
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Mikroskopische Techniken zur Interpretation der Pilzbesiedlung in mykoheterotrophen Pflanzengeweben und der symbiotischen Keimung von Samen
In diesem Artikel
Zusammenfassung
Dieses Protokoll zielt darauf ab, detaillierte Verfahren zum Sammeln, Fixieren und Pflegen mykoheterotropher Pflanzenproben bereitzustellen, wobei verschiedene Mikroskopietechniken wie Raster- und Transmissionselektronenmikroskopie, Licht-, Konfokal- und Fluoreszenzmikroskopie angewendet werden, um die Pilzbesiedlung in Pflanzengeweben und Samen zu untersuchen, die mit Mykorrhizapilzen gekeimt sind.
Zusammenfassung
Strukturelle Botanik ist eine unverzichtbare Perspektive, um die Ökologie, Physiologie, Entwicklung und Evolution von Pflanzen vollständig zu verstehen. Bei der Erforschung mykoheterotropher Pflanzen (d.h. Pflanzen, die Kohlenstoff aus Pilzen gewinnen) können bemerkenswerte Aspekte ihrer strukturellen Anpassungen, die Muster der Gewebebesiedlung durch Pilze und die Morphoanatomie unterirdischer Organe ihre Entwicklungsstrategien und ihre Beziehungen zu Hyphen, der Quelle von Nährstoffen, aufklären. Eine weitere wichtige Rolle symbiotischer Pilze hängt mit der Keimung von Orchideensamen zusammen; Alle Orchidaceae-Arten sind während der Keimung und des Keimlingsstadiums (anfängliche Mykoheterotrophie) mykoheterotroph, auch diejenigen, die in erwachsenen Stadien Photosynthese betreiben. Aufgrund des Mangels an Nahrungsreserven in Orchideensamen sind Pilzsymbionten unerlässlich, um Substrate bereitzustellen und die Keimung zu ermöglichen. Die Analyse der Keimungsstadien nach strukturellen Perspektiven kann auch wichtige Fragen zur Interaktion der Pilze mit den Samen beantworten. Verschiedene bildgebende Verfahren können angewendet werden, um Pilzendophyten in Pflanzengeweben zu enthüllen, wie in diesem Artikel vorgeschlagen. Freihändige und dünne Schnitte von Pflanzenorganen können gefärbt und anschließend lichtmikroskopisch beobachtet werden. Ein mit Weizenkeimagglutinin konjugiertes Fluorochrom kann auf die Pilze aufgetragen und mit Calcofluor White co-inkubiert werden, um Pflanzenzellwände in der konfokalen Mikroskopie hervorzuheben. Darüber hinaus werden die Methoden der Raster- und Transmissionselektronenmikroskopie für mykoheterotrophe Orchideen detailliert beschrieben und die Möglichkeiten der Anwendung solcher Protokolle in verwandten Pflanzen untersucht. Die symbiotische Keimung von Orchideensamen (d.h. in Gegenwart von Mykorrhizapilzen) wird im Protokoll ausführlich beschrieben, ebenso wie Möglichkeiten, die aus verschiedenen Keimungsstadien erhaltenen Strukturen für Analysen mit Licht-, Konfokal- und Elektronenmikroskopie vorzubereiten.
Einleitung
Strukturforschung in der Botanik, die Pflanzenmorphologie und Anatomie abdeckt, ist grundlegend für das Verständnis des gesamten Organismus1,2 und bietet unverzichtbare Perspektiven, um Wissen über die Ökologie, Physiologie, Entwicklung und Evolution von Pflanzen zu integrieren und dazu beizutragen3. Methoden in der Pflanzenmorphologie und Anatomie umfassen derzeit Protokolle, Geräte und Kenntnisse, die in jüngster Zeit sowie vor mehr als einem Jahrhundert entwickelt wurden2. Die kontinuierliche Durchführung und Anpassung klassischer Methoden (z.B. Lichtmikroskopie) sowie neuerer Techniken (z.B. konfokale Mikroskopie, Röntgenmikrotomographie) haben die gleiche wesentliche Grundlage: theoretisches Wissen, das die Entwicklung einer Methodik ermöglicht.
Das Hauptwerkzeug in der Pflanzenanatomie und -morphologie ist das Bild. Trotz des Missverständnisses, dass es sich bei solchen Analysen um einfache Beobachtungen handelt, die subjektiven Interpretationen Raum geben2, erfordert die Analyse und das Verständnis von Bildern in diesem Bereich Kenntnisse der angewandten Methoden (Ausrüstung, Art der Analyse, methodische Verfahren), der Zellbestandteile, der Histochemie und des Pflanzenkörpers (Gewebeorganisation und -funktion, Ontogenese, morphologische Anpassungen). Die Interpretation der mit einer Vielzahl von Methoden erhaltenen Bilder kann dazu führen, Form und Funktion zu korrelieren, die chemische Zusammensetzung einer Struktur zu entschlüsseln, die Beschreibung von Taxa zu bestätigen, Infektionen durch Phytopathogene zu verstehen und andere solche Bewertungen.
Bei der Erforschung mykoheterotropher (MH) Pflanzen (d.h. nicht-photosynthetischer Pflanzen, die Kohlenstoff aus Mykorrhizapilzen gewinnen4,5), können bemerkenswerte Aspekte ihrer strukturellen Anpassungen, die Muster der Gewebebesiedlung durch Pilze und die Morphoanatomie unterirdischer Organe ihre Entwicklungsstrategien und Beziehungen zu Hyphen, die die Quelle von Nährstoffen sind, aufklären. Die unterirdischen Organe von MH-Pflanzen zeigen in der Regel wichtige Anpassungen im Zusammenhang mit ihrer Assoziation mit Bodenpilzen, daher ist es wichtig, diese anatomischen und morphologischen Untersuchungen durchzuführen6. Die Luftorgane von MH-Arten sollten nicht ignoriert werden, da in diesen Geweben auch Endophyten vorhanden sein können, auch wenn es sich nicht um Mykorrhizapilze handelt (persönliche Beobachtungen, noch nicht veröffentlicht).
Neben der gut etablierten Wesentlichkeit der Assoziation von Mykorrhizapilzen mit MH-Arten während ihres gesamten Lebenszyklus7 hat jede Orchideenart, auch die autotrophen, ein initiales obligates mykoheterotrophes Stadium in natürlichen Umgebungen. Es tritt auf, weil der Embryo der Orchideen undifferenziert ist und kein Endosperm oder Keimblätter aufweist, so dass er ohne die Ernährungsunterstützung von Pilzpartnern nicht in der Lage ist, sich in natürlichen Umgebungen zu entwickeln und zu etablieren 4,8. In Anbetracht dessen können symbiotische Keimungsprotokolle nicht nur auf MH-Arten, sondern auch auf photosynthetisierende Orchideen angewendet werden, um die Spezifität von Orchideen und Pilzen bei der Keimung und Protocormentwicklung zu untersuchen, eine weitgehend angewandte Methodik in Initiativen zur Erhaltung bedrohter Arten 9,10,11.
In dieser Methodenmontage beschreiben wir wichtige Schritte zur Entnahme, Fixierung und Lagerung von MH-Pflanzenproben für anatomische Studien (Abschnitt 1), Oberflächenanalyse und Probenauswahl (Abschnitt 2), Schnittmethoden (Freihand: Abschnitt 3, Mikrotomie: Abschnitt 4, Kryomikrotomie: Abschnitt 5), Färbung und Montage (Abschnitt 6), Fluoreszenz- und Konfokalmikroskopie von Pilzendophyten (Abschnitt 7), Rasterelektronenmikroskopie (Abschnitt 8), und Transmissionselektronenmikroskopie (Abschnitt 9). Darüber hinaus beschreiben wir eine symbiotische Keimungsmethode für Orchideensamen (MH und autotroph, Abschnitt 10), da die zuvor genannten bildgebenden Verfahren erfolgreich angewendet werden können, um die Pilzbesiedlung von Samen, Protocormen und Keimlingen im Keimprozess zu analysieren.

