des essais de germination. Modifié à partir des stades décrits dans Otero et al.36.
Résultats
Suivre les étapes essentielles de la fixation des tissus végétaux donne des structures cellulaires aussi proches que possible de l’état vivant, compte tenu de la morphologie, du volume et de l’organisation spatiale des composants cellulaires et des tissus16. Observer de tels traits dans les échantillons après fixation chimique (Figure 4). La figure 4C-F représente des échantillons correctement...
Discussion
Les analyses d’images en anatomie et morphologie des plantes ont un potentiel important pour atteindre les objectifs et aider à comprendre les relations entre les plantes mycohétérotrophes et leurs endophytes fongiques indispensables, comme le démontrent les études sur les organes souterrains6,40, les analyses structurelles de la germination symbiotique des graines39 et les structures aériennes et reproductrices 41
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs remercient le financement de FAEPEX et FAPESP (2015/26479-6). MPP remercie Capes pour sa bourse de maîtrise (processus 88887.600591/2021-00) et CNPq. JLSM remercie CNPq pour les subventions de productivité (303664/2020-7). Les auteurs remercient également l’accès aux équipements et à l’assistance fournis par le LME (Laboratoire de Microscopie Electronique - IB/Unicamp), l’INFABiC (Institut National des Sciences et Technologies de la Photonique Appliquée à la Biologie Cellulaire - Unicamp) et le LaBiVasc (Laboratoire de Biologie Vasculaire - DBEF/IB/Unicamp) ; LAMEB (UFSC) et Eliana de Medeiros Oliveira (UFSC) pour leurs contributions au protocole de cryoprotection ; LME pour les contributions au protocole TEM.
matériels
| Name | Company | Catalog Number | Comments |
| Acetone | Sigma-Aldrich | 179124 | (for SEM stubs mounting) |
| Agar-agar (AA) | Sigma-Aldrich | A1296 | (for seeds germination tests) |
| Calcofluor White Stain | Sigma-Aldrich | 18909 | fluorescent dye (detects cellulose) |
| Citrate Buffer Solution, 0.09M pH 4.8 | Sigma-Aldrich | C2488 | (for toluidine blue O staining) |
| Conductive Double-Sided Carbon Tape | Fisher Scientific | 50-285-81 | (for SEM) |
| Confocal Microscope | Zeiss | (any model) | |
| Copper Grids | Sigma-Aldrich | G4776 | (for TEM) |
| Critical-point dryer | Balzers | (any model) | |
| Cryostat | Leica Biosystems | (any model) | |
| Dissecting microscope | Leica Biosystems | (= stereomicroscope, any model) | |
| Entellan | Sigma-Aldrich | 107960 | rapid mounting medium for microscopy |
| Ethyl alcohol, pure (≥99.5%) | Sigma-Aldrich | 459836 | (= ethanol, for dehydration processes) |
| Formaldehyde solution, 37% | Sigma-Aldrich | 252549 | (for NBF solution preparation) |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128 | histological tissue fixative |
| Gelatin capsules for TEM | Fisher Scientific | 50-248-71 | (for resin polymerisation in TEM) |
| Gelatin solution, 2% in H2O | Sigma-Aldrich | G1393 | (dilute for slides preparation - OCT adherence) |
| Glutaraldehyde solution, 25% | Sigma-Aldrich | G6257 | (for Karnovsky’s solution preparation) |
| HistoResin | Leica Biosystems | 14702231731 | glycol methacrylate (GMA) embedding kit |
| Iodine | Sigma-Aldrich | 207772 | (for Lugol solution preparation) |
| Lead(II) nitrate | Sigma-Aldrich | 228621 | Pb(NO3)2 (for TEM contrast staining) |
| Light Microscope | Olympus | (any model) | |
| LR White acrylic resin | Sigma-Aldrich | L9774 | hydrophilic acrylic resin for TEM |
| Lugol solution | Sigma-Aldrich | 62650 | (for staining) |
| Metal stubs for specimen mounts | Rave Scientific | (for SEM, different models) | |
| Microtome | Leica Biosystems | manual rotary microtome or other model | |
| Oatmeal agar (OMA) | Millipore | O3506 | (for seeds germination tests) |
| OCT Compound, Tissue-Tek | Sakura Finetek USA | 4583 | embedding medium for frozen tissues |
| Osmium tetroxide | Sigma-Aldrich | 201030 | OsO4 (for TEM postfixation) |
| Parafilm M | Sigma-Aldrich | P7793 | sealing thermoplastic film |
| Paraformaldehyde | Sigma-Aldrich | 158127 | (for Karnovsky’s solution preparation) |
| Poly-L-lysine solution, 0.1% in H2O | Sigma-Aldrich | P8920 | (for slides preparation - OCT adherence) |