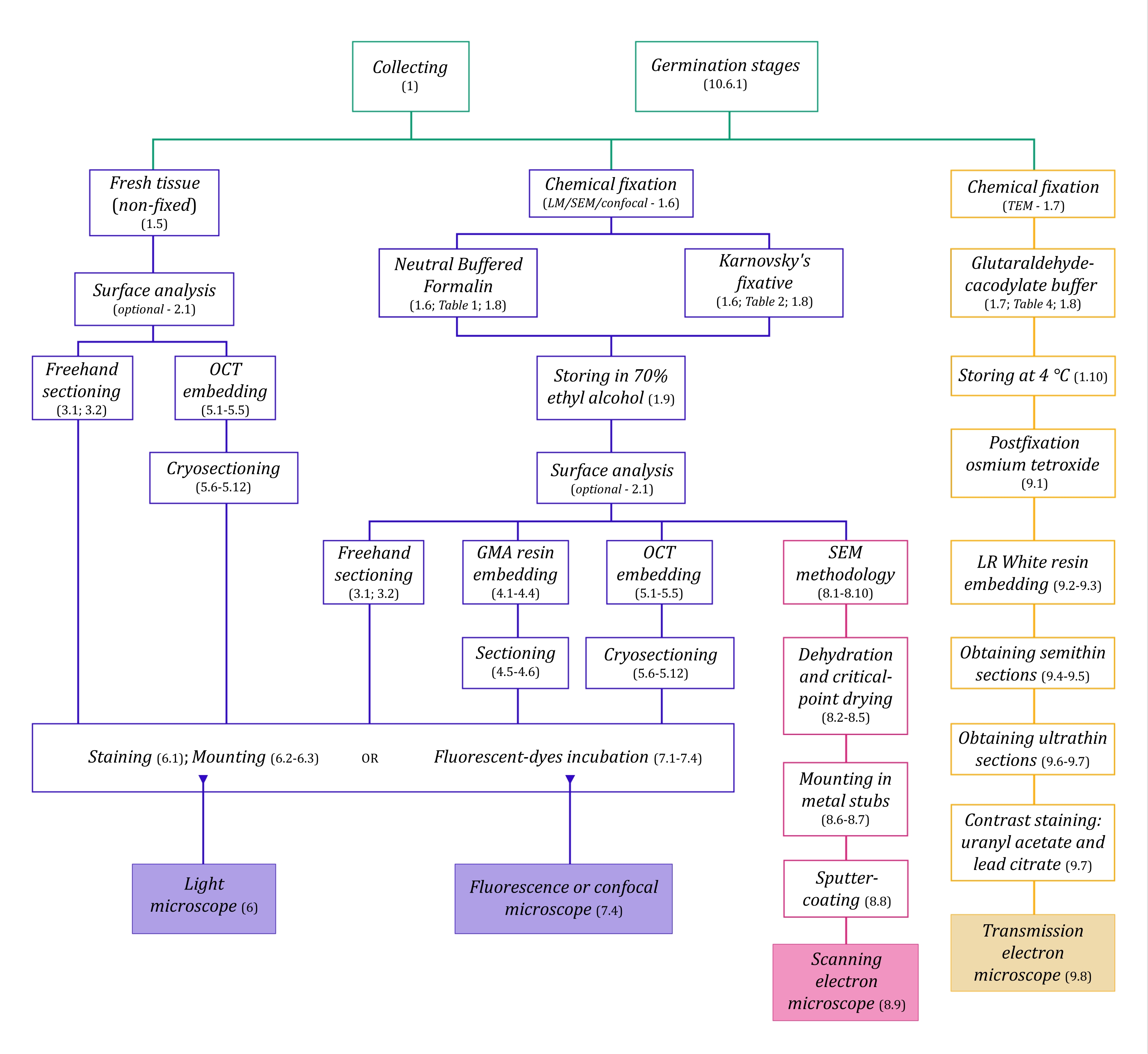
Abbildung 1: Schematische Zusammenfassung bildgebender Verfahren. Die Schaltpläne geben Hinweise auf Protokollschritte, in denen sie detailliert sind. Abkürzungen: GMA = Glykolmethacrylat, OCT = optimale Schnitttemperaturverbindung, REM = Rasterelektronenmikroskopie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Den hier im Detail beschriebenen Mikroskopietechniken (Abbildung 1) gehen folgende wesentliche Schritte voraus: Sammeln, Fixieren, Dehydrieren, Einbetten und Schneiden von Proben. Da die Schritte je nach gewählter Technik(en) unterschiedlich sind (Abbildung 1), ist es wichtig, vorausschauend zu denken und zu berücksichtigen, welche Fixiermittel vorbereitet und zur Sammelstelle transportiert werden sollen, wie die Proben vor der Fixierung vorbereitet werden müssen, welche Dehydratisierungsverfahren anzuwenden sind (Abschnitt 1) und welche Möglichkeiten und Schnittmethoden es gibt (Abschnitte 4, 5, und 9). Abbildung 1 fasst nacheinander alle Schritte zusammen, die für jede unten beschriebene Mikroskopietechnik erforderlich sind.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Sammeln, Reparieren und Pflegen von Mustern
ANMERKUNG: Voll-MH-Pflanzen können normalerweise im dunklen Waldunterholz 12,13 gefunden werden, hauptsächlich in feuchten und streureichen Gebieten, während MH-Pflanzen teilweise in offeneren Wäldern 12,13 gefunden werden können. MH-Pflanzen haben in der Regel gut entwickelte unterirdische Organe in einer Vielzahl von Formen und Größen.
- Erkunden Sie beim Sammeln von MH-Arten den Boden um die Pflanzenbasis, achten Sie darauf, die unterirdischen Organe nicht zu beschädigen, und vermeiden Sie es, die Pflanzen vom Boden zu ziehen, um zu verhindern, dass die Luftorgane von den unterirdischen getrennt werden.
- Graben Sie vorsichtig mit einer Gartenkelle um Luftstrukturen herum, während Sie die unterirdischen Organe wie Wurzeln, Stängel, Rhizome und Speicherorgane erkunden, ohne diese Strukturen zu beschädigen.
- Entfernen Sie Bodenpartikel, um zerbrechliche Strukturen zu erhalten, und waschen Sie diese Organe vorsichtig mit Leitungswasser, um die verbleibenden Bodenpartikel abzuspülen, bevor Sie die Proben fixieren.
- MH-Pflanzen, die mit Laubstreu verbunden sind, erfordern besondere Aufmerksamkeit; Sammeln Sie sorgfältig Organe, die durch ihre Hyphen mit dem zersetzenden Material verbunden sind, vermeiden Sie es, diese Organe aus den verbundenen Strukturen zu ziehen, und sammeln Sie sie sorgfältig, da diese Teile sehr empfindlich sind. Bewahren Sie Strukturen mit solchen Verbindungen und sammeln Sie auch die Einstreu zur Analyse.
- Wenn Sie sich für die Analyse von frischem Material mit bildgebenden Verfahren entscheiden, bewahren Sie die Proben in geschlossenen Plastikbeuteln mit ausreichender Feuchtigkeit auf, wobei genügend Wasser verdunstet und die Pflanze befeuchtet, um zu verhindern, dass übermäßiges Wasser mit den Proben in Kontakt kommt. Transportieren Sie sie sofort ins Labor und analysieren Sie die Proben noch am selben Tag, an dem sie entnommen wurden, wobei darauf geachtet wird, ob die Proben bei der Analyse noch erhalten sind.
- Tragen Sie Fixiermittel in gut verschlossenen Behältern zur Sammelstelle. Fixieren Sie die Proben schnell nach der Entnahme für die Lichtmikroskopie (LM) und Rasterelektronenmikroskopie (REM) in einem der folgenden Fixiermittel: 10% neutrales gepuffertes Formalin (NBF14, Tabelle 1) oder Karnovsky-Lösung (modifiziert15, Tabelle 2). Karnovskys Lösung kann mit 0,2 M Phosphatpuffer15 hergestellt werden, dessen Rezeptur in Tabelle 3 beschrieben ist.
- Für die Analyse mittels Transmissionselektronenmikroskopie (TEM) schneiden Sie die Proben mit einer Dicke von 4-3 mm in einem Tropfen Glutaraldehyd-Natrium-Cacodylat-Puffer (modifiziert16, Tabelle 4) in kleinere Abschnitte von 1-2 mm Dicke. Verwerfen Sie die außerhalb des Tropfens geschnittenen Kanten. Übertragen Sie die Abschnitte sofort in ein Sammelröhrchen mit einem Fixiermittelvolumen, das mehr als 10-mal größer ist als das Volumen der Proben, da es sich um ein additives Fixiermittel handelt (dh seine Moleküle werden chemisch zu den fixierten Proteinenhinzugefügt 16).
ACHTUNG: Die drei beschriebenen Fixiermitteln sind hochgiftig. Vermeiden Sie die Inhalation, besonders während der Verwendung auf dem Feld. Bereiten Sie alle Fixiermittel in einem Abzug mit Handschuhen vor. Cacodylat und Säuren nicht mischen, da Arsengas gebildet werden kann16. - Wenn die fixierten Proben im Fixiermittel schwimmen, deutet dies auf das Vorhandensein von Gas in Pflanzengeweben hin. Luft und andere Gase verhindern, dass das Fixiermittel in die gesamte Probe eindringt2. Entfernen Sie Gas aus Geweben, indem Sie es in kleineren Teilen erneut beproben und eine Vakuumpumpe (-300 bis -400 inHg Druck) verwenden, bis alle Proben auf den Boden der Lösungsinken 17. Seien Sie vorsichtig, da ein übermäßiger Druck durch die Pumpe die Proben beschädigen kann.
- Nach mindestens 48 h in Karnovsky-Lösung oder 10% NBF waschen Sie die Proben in 0,2 M PB (Tabelle 3) und dehydrieren Sie sie mit einer Reihe von 10%, 30%, 50% und schließlich 70% Ethanol. Bei empfindlichen Proben 30 min in jeder Konzentration dehydrieren; Bei größeren Proben 1 h oder länger dehydrieren.
HINWEIS: Eine 70% ige Ethanollösung ist das ideale Medium für die Lagerung von Proben. Proben aus 70% Ethanol können jahrelang bei Raumtemperatur gelagert werden. Lagern Sie Pflanzenmaterial nicht über längere Zeit in den Fixiermitteln, da das Entfernen von Fixiermitteln ein wesentlicher Schritt nach der Fixierungist 2. - Lagern Sie die Proben in Glutaraldehyd-Cacodylat bei 4 °C, bevor Sie mit der Postfixierung fortfahren (Schritt 9.1).
| 10% neutral gepuffertes Formalin (NBF)14 | |
| Schritt 1 | 10 mL 37-40% ige Formaldehydlösung in 80 mL destilliertes Wasser geben |
| Schritt 2 | 0,4 g natriumphosphat monobasisches Monohydrat (NaH2PO4· H2O) zur Lösung |
| Schritt 3 | 0,65 g Natriumphosphat dibasisch, wasserfrei (Na2HPO4) zugeben |
| Schritt 4 | das Volumen auf 100 mL erhöhen |
Tabelle 1: 10% neutrales gepuffertes Formalin Rezept 14.
| Karnovskys Lösung (modifiziert15) | |
| Schritt 1 | in 20 mL destilliertem Wasser bei 60-70 °C |
| Schritt 2 | 0,8 g Paraformaldehyd zugeben (um 4 % w/v unter Rühren zu erhalten |
| Schritt 3 | 1-4 Tropfen 40% NaOH hinzufügen und umrühren, bis die Lösung klar wird |
| Schritt 4 | kühlen und 30 mL 0,2 M Phosphatpuffer pH 7,2 hinzufügen (Tabelle 3) |
| Schritt 5 | 25% Glutaraldehyd in 0,1 M PB (pH 7,2) verdünnen, um 1% Glutaraldehyd zu erhalten (Endvolumen: ~60 ml) |
| Schritt 6 | 1% Glutaraldehyd (Schritt 5) zu der in Schritt 4 erhaltenen Lösung hinzufügen, bis bis bis zu 100 ml Fixiermittel hergestellt werden |
Tabelle 2: Karnovskys Lösungsrezept (modifiziert15).
| 0,2 M Phosphatpuffer (PB) pH 7,2 | |
| Schritt 1 | 14,196 g natriumphosphat dibasisch, wasserfrei (Na2HPO4) zu 400 mL destilliertem Wasser hinzufügen |
| Schritt 2 | 13,8 g natriumphosphat einbasisches Monohydrat (NaH2PO4· H2O) |
| Schritt 3 | Umrühren, bis die Lösung klar ist |
| Schritt 4 | Stellen Sie das Endvolumen mit destilliertem Wasser auf 500 ml ein |
| Schritt 5 | pH-Wert auf 7,2 einstellen |
| Schritt 6 | bei 0,1 M PB 1:1 verdünnen |
Tabelle 3: 0,2 M Phosphatpufferrezeptur.
| 3% Glutaraldehyd 0,2 M Natriumcacodylatpuffer (modifiziert16) | |
| Schritt 1 | 0,2 M Cacodylatpuffer: 4,28 g Natriumcacodylattrihydrat in 100 ml destilliertes Wasser geben |
| Schritt 2 | pH-Wert auf 7,2 einstellen |
| Schritt 3 | 12 mL 25% Glutaraldehyd in 25 mL der Lösung in Schritt 2 zugeben (0,2 M Cacodylatpuffer pH 7,2) |
| Schritt 4 | das Volumen mit destilliertem Wasser auf 100 ml auffüllen |
Tabelle 4: 3% Glutaraldehyd 0,2 M Natriumcacodylatpufferrezeptur (modifiziert16).
2. Oberflächenanalyse von Organen in fixiertem und nicht fixiertem Material
- Um oberflächliche Hyphen in Organen, insbesondere unterirdischen, und solchen, die mit Blattstreu in Kontakt kommen, zu analysieren, beobachten Sie festes oder frisches Material in einem Seziermikroskop (Stereomikroskop) bei einer Vergrößerung von 7,5x oder höher, abhängig von den analysierten Proben.
- Visualisieren Sie die Proben, die in das Fixiermittel 70% Ethanol (falls darin gespeichert) oder Leitungswasser bei frischem Material eingetaucht sind. Verhindern Sie direktes Licht aus dem Seziermikroskop, da es Proben trocknen und beschädigen kann.
- Suchen Sie nach Interessengebieten in den Proben, geleitet von den oberflächlichen Hyphen und Rhizomorphen. Wählen Sie Proben aus, die Bereiche mit oberflächlichen Rhizomorphen enthalten, da diese geschnitten werden können, um Pelotone und Hyphenspiralen innerhalb kortikaler Zellen in Wurzeln und Stängeln sichtbar zu machen.
- Führen Sie nach der Auswahl die Schritte 1.6 und 1.9 aus, wenn die Stichproben noch nicht festgelegt sind. Falls gewünscht, fotografieren Sie frische Proben mit einem Lichtmikroskop ohne Fixierung, wie in Abschnitt 3 beschrieben.
- Verwenden Sie die Kamera, die mit dem Stereomikroskop gekoppelt ist, um Bilder von Organoberflächen, Rhizomorphen und anderen beobachteten Strukturen zu sammeln. Achten Sie in solchen Fällen auf eine angemessene Hintergrundfarbe, um einen guten Kontrast zum Material zu bilden, und wählen Sie, wenn möglich, ein Hintergrundmaterial mit einer weniger rauen Oberfläche (z. B. Papier).
3. Freihändige Abschnitte von Pflanzenorgeln
HINWEIS: Freihandabschnitte von Pflanzenorganen können eine Herausforderung darstellen, insbesondere für kleine und dünne Strukturen. Diese Abschnitte von Geweben mit Pilzendophyten können jedoch in einigen Fällen Hyphen und andere Merkmale im Vergleich zu dünnen Schnitten besser aufweisen.
- Schneiden Sie frische oder fixierte Proben mit einer scharfen Klinge, schneiden Sie sie so dünn wie möglich und legen Sie sie sofort in eine kleine Petrischale mit Wasser (wenn frisch) oder 70% Ethanol (falls fixiert). Verwenden Sie einen kleinen Pinsel, um die Abschnitte zu manipulieren, ohne sie zu beschädigen.
- Um das Schneiden anspruchsvollerer Materialien (d. H. Kleine, dünne, flexible Organe) zu erleichtern, umgeben Sie die Probe in einer Struktur, z. B. Polystyrol oder Cecropia petiole. Schnitzen Sie die Stütze, um die Probe aufzunehmen, und machen Sie einen dünnen Schnitt der Probe und der Stütze insgesamt.
- Färben und montieren Sie die Proben wie in Abschnitt 6 beschrieben.
4. Einbetten von Pflanzenproben in Harz und Schneiden
- Dehydrieren Sie die Proben, die in 70% Ethanol in 80%, 96% und 2x in 100% Ethanol gelagert sind, für 30 min bis 2 h, abhängig von der Größe und Zusammensetzung der Proben.
- Verwenden Sie ein Glykolmethacrylat (GMA) Harzkit gemäß den Anweisungen des Herstellers. Siehe Gerrits und Horobin (1996)18 für weitere Überlegungen. Befolgen Sie die Infiltrations- und Einbettungsschritte entsprechend.
ACHTUNG: GMA-Harz ist giftig, es kann allergische Reaktionen und Haut-, Augen- und Schleimhautreizungen verursachen. Verwenden Sie die Reagenzien in einem Abzug und verwenden Sie Handschuhe. - Verwenden Sie Polyethylen-Formschalen zum Einbetten, die nach Probengröße ausgewählt sind (z. B. 13 mm x 19 mm x 5 mm für größere Proben, 6 mm x 8 mm x 5 mm für kleinere). Achten Sie auf die gewünschte Probenorientierung in der Form und verwenden Sie eine Nadel zur Orientierung.
- Zur Polymerisation, vorzugsweise bei Raumtemperatur, bis sie vollständig erstarrt ist. Der Aushärtungsprozess dauert in der Regel einige Stunden, obwohl es empfohlen wird, die Blöcke am nächsten Tag zu entformen. Lösen Sie nach der Polymerisation die Harzblöcke vorsichtig von den Formen und befestigen Sie die Blöcke so schnell wie möglich, um eine Blockkrümmung zu vermeiden.
- Schleifen Sie die Vorderseite des Harzblocks, der befestigt wird, und erzeugen Sie eine flache Oberfläche. Kleben Sie den Harzblock mit einem flüssigen Cyanacrylatkleber mittlerer Viskosität auf einen Holzquader (2 cm x 2 cm x 3 cm empfohlen) (siehe Materialtabelle). Stellen Sie sicher, dass das Harz vollständig befestigt ist, um eine Beeinträchtigung der Schnitte zu vermeiden.
- Führen Sie die Schnitte in einem rotierenden Mikrotom wie unten beschrieben durch.
ACHTUNG: Die Klingen von Mikrotommessern sind sehr scharf und können Unfälle verursachen. Achten Sie darauf, sie nach allen Sicherheitsmaßnahmen zu behandeln. Bevor Sie sich dem Messer nähern (z. B. um den Block zu wechseln, das Harz zu befeuchten), verriegeln Sie das grobe Handrad und setzen Sie die Klingensicherheitsabdeckung auf. Lagern Sie Einwegklingen in geeigneten Koffern. Seien Sie äußerst vorsichtig, wenn Sie die Klingen austauschen.
HINWEIS: Verschiedene Arten von Messern (z. B. Einwegmesser oder fest; Glas oder Stahl) können verwendet werden, um GMA-Harz18 zu schneiden. Die Qualität der Abschnitte hängt davon ab, wie scharf das Messer ist. Stellen Sie sicher, dass das Messer gut befestigt ist und sich nicht bewegen kann. Einwegmesser müssen möglicherweise regelmäßig gewechselt werden, um eine bessere Aufteilung zu erreichen.- Befestigen Sie den Holzquader fest am Blockhalter. Stellen Sie die Ausrichtung der Schnitte mit den Orientierungsschrauben ein und sorgen Sie mit der Messerneigung für einen angemessenen Winkel des Messers. Wählen Sie die Dicke der Abschnitte aus; Verwenden Sie eine dickere Einstellung für das Trimmen und eine dünnere Einstellung für ausgewählte Abschnitte, da GMA-Abschnitte besser am Glasobjektträger haften, wenn sie dünner sind. Die empfohlene Dicke für Pflanzengewebe beträgt 5-8 μm.
- Bevor Sie beginnen, kühlen Sie den Raum, falls erforderlich, da höhere Temperaturen die Qualität der Abschnitte verschlechtern. Bereiten Sie Folgendes zum Schneiden vor: ein Becherglas mit destilliertem Wasser, eine Kochplatte, Pinsel (mindestens zwei), eine Feinspitzenpinzette, eine Pasteur-Pipette, Glasobjektträger, Filterpapier (oder Seidenpapier) und einen Bleistift (zur Identifizierung der zu schneidenden Proben).
- Stellen Sie die Heizplatte auf 50 °C ein und stellen Sie das Becherglas darauf. Achten Sie auf Unterschiede in der Erwärmung in Abhängigkeit von der Fläche der Heizplatte (normalerweise erwärmt sich der mittlere Bereich mehr als die Kanten; erwärmen Sie das Wasser vorzugsweise in der Mitte).
- Wählen Sie einen Glasobjektträger, identifizieren Sie ihn mit einem Bleistift und pipetten Sie das warme destillierte Wasser auf der gesamten Objektträgeroberfläche. Verwenden Sie bei Bedarf eine Lösung (z. B. Reinigungsmittel und Wasser oder 70% Ethanol), um die Spannung zwischen Wasser und Glas zu brechen, so dass der gesamte Objektträger gleichmäßig bedeckt ist. Einige Objektträgertypen müssen mit 70% Ethanol vorgereinigt werden, um eine ausreichende Schnitthaftung zu erreichen.
- Beginnen Sie, indem Sie die Vorderseite des Harzblocks allmählich in Richtung der Messerklinge vorschieben. Versuchen Sie nicht, den Block zu trennen, ohne zuerst den Block voranzubringen, da sonst die Ausrüstung und der Block beschädigt werden können. Falls erforderlich, trimmen Sie den Block mit einer höheren Dicke (10 μm und mehr).
- Wenn Sie sich einem geeigneten Abschnitt nähern, machen Sie eine feste Einwegbewegung mit dem Handrad, so dass der Abschnitt sofort gemacht wird. Kontrollieren Sie die Harzfeuchtigkeit. Befeuchten Sie während des Schneidens regelmäßig die Vorderseite des zu schneidenden Blocks mit einem Pinsel, der in destilliertes Wasser getaucht wird, wenn es ein Problem mit Curling-Abschnitten gibt. Entfernen Sie überschüssiges Wasser mit einem Seidenpapier.