| Poly-Prep Slides | Sigma-Aldrich | P0425 | poly-L-lysine coated glass slides |
| Polyethylene Molding Cup Trays | Polysciences | 17177A-3 | (6x8x5 mm, for embbeding samples in GMA resin) |
| Polyethylene Molding Cup Trays | Polysciences | 17177C-3 | (13x19x5 mm, for embbeding samples in GMA resin) |
| Potassium iodide | Sigma-Aldrich | 221945 | (for Lugol solution preparation) |
| Potato Dextrose Agar (PDA) | Millipore | 70139 | (for seeds germination tests) |
| Scanning Electron Microscope | Jeol | (any model) | |
| Silane [(3-Aminopropyl)triethoxysilane] | Sigma-Aldrich | A3648 | (for slides preparation - OCT adherence) |
| Silane-Prep Slides | Sigma-Aldrich | S4651 | glass slides coated with silane |
| Silica gel orange, granular | Supelco | 10087 | (for dessicating processes) |
| Sodium cacodylate trihydrate | Sigma-Aldrich | C0250 | (for glutaraldehyde-sodium cacodylate buffer) |
| Sodium hydroxide | Sigma-Aldrich | S5881 | NaOH (for Karnovsky’s solution preparation and TEM contrast staining) |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044 | NaClO (for seeds surface disinfection) |
| Sodium phosphate dibasic, anhydrous | Sigma-Aldrich | 71640 | Na2HPO4 (for NBF solution and PB preparation) |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638 | NaH2PO4·H2O (for NBF and PB) |
| Sputter coater | Balzers | (any model) | |
| Sucrose | Sigma-Aldrich | S0389 | C12H22O11 (for cryoprotection and germination test) |
| Sudan III | Sigma-Aldrich | S4131 | (for staining) |
| Sudan IV | Sigma-Aldrich | 198102 | (for staining) |
| Sudan Black B | Sigma-Aldrich | 199664 | (for staining) |
| Syringe | (3 mL, any brand, for TEM contrast staining) | ||
| Syringe Filter Unit, Millex-GV 0.22 µm | Millipore | SLGV033R | PVDF, 33 mm, gamma sterilized (for TEM contrast staining) |
| Tek Bond Super Glue 793 | Tek Bond Saint-Gobain | 78072720018 | liquid cyanoacrylate adhesive, medium viscosity |
| Toluidine Blue O | Sigma-Aldrich | T3260 | (for staining) |
| Transmission Electron Microscope | Jeol | (any model) | |
| Triphenyltetrazolium chloride | Sigma-Aldrich | T8877 | (for the tetrazolium test in seeds germination) |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804 | Na3(C6H5O7)·2H2O (for TEM contrast staining) |
| Ultramicrotome | Leica Biosystems | (any model) | |
| Uranyl acetate | Fisher Scientific | 18-607-645 | UO2(CH3COO)2 (for TEM contrast staining) |
| Vacuum pump | (any model) | ||
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | TermoFisher Scientific | W11261 | fluorescent dye-conjugated lectin (detects sialic acid and N-acetylglucosaminyl residues) |
Références
- Evert, R. F. . Esau’s Plant Anatomy: Meristems, Cells, and Tissues of the Plant Body: Their Structure, Function, and Development. , (2006).
- Yeung, E. C. T., Stasolla, C., Sumner, M. J., Huang, B. Q. . Plant Microtechniques and Protocols. , (2015).
- Sokoloff, D. D., Jura-Morawiec, J., Zoric, L., Fay, M. F. Plant anatomy: at the heart of modern botany. Botanical Journal of the Linnean Society. 195 (3), 249-253 (2021).
- Leake, J. R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytologist. 127 (2), 171-216 (1994).
- Bidartondo, M. I. The evolutionary ecology of myco-heterotrophy. New Phytologist. 167 (2), 335-352 (2005).
- Imhof, S., Massicotte, H. B., Melville, L. H., Peterson, R. L. Subterranean morphology and mycorrhizal structures. Mycoheterotrophy. , 157-214 (2013).
- Rasmussen, H. N., Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos. 118 (3), 334-345 (2009).
- Rasmussen, H. N., Dixon, K. W., Jersáková, J., Těšitelová, T. Germination and seedling establishment in orchids: a complex of requirements. Annals of Botany. 116 (3), 391-402 (2015).
- Zettler, L. W. Terrestrial orchid conservation by symbiotic seed germination: techniques and perspectives. Selbyana. 18 (2), 188-194 (1997).
- Stewart, S. L., Kane, M. E. Symbiotic seed germination and evidence for in vitro mycobiont specificity in Spiranthes brevilabris (Orchidaceae) and its implications for species-level conservation. In Vitro Cellular & Developmental Biology - Plant. 43 (3), 178-186 (2007).