- Legen Sie den erhaltenen Abschnitt mit einer feinen Pinzette über die Rutsche ins Wasser. Bei Kontakt mit Wasser dehnt sich GMA-Harz aus. Verwenden Sie bei Bedarf einen Pinsel, um die Abschnitte vorsichtig zu entfalten und zu dehnen. Verwenden Sie einen anderen Pinsel, um die Klinge ständig frei von Harzablagerungen zu halten. Wechseln Sie nicht zwischen den Pinseln, um eine Benetzung der Klinge zu vermeiden.
- Nachdem Sie alle gewünschten Abschnitte in der Folie aufgereiht haben, trocknen Sie die Unterseite der Folie und legen Sie sie über die Heizplatte. Entfernen Sie das überschüssige Wasser von der Oberseite des Objektträgers, indem Sie es vorsichtig mit einem Filterpapier tupfen (optional). Die Abschnitte haften, wenn das Wasser von der Rutsche verdunstet. Lassen Sie die Dias nicht zu lange, um zu verhindern, dass die Abschnitte durch übermäßige Hitze beschädigt werden.
- Bewahren Sie die Dias in einer Diabox auf, fern von Staub und Sonne, und verwenden Sie sie zum Färben und anderen Verfahren. Die Dias können mehrere Jahre gelagert werden.
5. Einfrieren von Pflanzenproben und Schneiden mit einem Kryostaten
HINWEIS: Die wesentliche Überlegung beim Kryoschneiden von biologischem Gewebe besteht darin, Schäden durch Eiskristallbildung beim Einfrieren von Proben zu reduzieren. Kryoprotektion wird normalerweise durch Infusion chemisch inerter Lösungen wie Glycerin oder Saccharose19,20 durchgeführt.
- Führen Sie einen Tag vor dem Probenschneiden die folgenden Schritte aus.
- 100 ml 0,2 M PB (Tabelle 3) werden in 100 ml destilliertem Wasser verdünnt, um 200 mL 0,1 M PB zu erhalten. Bereiten Sie 10%, 20% und 30% Saccharoselösungen in 0,1 M PB vor (z. B. für eine 10% ige Lösung 2 g Saccharose in 20 ml Puffer hinzufügen).
- Für frische Proben waschen Sie sie in 0,1 M PB für 30 min. Für Proben in einem Fixiermittel waschen Sie sie in demselben Puffer, der für die Vorbereitung des Fixiermittels verwendet wurde, für 30 min. Für Proben aus 70% Ethanol hydratisieren Sie sie in 50% und 30% Ethanol und waschen Sie sie in 0,1 M PB für 1 h in jeder Lösung.
- Die Proben werden bei Raumtemperatur für 2 h in 10% Saccharose, 2 h in 20% Saccharose und 2 h in 30% Saccharose inkubiert. Anschließend über Nacht in 30%iger Saccharose bei 4 °C (oder mindestens 3 h; maximale Zeit beträgt 48 h) inkubieren.
- Am Tag des Schneidens 40% und 50% Saccharose in 0,1 M PB vorbereiten; Bereiten Sie Saccharoselösungen nicht mehr als 12 Stunden im Voraus vor. Für 2 h in jeder Saccharosekonzentration bei 4 °C inkubieren.
- Zum Einbetten in kleine Formen eine Schicht OCT-Compound (optimales Schnitttemperaturmedium, das zum Einbetten und Einfrieren der Proben verwendet wird) herstellen und bei -20 °C einfrieren. Die Formen können normale histologische Formen sein, obwohl Papier- oder Alufolienformen mit einem kleinen Quader als Rahmen und Klebeband hergestellt werden können, um das Ausformen der Blöcke zu erleichtern.
- Nachdem die untere Schicht der OCT-Verbindung in den Formen eingefroren wurde, arbeiten Sie in einer Kryostatkammer (ca. -27 °C). Legen Sie die in 50% iger Saccharose inkubierten Proben in die Formen, in der Orientierung, in der sie geschnitten werden. Die Oberseite eines quaderförmigen Blocks ist normalerweise eine bessere Seite des Schnitts. Markieren Sie in der Form, in der die Proben platziert werden, damit der Block leicht zugeschnitten und die korrekte Ausrichtung beibehalten werden kann.
- Umgeben Sie die Proben mit OCT-Verbindung und platzen Sie jede Luftblase, die die Proben berührt. Bei -20 °C einfrieren. Wenn die Blöcke vollständig gefroren sind, legen Sie sie in die Kryostatkammer (ca. -27 °C). Entformen Sie jeden einzelnen nur vor dem Gebrauch und achten Sie auf die Markierungen, die die Probenposition im Block anzeigen.
- Da OCT-Compound leicht mit einer Klinge geschnitten werden kann, trimmen Sie die Blöcke entsprechend, bevor Sie sie auf die Chucks positionieren. Geben Sie etwas OCT-Verbindung in das Kryostatfutter und positionieren Sie den Block so, dass die Oberseite geschnitten ist. Flächen mit kleineren Flächen bieten bessere Schnitte. Warten Sie, bis der Block gut am Spannfutter befestigt ist, und testen Sie ihn, bevor Sie mit dem Schneiden beginnen.
- Legen Sie das Spannfutter fest in den Futterhalter. Passen Sie die Ausrichtung der Schnitte mit den Ausrichtungsschrauben an. Stellen Sie den Winkel des Messers mit der Messerneigung ein. Wählen Sie die Dicke der Schnitte aus. Die Proben können dicker geschnitten werden als übliche Harzabschnitte. Es werden erfolgreich Abschnitte in einem Bereich zwischen 5-20 μm erzielt, wobei dickere Abschnitte leichter herzustellen sind (weniger Kräuseln und weniger Beschädigung der Strukturen).
- Schieben Sie die Vorderseite des gefrorenen Blocks in Richtung Messerklinge. Versuchen Sie nicht, ohne dies zu tun, sonst kann sich der Block vom Spannfutter lösen und beschädigt werden. Falls erforderlich, trimmen Sie den Block mit einer höheren Dicke (10 μm und mehr).
- Wenn Sie sich einem geeigneten Abschnitt nähern, platzieren Sie die Anti-Roll-Platte (dh eine transparente Platte, die den Abschnitt hält) über dem Messer und machen Sie eine feste Einwegbewegung mit dem Handrad, so dass der Abschnitt sofort gemacht wird. Kräuselprobleme können verursacht werden, wenn die Stabilisatorplatte eingestellt werden muss (sie ist normalerweise in Bezug auf die Klinge einstellbar) oder wenn sich Schmutz in der Klinge befindet. Reinigen Sie die Klinge ständig mit einem Pinsel, um Schmutz zu entfernen.
- Verwenden Sie spezielle Objektträger, damit die Abschnitte leicht befestigt werden können, wie silanisierte Objektträger (kommerziell oder mit 2% Aminoalkylsilan in Aceton 21 hergestellt) oder Objektträger, die mit 500 μg / ml Poly-L-Lysin in destilliertem Wasser 21 oder 0,2% Gelatine hergestellt wurden (siehe Details21). Halten Sie die Objektträger auf Raumtemperatur.
- Um den Abschnitt an einem Schieber zu befestigen, heben Sie die Stabilisatorplatte an und lassen Sie den Schieber den Abschnitt schnell berühren. Da der Schlitten Raumtemperatur hat, schmilzt der Abschnitt sofort und haftet am Objektträger. Achten Sie darauf, die behandelte Seite des Objektträgers auf den Abschnitt zu drehen, der über dem Messer oder auf der Innenseite der Stabilisatorplatte verbleiben kann. Um ein Kräuseln der Abschnitte zu vermeiden, führen Sie diesen Schritt schnell durch, sobald die Platte angehoben wird, und achten Sie darauf, den Abschnitt nicht zu verzerren.
- Lassen Sie den Objektträger außerhalb der Kryostatkammer (bei Raumtemperatur), wenn neue Abschnitte hinzugefügt werden sollen. Nachdem Sie alle gewünschten Abschnitte auf den Objektträger geklebt haben, bewahren Sie ihn in der Kryostatkammer oder im Gefrierschrank auf (-20 °C oder darunter). Setzen Sie die Objektträger keiner Feuchtigkeit aus. Bewahren Sie sie in einer Folienbox auf und denken Sie daran, die Folien mit einem Bleistift zu identifizieren.
HINWEIS: Dias und OCT-Blöcke können bei -20 °C gelagert werden, wenn auch nicht zu lange. Um bessere Ergebnisse zu erzielen, verwenden Sie die Folien und die Blöcke innerhalb weniger Tage.
6. Färbung von Pflanzenschnitten und Endophyten für die Lichtmikroskopie
HINWEIS: Viele Arten von Flecken können für Pflanzenabschnitte verwendet werden. Es ist schwierig, endophytische Pilze und Pflanzengewebe unterschiedlich zu färben. Obwohl es sich nicht um ein Färbeverfahren handelt, wird in Abschnitt 7 ein Verfahren zur Markierung von Pilzstrukturen vorgestellt (Fluoreszenz mit einem Weizenkeimagglutinin-Konjugat). Freihandschnitte (erklärt in Abschnitt 3), Harzschnitte (Abschnitt 4) und Kryosektionen (Abschnitt 5) können gefärbt werden, obwohl Phenol- und Alkoholfärbungen für diese Proben eine Herausforderung darstellen, da GMA-Harz und OCT in diesen Fällen die Haftung am Objektträger verlieren.
- Verwenden Sie eine oder kombinieren Sie die folgenden üblichen Färbemethoden für Pflanzenproben.
- Toluidinblau O22,23, eine weit verbreitete Methode zur allgemeinen Färbung von Pflanzenabschnitten. Bereiten Sie eine Lösung von 0,05% Toluidinblau O in 0,1 M Phosphat (pH 6,8) oder 0,09 M Citratpuffer (pH 4,5-4,8) je nach Art und Art des Gewebes vor. Inkubieren Sie GMA-Harzschnitte für 2-10 Minuten mit einem Objektträgerfärbeglas oder indem Sie einige Tropfen über die Abschnitte legen, wenn nur wenige Objektträger gefärbt sind. Nach der Inkubation vorsichtig mit destilliertem Wasser oder Puffer waschen und die Objektträger mit Wasser montieren oder auf einer Heizplatte trocknen, um dauerhafte Objektträger wie in Schritt 6.3 beschrieben herzustellen.
- Lugol-Reagenz2 zeigt das Vorhandensein von Stärke an. Eine 5%ige Jod- (I2) und 10%ige Kaliumiodidlösung (KI) in destilliertem Wasser herstellen. Schichten für 2 min einfärben, indem Sie einige Tropfen über dem Objektträger hinzufügen, und dann mit destilliertem Wasser waschen. Dieser histochemische Test wird normalerweise auf temporäre Objektträger angewendet.
- Sudan III, IV und schwarz B24,25 färben sich für verschiedene Lipide. Bereiten Sie eine Lösung von 0,3% Sudan (III, IV oder schwarz B) in 70% Ethanol vor, erwärmen Sie sie bis zum Kochen und lassen Sie sie abkühlen. Verwenden Sie den Überstand, filtern Sie ihn und inkubieren Sie Abschnitte für 15-30 min in einer geschlossenen Petrischale. Waschen Sie die Abschnitte sorgfältig mit 70% Ethanol und destilliertem Wasser. Montieren Sie die Objektträger mit Wasser (normalerweise nur auf temporäre Objektträger angewendet).
HINWEIS: Da Sudan ein Farbstoff auf Alkoholbasis ist, ist er besser für Freihandabschnitte geeignet. Führen Sie die GMA-Harzschnittfärbung sorgfältig durch, da sie sich normalerweise vom Objektträger lösen.
- Für temporäre Objektträger montieren Sie die Abschnitte in Wasser oder Glycerin und beobachten Sie sie anschließend. Versiegeln Sie das Deckglas mit Nagellack, um es etwas länger zu konservieren.
- Bei permanenten Objektträgern montieren Sie die Profile mit Kunstharzen (z.B. Schnellmontagemedium, siehe Materialtabelle). Tropfen Sie ein paar Tropfen des Montagemediums ab (es kann das Deckglas überlaufen), legen Sie das Deckglas vorsichtig, um Blasen zu vermeiden, und drücken Sie das Objektblatt mit Wäscheklammern gegen das Deckglas, bis es vollständig trocken ist. Entfernen Sie das überschüssige getrocknete Montagemedium mit einer Rasierklinge.
7. Anwendung eines an Weizenkeimagglutinin konjugierten Fluorochroms in der Fluoreszenz- und Konfokalmikroskopie
HINWEIS: Diese Methode kann auf Freihandschnitte (siehe Abschnitt 3), Harzschnitte (Abschnitt 4) und Kryosektionen (Abschnitt 5) angewendet werden. Kryosektionen können für konfokale Mikroskopiezwecke geeignet sein, da im Vergleich zu Harzschnitten dickere Proben bereitgestellt werden können, aber nicht so dick wie Freihandproben. Ein mit Weizenkeimagglutinin konjugiertes Fluorochrom (WGA, siehe Table of Materials) wird zur Pilzbildgebung in der Fluoreszenzmikroskopie26 angewendet. Ein konfokales Mikroskop ist nicht unbedingt erforderlich, obwohl es klare dreidimensionale Bilder von Pflanzenstrukturen liefern kann27.
- Es wird eine Lösung von 0,2 mg/ml WGA-Fluorochrom-Konjugat in 0,1 M PB28 hergestellt (pH 7,2, siehe Schritt 5.1.1 und Tabelle 3). Bereiten Sie eine Lösung von 1% Calcofluorweiß in 0,1 M PB (pH 7,2) vor. Bereiten Sie kleine Mengen dieser Lösungen vor, da die Abschnitte direkt mit ihnen inkubiert werden.
- Die Abschnitte in den Glasobjektträgern 30 min lang in der WGA-Fluorochrom-Konjugatlösung29 inkubieren, mit genügend Volumen zum Abdecken der Abschnitte, dann in 0,1 M PB waschen.
- Inkubieren Sie die Abschnitte in der Calcofluorlösung unter Verwendung eines ausreichenden Volumens als Montagemedium. Die Lösung kann während des Beobachtungszeitraums beibehalten werden.
- Deckgläser auf die Objektträger legen und in einem konfokalen Mikroskop oder einem Fluoreszenzlichtmikroskop mit folgenden Filtern beobachten: TC/GFP (Anregung: 470-440, Emission: 525-550, für WGA-Fluorochrom in der Materialtabelle - Pilzzellwand fluoresziert grün unter diesem Filter29) und DAPI (Anregung: 358, Emission: 463, für Calcofluorweiß)30.
HINWEIS: Dreidimensionale Bilder können mit der Funktion der Z-Serie im konfokalen Mikroskop27 erhalten werden.
8. Rasterelektronenmikroskopie pflanzlicher Organe
- Nach der Fixierung der Proben, der Dehydratisierung und der Lagerung in 70% Ethanol (Abschnitt 1) besteht eine Möglichkeit darin, Proben zu schneiden, um bei Bedarf jede gewünschte Oberfläche für die REM-Analyse freizulegen (z. B. innere Gewebe, Eierstockstruktur). Verwenden Sie eine scharfe und neue Rasierklinge und machen Sie Schnitte mit einer Einwegbewegung, um ein beschädigtes Aussehen dieser Bereiche im REM zu vermeiden. Verwenden Sie bei Bedarf ein Stereomikroskop, um die Proben auszuwählen, und berücksichtigen Sie den Bereich der Metallstummeln, um die Probengrößen zu bestimmen.
- Weitere Dehydratproben für REM in einer ethanolischen Reihe: 80%, 96% und 2x in absolutem Ethylalkohol (≥99,8%). Bewahren Sie kleine und empfindliche Proben für 30 min in jeder Konzentration und größere und dichtere Proben für 1 h auf.
- Falten Sie kleine Umschläge mit Seidenpapier, um Proben für die nächsten Schritte zu organisieren, größere Proben können ohne Umschlag gehandhabt werden. Identifizieren Sie die Umschläge mit einem Bleistift, indem Sie einen Buchstaben oder eine Zahl schreiben, und führen Sie ein Protokoll aller Proben in jedem einzelnen. Bewahren Sie die Proben in absolutem Ethanol auf, wenn auch nicht lange, und fahren Sie so bald wie möglich mit Schritt 8.4 fort.
- Fahren Sie mit der Trocknung am kritischen Punkt (CP) fort. Betreiben Sie einen CP-Trockner gemäß den Standardarbeitsanweisungen. Legen Sie Proben in absolutes Ethanol (Zwischenflüssigkeit) in eine Druckkammer. AmCO2-kritischen Punkt (31 °C, 7,3 x 106 Pa) löst sich die Zwischenflüssigkeit in die Übergangsflüssigkeit (flüssiges Kohlendioxid) und die Proben werden getrocknet31.
- Lagern Sie die Proben nach der CP-Trocknung so schnell wie möglich in einem Austrocknungsbehälter, z. B. in einem verschlossenen Kolben mit Silikagel. Luftfeuchtigkeit kann die Proben zerstören, wenn sie resorbiertwird 31.
- Verwenden Sie Metallstummeln, um die Proben zu montieren. Vor der Montage Handschuhe anziehen, um die Stummel zu manipulieren, sie 5 Minuten lang in Aceton eintauchen, um Fett zu entfernen, und trocknen lassen. Verwenden Sie ein leitfähiges doppelseitiges Kohleklebeband, um Proben auf dem Stummel zu fixieren, und ein Stereomikroskop, um die Proben zu positionieren, wobei zu beachten ist, dass der Blick von oben die einzig mögliche Perspektive in REM-Bildern ist.
- Bearbeiten Sie Proben mit einer Feinpunktpinzette, seien Sie vorsichtig, da das von der Pinzette berührte Probenteil normalerweise beschädigt ist, also versuchen Sie, Teile zu berühren, die von den interessierenden Bereichen entfernt sind (z. B. Bereiche, die mit dem Band in Berührung kommen). Pflegen Sie die Stubs mit Proben in einer verschlossenen Petrischale mit Silikagel. Fahren Sie so schnell wie möglich mit Schritt 8.8 fort.
- Verwenden Sie einen Sputterbeschichter, um eine Metallschicht, normalerweise Gold oder Platin, auf der Oberfläche der Proben in einer Niederdruckatmosphäre eines Inertgases, häufig Argon31, abzuscheiden. Befolgen Sie die Standardarbeitsanweisungen, wenn Sie einen Sputterbeschichter verwenden. Die Schichtdicke hängt von der Topographie der Proben ab, in der Regel zwischen 15-40 nm32.
- Bewahren Sie die beschichteten Stummel in einer verschlossenen Petrischale mit Silikagel auf, und vorausgesetzt, dass das Silikagel Feuchtigkeit speichert, können Proben auf diese Weise wochenlang gelagert werden. Verwenden Sie ein Rasterelektronenmikroskop, um die Proben zu analysieren. Die Proben im Vakuum werden von einem Elektronenstrahl getroffen, und die Emission von Signalen aus einer solchen Wechselwirkung wird als Bild31 interpretiert. Einzelheiten zum Betrieb eines Rasterelektronenmikroskops finden Sie in Jeffree und Read (1991) 31 und Bozzola und Russell (1999) 32.
- Um die Stummel wiederzuverwenden, ziehen Sie das Klebeband und schrubben Sie sie mit Drahtwolle. In Leitungswasser waschen, in absolutes Ethanol eintauchen und angemessen trocknen, um eine Oxidation des Metallbestandteils zu verhindern.
9. Transmissionselektronenmikroskopie
- Präfix Proben mit Glutaraldehyd-Cacodylat-Puffer, wie in den Schritten 1.7 und 1.10 erläutert. Nach 12-24 h Vorfixierung waschen Sie die Proben 3x in 0,2 M Cacodylatpuffer (pH 7,25) für 10 min. Die Postfixation mit 1% Osmiumtetroxid (OsO4) in 0,2 M Cacodylatpuffer für 12 h im Dunkeln bei Raumtemperatur durchführen. 3x mit destilliertem Wasser 5 min waschen.
ACHTUNG: Cacodylat und Osmiumtetroxid sind hochgiftig und sollten nicht inhaliert werden. Verwenden Sie sie in Abzügen gemäß den jeweiligen Sicherheitsdatenblättern. - Dehydrieren Sie die Proben mit 30%, 50%, 70% und 96% Ethanol, 2x in jeder Konzentration, für 10 min. Dann dehydrieren Sie 3x in absolutem Alkohol, jeweils für 15 Minuten.