- Zhao, D. -. K., et al. Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Frontiers in Plant Science. 12, 701152 (2021).
- Selosse, M. A., Roy, M. Green plants that feed on fungi: facts and questions about mixotrophy. Trends in Plant Science. 14 (2), 64-70 (2009).
- Merckx, V. S. F. T., Mennes, C. B., Peay, K. G., Geml, J. Evolution and diversification. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 215-244 (2013).
- Boon, M. E., Drijver, J. Routine Cytological Staining Techniques: Theoretical Background and Practice. Macmillan International Higher Education. , (1986).
- Karnovsky, M. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. Journal of Cell Biology. 27 (2), 137-138 (1964).
- Hayat, M. . Fixation for Electron Microscopy. , (1981).
- Roland, J. C., Vian, B. General preparation and staining of thin sections. Electron Microscopy of Plant Cells. 1, 675 (1991).
- Gerrits, P. O., Horobin, R. W. Glycol methacrylate embedding for light microscopy: basic principles and trouble-shooting. Journal of Histotechnology. 19 (4), 297-311 (1996).
- Zhang, Z., Niu, L., Chen, X., Xu, X., Ru, Z. Improvement of plant cryosection. Frontiers in Biology. 7 (4), 374-377 (2012).
- BeneŠ, K. On the media improving freeze-sectioning of plant material. Biologia Plantarum. 15 (1), 50-56 (1973).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. Cold Spring Harbor Protocols. 2008 (5), (2008).
- Sakai, W. S. Simple method for differential staining of paraffin embedded plant material using toluidine blue O. Stain Technology. 48 (5), 247-249 (1973).
- O’Brien, T., Feder, N., McCully, M. E. Polychromatic staining of plant cell walls by toluidine blue O. Protoplasma. 59 (2), 368-373 (1964).
- Ventrella, M. C., Almeida, A. L., Nery, L. A., Coelho, V. P. d. e. M. Métodos Histoquímicos Aplicados às Sementes. Universidade Federal de Viçosa. , (2013).
- Pearse, A. G. E. . Histochemistry, Theoretical and Applied. , (1960).
- Andrade-Linares, D. R., Franken, P. Fungal endophytes in plant roots: taxonomy, colonization patterns, and functions. Symbiotic Endophytes. , 311-334 (2013).
- Wymer, C. L., Beven, A. F., Boudonck, K., Lloyd, C. W. Confocal microscopy of plant cells. Confocal Microscopy Methods and Protocols. , 103-130 (1999).
- Marques, J. P. R., Soares, M. K. M. Manual de Técnicas Aplicadas à Histopatologia Vegetal. FEALQ. , (2021).
- Navarro, B. L., Marques, J. P. R., Appezzato-da-Glória, B., Spósito, M. B. Histopathology of Phakopsora euvitis on Vitis vinifera. European Journal of Plant Pathology. 154 (4), 1185-1193 (2019).
- Marques, J. P. R., et al. Sugarcane cell wall-associated defense responses to infection by Sporisorium scitamineum. Frontiers in Plant Science. 9, 698 (2018).
- Jeffree, C. E., Read, N. D. Ambient-and low-temperature scanning electron microscopy. Electron Microscopy of Plant Cells. , 313-413 (1991).
- Bozzola, J. J., Russell, L. D. . Electron Microscopy: Principles and Techniques for Biologists. , (1999).
- Murray, S. Basic transmission and scanning electron microscopy. Introduction to electron Microscopy for Biologists. , 3-18 (2008).
- . Glossary of TEM terms Available from: https://www.jeol.co.jp/en/words/emterms/ (2021)
- Seaton, P. T., et al. Orchid seed and pollen: a toolkit for long-term storage, viability assessment and conservation. Orchid Propagation: From Laboratories to Greenhouses—Methods and Protocols. , 71-98 (2018).
- Otero, J. T., Ackerman, J. D., Bayman, P. Differences in mycorrhizal preferences between two tropical orchids. Molecular Ecology. 13 (8), 2393-2404 (2004).
- Koch, R. A., et al. Marasmioid rhizomorphs in bird nests: Species diversity, functional specificity, and new species from the tropics. Mycologia. 112 (6), 1086-1103 (2020).
- Webster, J., Weber, R. . Introduction to Fungi. , (2007).
- Sisti, L. S., et al. The role of non-mycorrhizal fungi in germination of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. Frontiers in Plant Science. 10, 1589 (2019).
- Martos, F., et al. Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytologist. 184 (3), 668-681 (2009).
- Alves, M. F., et al. Reproductive development and genetic structure of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. BMC Plant Biology. 21 (1), 332 (2021).
- Merckx, V. S. F. T. Mycoheterotrophy: an introduction. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 1-17 (2013).
- Hall, J. L., Hawes, C. . Electron Microscopy of Plant Cells. , (1991).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.