- Infiltrieren Sie die Proben in hydrophilen Acrylharzen (siehe Materialtabelle), einmal mit 1:1 Harz + absolutem Ethanol und 3x mit reinem Harz für jeweils 8-12 h. Die Polymerisation wird in Gelatinekapseln bei 60 °C durchgeführt, bis sie vollständig erstarrt ist (12 h maximal17).
- Bewerten Sie die Ausrichtung der Proben innerhalb des Harzblocks genau; Schneiden Sie den oberen Teil des Blocks mit einer Rasierklinge ab und bilden Sie eine pyramidenförmige Form, die die Probe im Schnittbereich konzentriert. Halbdünne Schnitte (250-500 nm)33 in einem Ultramikrotom mit einem Diamantmesser erhalten und in wenigen Tropfen Wasser auf Glasobjektträger legen.
- Bewahren Sie die Dias auf einer Heizplatte bei 60 °C auf. Die Abschnitte wie in Schritt 6.1.1 mit Toluidinblau O färben und den Fleck vollständig trocknen lassen. Vorsichtig mit Leitungswasser waschen. Bewerten Sie den erhaltenen Schnitt, indem Sie vier Quadranten zeichnen und den am besten geeigneten Quadranten für die Analyse auswählen.
- Schneiden Sie den Block so, dass die pyramidenförmige Form den ausgewählten Quadranten auf die Oberseite des Blocks konzentriert. Ultradünne Schnitte (50-100 nm) erzeugen33,34. Die Dicke wird nach der Interferenzfarbe der Schnitte bewertet: Abschnitte mit etwa 70 nm erscheinen silber-gold, mit etwa 100 nm erscheinen Gold und mit etwa 200 nm erscheinen blau34.
- Sammeln Sie die ultradünnen Schnitte aus Wasser mit Kupfergittern und fahren Sie mit der Kontrastfärbungsmethode mit Uranylacetat und Bleicitrat fort, wie unten beschrieben.
- Bereiten Sie eine Bleicitratlösung (Tabelle 5) vor und frieren Sie die endgültige Lösung in Mikrozentrifugenröhrchen mit jeweils 1 ml Lösung ein, wobei sie erst unmittelbar vor Gebrauch auftauen.
- Bereiten Sie eine Uranylacetatlösung vor: 0,625 g Uranylacetat [UO2(CH3COO)2] werden in 25 ml frisch gekochtem und abgekühltem destilliertem Wasser gelöst. In einem dunklen Kolben im Gefrierschrank aufbewahren.
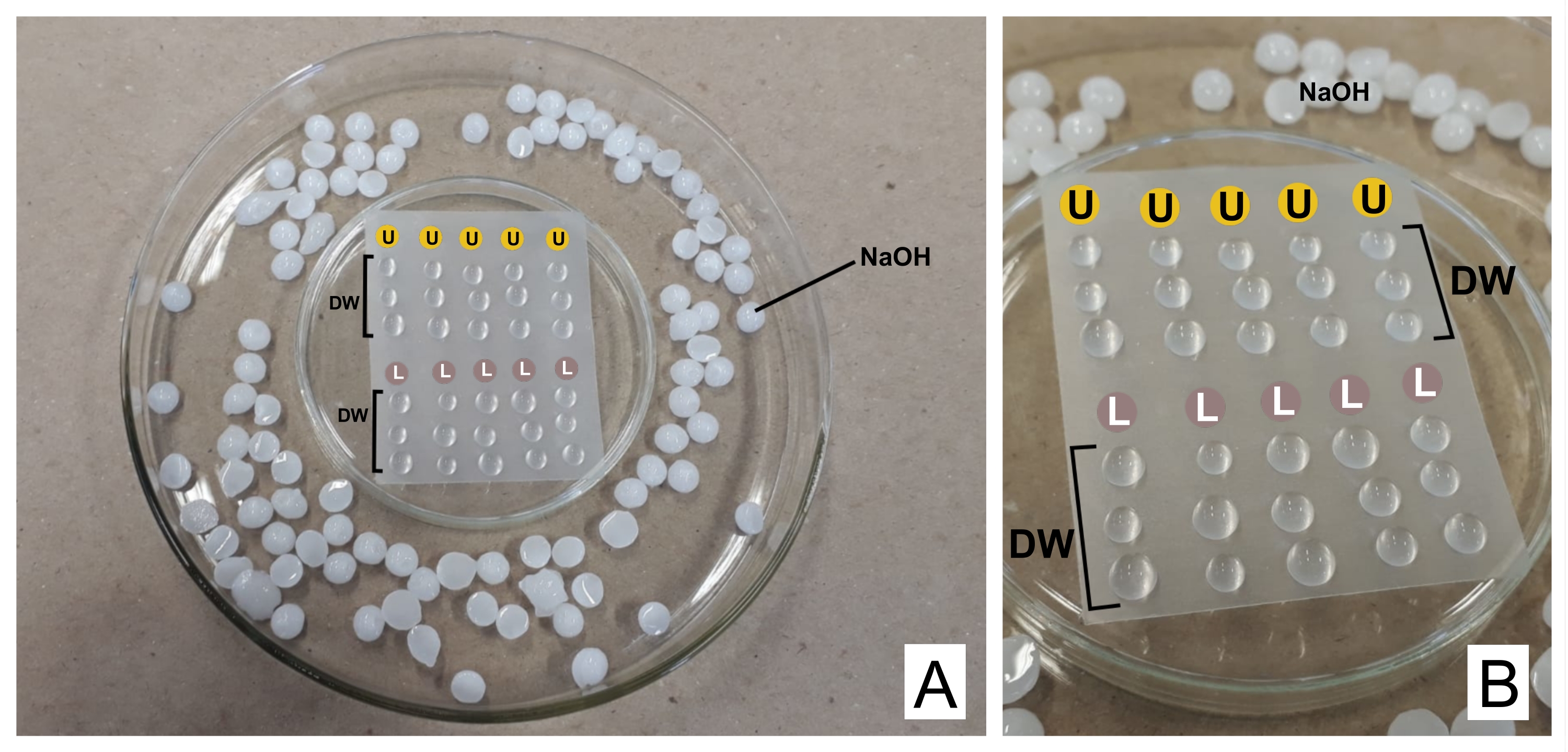
VORSICHT: Bleinitrat ist giftig, wenn es eingenommen wird. 1 N NaOH ist stark korrosiv. Uranylacetat ist radioaktiv und giftig. Es darf nicht eingenommen, eingeatmet oder mit der Haut in Berührung kommen. - Geben Sie beim Färben beide vorbereiteten Reagenzien in separate 3-ml-Spritzen mit Filtereinheiten (0,22 μm Pore, siehe Materialtabelle). Bereiten Sie eine auf den Kopf gestellte Petrischale mit einer versiegelnden thermoplastischen Folie darüber (siehe Materialtabelle) und in einer breiteren Schale mit NaOH-Pellets an den Rändern als Fangbehälter für CO232 vor (siehe Abbildung 2).
- Verwerfen Sie den ersten Tropfen und geben Sie einen Tropfen Uranylacetat und drei Tropfen destilliertes Wasser für jedes gefärbte Gitter über den Film. Machen Sie dasselbe mit Bleicitrat und fügen Sie drei weitere Tropfen destilliertes Wasser hinzu.
- Inkubieren Sie das Gitter (mit der undurchsichtigen Seite nach unten, wo sich die Abschnitte befinden) in Uranyl für 30 min (variable Zeit). Waschen Sie 3x in den destillierten Wassertropfen und trocknen Sie den Rost jedes Mal vorsichtig mit Filterpapier auf der brillanten Seite. Wiederholen Sie dies mit dem Bleicitrattropfen (30 min) und waschen Sie es.
- Nach mindestens 4 h analysieren Sie die Gitter in einem Transmissionselektronenmikroskop. In diesem Mikroskop durchläuft ein Elektronenstrahl die Abschnitte im Vakuum und das Bild wird auf eine fluoreszierende Leinwand projiziert. Einzelheiten zum Betrieb eines Transmissionselektronenmikroskops finden Sie unter Bozzola und Russell (1999)32.
| Bleicitratlösung (zur TEM-Kontrastfärbung) | |
| Schritt 1 | Einen Becher mit Alufolie umhüllen |
| Schritt 2 | 0,266 g Bleinitrat [Pb(NO3)2] werden in 6 ml frisch gekochtem und abgekühltem destilliertem Wasser gelöst |
| Schritt 3 | 2 min rühren |
| Schritt 4 | 0,352 g Trinatriumcitrat [Na3(C6H5O7).2H2O] zugeben (die Lösung muss milchig aussehen) |
| Schritt 5 | 15 min rühren, Becherglas mit Alufolie verschließen und die Lösung in ein 10-ml-Becherglas geben |
| Schritt 6 | 1,6 mL 1N NaOH und 2,4 mL destilliertes Wasser hinzufügen (die Lösung muss durchscheinend sein) |
| Schritt 7 | wenn nötig, stellen Sie den pH-Wert in der Nähe von 12 ein |
Tabelle 5: Rezept für Bleicitratlösung.

Abbildung 2: Kontrastfärbeschema mit Bleicitrat und Uranylacetatlösungen . (A) Bereiten Sie die Petrischalen vor, eine auf den Kopf gestellt (in der Mitte) mit thermoplastischer Folie, damit Tropfen darüber platziert werden können, in eine breitere. NaOH-Pellets werden um die zentrale Schale herum platziert. (B) Uranylacetat-Tropfen werden in die Kreise mit dem Buchstaben U gegeben, und Bleicitrat-Tropfen in den mit L gekennzeichneten Kreisen. DW bezeichnet Tropfen von destilliertem Wasser. Die Gitter werden sequentiell in der Spalte gefärbt, so dass fünf Gitter gleichzeitig wie dargestellt gefärbt werden können. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
10. Symbiotische Keimung von Orchideensamen
- Stellen Sie sicher, dass die Lösungen und alle Materialien, die bei der symbiotischen Keimung von Samen verwendet werden, steril sind, um eine Kontamination zu vermeiden. Beginnen Sie mit dem Autoklavieren für 20 Minuten bei 121 °C. Die symbiotischen Keimschritte sind in Abbildung 3 zusammengefasst.
- Desinfizieren Sie oberflächlich Früchte und Samen, indem Sie sie in Natriumhypochloritlösung mit 2% aktivem Chlor für 10-15 min für Früchte und 7-10 min für Samen tauchen, unter Berücksichtigung der Steifigkeit und Dicke der Samenhülle9. Schlanke und zerbrechliche Samen können in eine 1:1 verdünnte Natriumhypochloritlösung getaucht werden. Anschließend 3x in autoklaviertem destilliertem Wasser waschen, um die Hypochloritlösung zu entfernen.
- Rekuperieren Sie die Samen durch Filtern in siebgrafischem Gewebe und verwenden Sie die Samen, um mit Keimungstests fortzufahren (vorzugsweise). Gegebenenfalls in Filterpapierhüllen in Glaskolben mit Silikagel bei 4 °C aufbewahren, die Kolben hermetisch verschließen und mit Frischhaltefolie verschließen. Einige Tropfen Wasser aus der letzten Wäsche in Kartoffeldextrose-Agar (PDA, 39 g / L) geben, um die Wirksamkeit des Waschprozesses zu bewerten.
- Vor der Aussaat von Samen im Kulturmedium ist ihre Lebensfähigkeit durch den Tetrazoliumtest (fakultativ) wie unten beschriebenzu bewerten 35.
- Etwa 10 mg Samen in einem Mikrozentrifugenröhrchen mit 1 ml 10%iger Saccharose in destilliertem Wasser für 24 h bei Raumtemperatur (ca. 25 °C) bei Licht bebrüten.
- Entfernen Sie die Saccharoselösung mit einer Mikropipette und fügen Sie 1 ml 1% ige Tetrazoliumlösung (Triphenyltetrazoliumchlorid) in destilliertes Wasser hinzu. Bei 40 °C in einem Thermoblock für 24 h im Dunkeln inkubieren.
- Entfernen Sie die Tetrazoliumlösung mit einer Mikropipette und waschen Sie die Samen mit destilliertem Wasser 2x oder bis die Lösung entfernt ist. Entfernen Sie alle Flüssigkeiten. Bei Bedarf können die Samen bis zu einer Woche im Gefrierschrank gelagert werden (wie in Schritt 10.3), bevor sie analysiert werden.
- Resuspendieren Sie die Samen in destilliertem Wasser und analysieren Sie sie unter einem Lichtmikroskop. Lebensfähige Samen erhalten eine hell- bis dunkelrote Farbe, während nicht lebensfähige Samen ihre natürliche Farbe behalten.
- Führen Sie das folgende angepasste9-Protokoll für die symbiotische Keimung von Orchideensamen durch.
- Inkubieren Sie die Samen über autoklavierte Filterpapierscheiben (1-2 cm Durchmesser), die in Petrischalen mit Haferflocken-Agar (OMA) Kulturmedium (2,5 g / L Haferflocken und 7 g / L Agar, pH 6) platziert werden.
- In der Mitte der Petrischale ein Fragment des Kulturmediums (ca. 1 cm2) mit Myzel aus dem gewählten isolierten Pilz für den Keimvorgang beimpfen. Verschließen Sie die Petrischalen mit Frischhaltefolie und brüten Sie sie im Dunkeln bei ca. 25 °C oder Raumtemperatur aus, da dies für das Pilzwachstum besser geeignet ist.
- Bereiten Sie einige Gerichte mit Samen und ohne Pilzimpfung als Negativkontrolle für den Keimtest zu.
- Analysieren Sie die Keimergebnisse wöchentlich, indem Sie quantitative und qualitative Daten sammeln und Protocorms und Sämlinge fotografieren. Die Beobachtung von Samen und Protocormen sollte mit einem Stereomikroskop durchgeführt werden, um die Keimung genauer beurteilen zu können. Verwenden Sie eine Lichtquelle, die von unten kommt, da sie einen größeren Kontrast ermöglicht und es ermöglicht, das Pilzmyzel leichter von Protocormen zu unterscheiden.
- Proben in verschiedenen Entwicklungsstadien sammeln und für anatomische Analysen fixieren (Abschnitt 1). Wenden Sie alle zuvor beschriebenen Bildanalysen an, um Pilzendophyten in Samen, Protocormen und Keimlingen während der Keimung zu untersuchen (Lichtmikroskopie - Abschnitte 4, 5 und 6; konfokal und Fluoreszenz - Abschnitt 7; SEM und TEM - Abschnitte 8 und 9).
- Generieren Sie quantitative Ergebnisse nach der Klassifizierung der Stufen gemäß Tabelle 6. Die Stadien beschreiben die übliche Entwicklung von Samen aus mykoheterotrophen Orchideen. Sammeln Sie wöchentlich Daten und Tabelle mit den Anfangsdaten jeder beobachteten Phase.
- Sammeln Sie außerdem quantitative Daten, die den Prozentsatz und die Rate der Keimung schätzen. Zählen Sie mindestens 100 Samen oder definieren Sie Zählfelder35. Markieren Sie drei oder mehr Zählfelder pro Petrischale, bestehend aus festen Regionen mit einer standardisierten Fläche, und werten Sie wöchentlich aus. Berechnen Sie die gesammelten Daten gemäß der Wachstumsindexgleichung (GI):

wobei N 0 die Anzahl der gezählten Samen in Stufe0 ist, N 1 sich auf Stufe1 bezieht und bis Stufe 6 (registriert als N6) 36 folgt.

Abbildung 3: Schematische Zusammenfassung der Methodik der symbiotischen Keimung von Saatgut. Die Schaltpläne geben Hinweise auf detaillierte Schritte im Protokoll. Abkürzungen: OMA = Haferflocken-Agar, PDA = Kartoffel-Dextrose-Agar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Keimphase | Beschreibung |
| 0 | Keine Keimung |
| 1 | Schwellung des Embryos |
| 2 | Testa-Ruptur |
| 3 | Saugfähige Haare entwickeln sich |
| 4 | Stielprojektion entwickelt sich |
| 5 | Schützende Schuppen (Hochblätter) entwickeln sich |
| 6 | Erste Wurzeln entstehen |
Tabelle 6: Beschreibung der Protocorm-Entwicklungsstadien für periodische Analysen von Keimungstests. Modifiziert von den in Otero et al.36 beschriebenen Stadien.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Nach den wesentlichen Stadien der Fixierung von Pflanzengewebe ergeben Zellstrukturen, die dem Lebenszustand unter Berücksichtigung der Morphologie, des Volumens und der räumlichen Organisation von Zellkomponenten und Geweben so ähnlich wie möglich sind16. Beobachten Sie solche Merkmale in den Proben nach der chemischen Fixierung (Abbildung 4). Abbildung 4C-F stellt ausreichend fixierte Proben unter L...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Bildanalysen in der Pflanzenanatomie und -morphologie haben ein wichtiges Potenzial, Ziele zu erreichen und zum Verständnis der Beziehungen zwischen mykoheterotrophen Pflanzen und ihren unverzichtbaren Pilzendophyten beizutragen, wie Untersuchungen unterirdischer Organe6,40, Strukturanalysen der symbiotischen Keimung von Samen39 sowie Luft- und Fortpflanzungsstrukturen 41 zeigen. . Obwohl die strukturelle Botanik i...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Die Autoren danken der Finanzierung durch FAEPEX und FAPESP (2015/26479-6). MPP dankt Capes für sein Masterstipendium (Prozess 88887.600591/2021-00) und CNPq. JLSM dankt CNPq für Produktivitätszuschüsse (303664/2020-7). Die Autoren danken auch dem Zugang zu Ausrüstung und Unterstützung durch LME (Laboratory of Electron Microscopy - IB / Unicamp), INFABiC (National Institute of Science and Technology on Photonics Applied to Cell Biology - Unicamp) und LaBiVasc (Laboratory of Vascular Biology - DBEF / IB / Unicamp); LAMEB (UFSC) und Eliana de Medeiros Oliveira (UFSC) für Beiträge zum Kryoprotektionsprotokoll; LME für Beiträge zum TEM-Protokoll.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Acetone | Sigma-Aldrich | 179124 | (for SEM stubs mounting) |
| Agar-agar (AA) | Sigma-Aldrich | A1296 | (for seeds germination tests) |
| Calcofluor White Stain | Sigma-Aldrich | 18909 | fluorescent dye (detects cellulose) |
| Citrate Buffer Solution, 0.09M pH 4.8 | Sigma-Aldrich | C2488 | (for toluidine blue O staining) |
| Conductive Double-Sided Carbon Tape | Fisher Scientific | 50-285-81 | (for SEM) |
| Confocal Microscope | Zeiss | (any model) | |
| Copper Grids | Sigma-Aldrich | G4776 | (for TEM) |
| Critical-point dryer | Balzers | (any model) | |
| Cryostat | Leica Biosystems | (any model) | |
| Dissecting microscope | Leica Biosystems | (= stereomicroscope, any model) | |
| Entellan | Sigma-Aldrich | 107960 | rapid mounting medium for microscopy |
| Ethyl alcohol, pure (≥99.5%) | Sigma-Aldrich | 459836 | (= ethanol, for dehydration processes) |
| Formaldehyde solution, 37% | Sigma-Aldrich | 252549 | (for NBF solution preparation) |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128 | histological tissue fixative |
| Gelatin capsules for TEM | Fisher Scientific | 50-248-71 | (for resin polymerisation in TEM) |
| Gelatin solution, 2% in H2O | Sigma-Aldrich | G1393 | (dilute for slides preparation - OCT adherence) |
| Glutaraldehyde solution, 25% | Sigma-Aldrich | G6257 | (for Karnovsky’s solution preparation) |
| HistoResin | Leica Biosystems | 14702231731 | glycol methacrylate (GMA) embedding kit |
| Iodine | Sigma-Aldrich | 207772 | (for Lugol solution preparation) |
| Lead(II) nitrate | Sigma-Aldrich | 228621 | Pb(NO3)2 (for TEM contrast staining) |
| Light Microscope | Olympus | (any model) | |
| LR White acrylic resin | Sigma-Aldrich | L9774 | hydrophilic acrylic resin for TEM |
| Lugol solution | Sigma-Aldrich | 62650 | (for staining) |
| Metal stubs for specimen mounts | Rave Scientific | (for SEM, different models) | |
| Microtome | Leica Biosystems | manual rotary microtome or other model | |
| Oatmeal agar (OMA) | Millipore | O3506 | (for seeds germination tests) |
| OCT Compound, Tissue-Tek | Sakura Finetek USA | 4583 | embedding medium for frozen tissues |
| Osmium tetroxide | Sigma-Aldrich | 201030 | OsO4 (for TEM postfixation) |
| Parafilm M | Sigma-Aldrich | P7793 | sealing thermoplastic film |
| Paraformaldehyde | Sigma-Aldrich | 158127 | (for Karnovsky’s solution preparation) |
| Poly-L-lysine solution, 0.1% in H2O | Sigma-Aldrich | P8920 | (for slides preparation - OCT adherence) |
| Poly-Prep Slides | Sigma-Aldrich | P0425 | poly-L-lysine coated glass slides |
| Polyethylene Molding Cup Trays | Polysciences | 17177A-3 | (6x8x5 mm, for embbeding samples in GMA resin) |
| Polyethylene Molding Cup Trays | Polysciences | 17177C-3 | (13x19x5 mm, for embbeding samples in GMA resin) |
| Potassium iodide | Sigma-Aldrich | 221945 | (for Lugol solution preparation) |
| Potato Dextrose Agar (PDA) | Millipore | 70139 | (for seeds germination tests) |
| Scanning Electron Microscope | Jeol | (any model) | |
| Silane [(3-Aminopropyl)triethoxysilane] | Sigma-Aldrich | A3648 | (for slides preparation - OCT adherence) |
| Silane-Prep Slides | Sigma-Aldrich | S4651 | glass slides coated with silane |
| Silica gel orange, granular | Supelco | 10087 | (for dessicating processes) |
| Sodium cacodylate trihydrate | Sigma-Aldrich | C0250 | (for glutaraldehyde-sodium cacodylate buffer) |
| Sodium hydroxide | Sigma-Aldrich | S5881 | NaOH (for Karnovsky’s solution preparation and TEM contrast staining) |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044 | NaClO (for seeds surface disinfection) |
| Sodium phosphate dibasic, anhydrous | Sigma-Aldrich | 71640 | Na2HPO4 (for NBF solution and PB preparation) |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638 | NaH2PO4·H2O (for NBF and PB) |
| Sputter coater | Balzers | (any model) | |
| Sucrose | Sigma-Aldrich | S0389 | C12H22O11 (for cryoprotection and germination test) |
| Sudan III | Sigma-Aldrich | S4131 | (for staining) |
| Sudan IV | Sigma-Aldrich | 198102 | (for staining) |
| Sudan Black B | Sigma-Aldrich | 199664 | (for staining) |
| Syringe | (3 mL, any brand, for TEM contrast staining) | ||
| Syringe Filter Unit, Millex-GV 0.22 µm | Millipore | SLGV033R | PVDF, 33 mm, gamma sterilized (for TEM contrast staining) |
| Tek Bond Super Glue 793 | Tek Bond Saint-Gobain | 78072720018 | liquid cyanoacrylate adhesive, medium viscosity |
| Toluidine Blue O | Sigma-Aldrich | T3260 | (for staining) |
| Transmission Electron Microscope | Jeol | (any model) | |
| Triphenyltetrazolium chloride | Sigma-Aldrich | T8877 | (for the tetrazolium test in seeds germination) |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804 | Na3(C6H5O7)·2H2O (for TEM contrast staining) |
| Ultramicrotome | Leica Biosystems | (any model) | |
| Uranyl acetate | Fisher Scientific | 18-607-645 | UO2(CH3COO)2 (for TEM contrast staining) |
| Vacuum pump | (any model) | ||
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | TermoFisher Scientific | W11261 | fluorescent dye-conjugated lectin (detects sialic acid and N-acetylglucosaminyl residues) |
Referenzen
- Evert, R. F. Esau’s Plant Anatomy: Meristems, Cells, and Tissues of the Plant Body: Their Structure, Function, and Development. , John Wiley & Sons. Hoboken, NJ, USA. (2006).
- Yeung, E. C. T., Stasolla, C., Sumner, M. J., Huang, B. Q. Plant Microtechniques and Protocols. , Springer International Publishing. Cham, Switzerland. (2015).
- Sokoloff, D. D., Jura-Morawiec, J., Zoric, L., Fay, M. F. Plant anatomy: at the heart of modern botany. Botanical Journal of the Linnean Society. 195 (3), 249-253 (2021).
- Leake, J. R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytologist. 127 (2), 171-216 (1994).
- Bidartondo, M. I. The evolutionary ecology of myco-heterotrophy. New Phytologist. 167 (2), 335-352 (2005).
- Imhof, S., Massicotte, H. B., Melville, L. H., Peterson, R. L. Subterranean morphology and mycorrhizal structures. Mycoheterotrophy. , Springer. New York, NY. 157-214 (2013).
- Rasmussen, H. N., Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos. 118 (3), 334-345 (2009).
- Rasmussen, H. N., Dixon, K. W., Jersáková, J., Těšitelová, T. Germination and seedling establishment in orchids: a complex of requirements. Annals of Botany. 116 (3), 391-402 (2015).
- Zettler, L. W. Terrestrial orchid conservation by symbiotic seed germination: techniques and perspectives. Selbyana. 18 (2), 188-194 (1997).
- Stewart, S. L., Kane, M. E. Symbiotic seed germination and evidence for in vitro mycobiont specificity in Spiranthes brevilabris (Orchidaceae) and its implications for species-level conservation. In Vitro Cellular & Developmental Biology - Plant. 43 (3), 178-186 (2007).
- Zhao, D. -K., et al. Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Frontiers in Plant Science. 12, 701152(2021).
- Selosse, M. A., Roy, M. Green plants that feed on fungi: facts and questions about mixotrophy. Trends in Plant Science. 14 (2), 64-70 (2009).
- Merckx, V. S. F. T., Mennes, C. B., Peay, K. G., Geml, J. Evolution and diversification. Mycoheterotrophy: The Biology of Plants Living on Fungi. , Springer. New York, NY. 215-244 (2013).
- Boon, M. E., Drijver, J. Routine Cytological Staining Techniques: Theoretical Background and Practice. Macmillan International Higher Education. , Hampshire, UK. (1986).
- Karnovsky, M. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. Journal of Cell Biology. 27 (2), 137-138 (1964).
- Hayat, M. Fixation for Electron Microscopy. , Academic Press. New York, USA. (1981).
- Roland, J. C., Vian, B. General preparation and staining of thin sections. Electron Microscopy of Plant Cells. 1, 675(1991).
- Gerrits, P. O., Horobin, R. W. Glycol methacrylate embedding for light microscopy: basic principles and trouble-shooting. Journal of Histotechnology. 19 (4), 297-311 (1996).
- Zhang, Z., Niu, L., Chen, X., Xu, X., Ru, Z. Improvement of plant cryosection. Frontiers in Biology. 7 (4), 374-377 (2012).
- BeneŠ, K. On the media improving freeze-sectioning of plant material. Biologia Plantarum. 15 (1), 50-56 (1973).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. Cold Spring Harbor Protocols. 2008 (5), (2008).
- Sakai, W. S. Simple method for differential staining of paraffin embedded plant material using toluidine blue O. Stain Technology. 48 (5), 247-249 (1973).
- O’Brien, T., Feder, N., McCully, M. E. Polychromatic staining of plant cell walls by toluidine blue O. Protoplasma. 59 (2), 368-373 (1964).
- Ventrella, M. C., Almeida, A. L., Nery, L. A., Coelho, V. P. deM. Métodos Histoquímicos Aplicados às Sementes. Universidade Federal de Viçosa. , Viçosa, Brazil. (2013).
- Pearse, A. G. E. Histochemistry, Theoretical and Applied. , J & A Churchill. London, UK. (1960).
- Andrade-Linares, D. R., Franken, P. Fungal endophytes in plant roots: taxonomy, colonization patterns, and functions. Symbiotic Endophytes. , Springer. Berlin. 311-334 (2013).
- Wymer, C. L., Beven, A. F., Boudonck, K., Lloyd, C. W. Confocal microscopy of plant cells. Confocal Microscopy Methods and Protocols. , Humana Press. Totowa. 103-130 (1999).
- Marques, J. P. R., Soares, M. K. M. Manual de Técnicas Aplicadas à Histopatologia Vegetal. FEALQ. , Piracicaba, Brazil. (2021).
- Navarro, B. L., Marques, J. P. R., Appezzato-da-Glória, B., Spósito, M. B. Histopathology of Phakopsora euvitis on Vitis vinifera. European Journal of Plant Pathology. 154 (4), 1185-1193 (2019).
- Marques, J. P. R., et al. Sugarcane cell wall-associated defense responses to infection by Sporisorium scitamineum. Frontiers in Plant Science. 9, 698(2018).
- Jeffree, C. E., Read, N. D. Ambient-and low-temperature scanning electron microscopy. Electron Microscopy of Plant Cells. , 313-413 (1991).
- Bozzola, J. J., Russell, L. D. Electron Microscopy: Principles and Techniques for Biologists. , Jones & Bartlett Learning. Sudbury, MA, USA. (1999).
- Murray, S. Basic transmission and scanning electron microscopy. Introduction to electron Microscopy for Biologists. , Academic Press. 3-18 (2008).
- Tanaka, M. Glossary of TEM terms. , Available from: https://www.jeol.co.jp/en/words/emterms/ (2021).
- Seaton, P. T., et al. Orchid seed and pollen: a toolkit for long-term storage, viability assessment and conservation. Orchid Propagation: From Laboratories to Greenhouses—Methods and Protocols. , Humana Press. New York, NY. 71-98 (2018).
- Otero, J. T., Ackerman, J. D., Bayman, P. Differences in mycorrhizal preferences between two tropical orchids. Molecular Ecology. 13 (8), 2393-2404 (2004).
- Koch, R. A., et al. Marasmioid rhizomorphs in bird nests: Species diversity, functional specificity, and new species from the tropics. Mycologia. 112 (6), 1086-1103 (2020).
- Webster, J., Weber, R. Introduction to Fungi. , Cambridge University Press. Cambridge, UK. (2007).
- Sisti, L. S., et al. The role of non-mycorrhizal fungi in germination of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. Frontiers in Plant Science. 10, 1589(2019).
- Martos, F., et al. Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytologist. 184 (3), 668-681 (2009).
- Alves, M. F., et al. Reproductive development and genetic structure of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. BMC Plant Biology. 21 (1), 332(2021).
- Merckx, V. S. F. T. Mycoheterotrophy: an introduction. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 1-17 (2013).
- Hall, J. L., Hawes, C. Electron Microscopy of Plant Cells. , Academic Press. Cambridge, UK. (1991).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten