
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Tecniche di microscopia per l'interpretazione della colonizzazione fungina nei tessuti delle piante micoeterotrofiche e della germinazione simbiotica dei semi
In questo articolo
Riepilogo
Questo protocollo mira a fornire procedure dettagliate per la raccolta, la fissazione e il mantenimento di campioni di piante micoeterotrofiche, applicando diverse tecniche di microscopia come la microscopia elettronica a scansione e trasmissione, la microscopia ottica, confocale e a fluorescenza per studiare la colonizzazione fungina nei tessuti delle piante e nei semi germinati con funghi micorrizici.
Abstract
La botanica strutturale è una prospettiva indispensabile per comprendere appieno l'ecologia, la fisiologia, lo sviluppo e l'evoluzione delle piante. Quando si studiano piante micoeterotrofiche (cioè piante che ottengono carbonio dai funghi), aspetti notevoli dei loro adattamenti strutturali, i modelli di colonizzazione dei tessuti da parte dei funghi e la morfoanatomia degli organi sotterranei possono illuminare le loro strategie di sviluppo e le loro relazioni con le ife, la fonte di nutrienti. Un altro ruolo importante dei funghi simbionti è legato alla germinazione dei semi di orchidea; tutte le specie di Orchidaceae sono micoeterotrofiche durante la germinazione e la fase di semina (micoeterotrofia iniziale), anche quelle che fotosintetizzano negli stadi adulti. A causa della mancanza di riserve nutrizionali nei semi di orchidee, i simbionti fungini sono essenziali per fornire substrati e consentire la germinazione. L'analisi delle fasi di germinazione da prospettive strutturali può anche rispondere a domande importanti riguardanti l'interazione dei funghi con i semi. Diverse tecniche di imaging possono essere applicate per svelare gli endofiti dei funghi nei tessuti vegetali, come proposto in questo articolo. Le sezioni a mano libera e sottili degli organi vegetali possono essere colorate e quindi osservate con microscopia ottica. Un fluorocromo coniugato all'agglutinina del germe di grano può essere applicato ai funghi e co-incubato con Calcofluor White per evidenziare le pareti cellulari delle piante in microscopia confocale. Inoltre, le metodologie di scansione e microscopia elettronica a trasmissione sono dettagliate per orchidee micoeterotrofe e vengono esplorate le possibilità di applicare tali protocolli in piante correlate. La germinazione simbiotica dei semi di orchidea (cioè in presenza di funghi micorrizici) è descritta nel protocollo in dettaglio, insieme alle possibilità di preparare le strutture ottenute da diversi stadi di germinazione per analisi con microscopia ottica, confocale ed elettronica.
Introduzione
La ricerca strutturale in botanica, che copre la morfologia e l'anatomia delle piante, è fondamentale per comprendere l'intero organismo1,2 e fornisce prospettive indispensabili per integrare e contribuire alla conoscenza riguardante l'ecologia, la fisiologia, lo sviluppo e l'evoluzione delle piante3. I metodi in morfologia e anatomia vegetale comprendono attualmente protocolli, attrezzature e conoscenze sviluppate di recente e più di un secolo fa2. L'esecuzione e l'adattamento continui di metodi classici (ad esempio, microscopia ottica) insieme a tecniche più recenti (ad esempio, microscopia confocale, microtomografia a raggi X) hanno la stessa base essenziale: conoscenze teoriche che consentono lo sviluppo di una metodologia.
Lo strumento principale nell'anatomia e nella morfologia delle piante è l'immagine. Nonostante l'idea errata che tali analisi siano semplici osservazioni, dando spazio a interpretazioni soggettive2, l'analisi e la comprensione delle immagini in quest'area richiede la conoscenza dei metodi applicati (l'attrezzatura, il tipo di analisi, le procedure metodologiche), i componenti cellulari, l'istochimica e il corpo vegetale (organizzazione e funzione dei tessuti, ontogenesi, adattamenti morfologici). L'interpretazione delle immagini ottenute attraverso una varietà di metodi può portare a correlare forma e funzione, decifrare la composizione chimica di una struttura, corroborare nella descrizione dei taxa, comprendere le infezioni da fitopatogeni e altre valutazioni simili.
Quando si studiano piante micoeterotrofe (MH) (cioè piante non fotosintetiche che ottengono carbonio dai funghi micorrizici4,5), aspetti notevoli dei loro adattamenti strutturali, i modelli di colonizzazione dei tessuti da parte dei funghi e la morfoanatomia degli organi sotterranei possono illuminare le loro strategie di sviluppo e le relazioni con le ife, che sono la fonte di nutrienti. Gli organi sotterranei delle piante MH di solito mostrano importanti adattamenti legati alla loro associazione con i funghi del suolo, quindi è essenziale eseguire queste indagini anatomiche e morfologiche6. Gli organi aerei delle specie MH non devono essere ignorati, poiché gli endofiti possono essere presenti anche in questi tessuti, anche se non sono funghi micorrizici (osservazioni personali, non ancora pubblicate).
Oltre alla consolidata essenzialità dell'associazione dei funghi micorrizici con le specie MH durante il loro intero ciclo vitale7, ogni specie di orchidea, anche quelle autotrofe, ha un primo stadio micoeterotrofico obbligato in ambienti naturali. Si verifica perché l'embrione delle orchidee è indifferenziato e manca di un endosperma o di cotiledoni, essendo quindi incapace di svilupparsi e stabilirsi in ambienti naturali senza il supporto nutrizionale di partner fungini 4,8. Considerando che, i protocolli di germinazione simbiotica possono essere applicati non solo alle specie MH ma anche alle orchidee fotosintetizzanti, con l'obiettivo di indagare la specificità orchidea-fungo nella germinazione e nello sviluppo del protocormo, una metodologia ampiamente applicata nelle iniziative per la conservazione delle specie minacciate 9,10,11.
In questo assemblaggio di metodi, descriviamo importanti passaggi coinvolti nella raccolta, fissazione e conservazione di campioni di piante MH per studi anatomici (sezione 1), analisi superficiale e selezione dei campioni (sezione 2), metodi di sezionamento (mano libera: sezione 3, microtomia: sezione 4, criomicrotomia: sezione 5), colorazione e montaggio (sezione 6), fluorescenza e microscopia confocale di endofiti fungini (sezione 7), microscopia elettronica a scansione (sezione 8), e microscopia elettronica a trasmissione (sezione 9). Inoltre, descriviamo un metodo di germinazione simbiotica per i semi di orchidea (MH e autotrofico, sezione 10), poiché i metodi di imaging precedentemente menzionati possono essere applicati con successo per analizzare la colonizzazione fungina di semi, protocormi e piantine nel processo di germinazione.

Figura 1: Riepilogo schematico dei metodi di imaging. Gli schemi forniscono indicazioni sui passaggi del protocollo in cui sono dettagliati. Abbreviazioni: GMA = glicole metacrilato, OCT = composto con temperatura di taglio ottimale, SEM = microscopia elettronica a scansione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
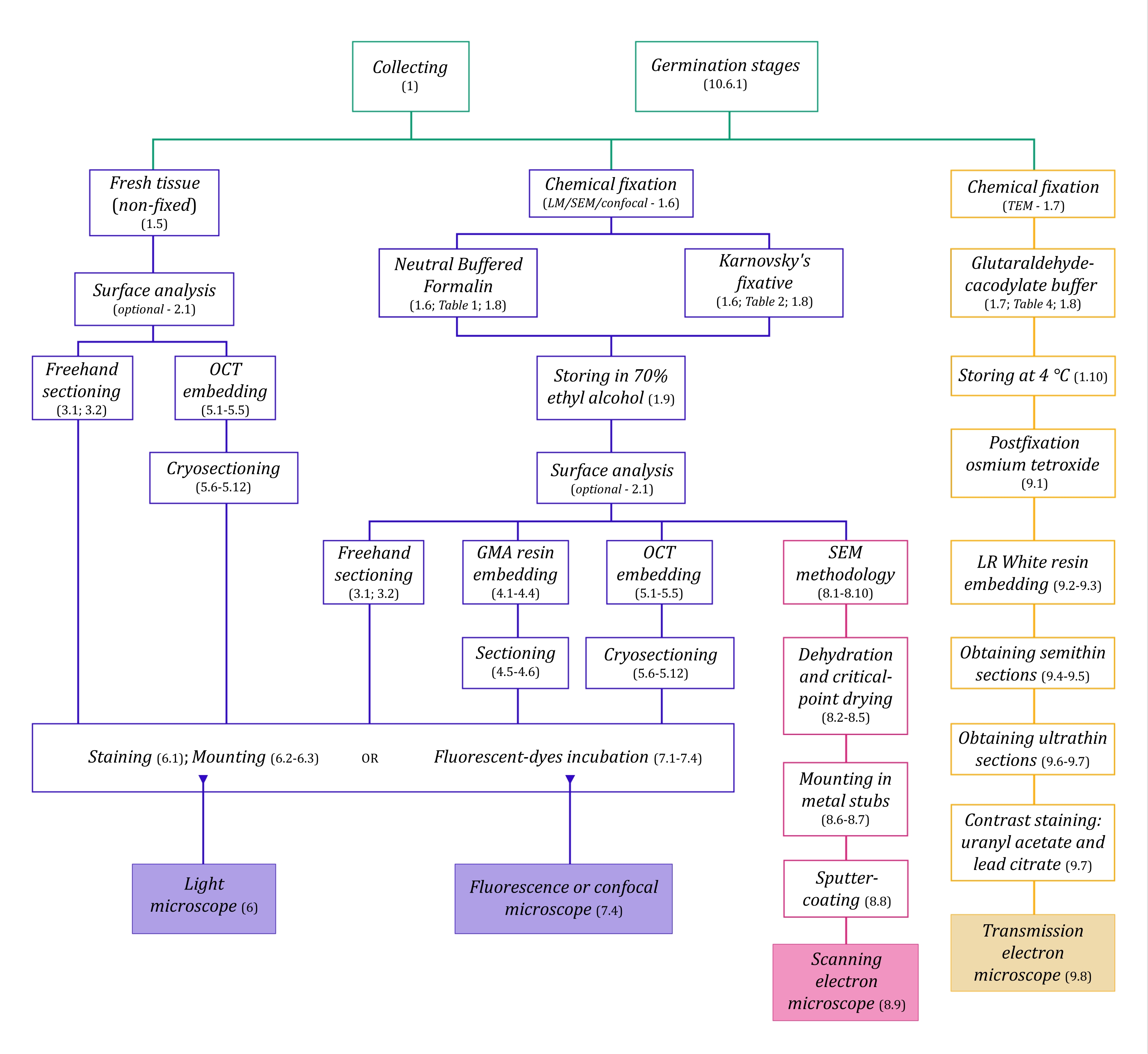{kind=link}
Le tecniche di microscopia qui descritte in dettaglio (Figura 1) sono precedute dalle seguenti fasi essenziali: raccolta, fissaggio, disidratazione, incorporamento e sezionamento dei campioni. Poiché le fasi sono variabili (Figura 1) a seconda della tecnica o delle tecniche scelte, è importante pensare in anticipo, considerando i fissativi da preparare e trasportare al sito di raccolta, come i campioni devono essere preparati prima del fissaggio, i processi di disidratazione da utilizzare (sezione 1) e le diverse possibilità di incorporamento e metodi di sezionamento (sezioni 4, 5, e 9). La Figura 1 riassume sequenzialmente tutti i passaggi necessari per ciascuna tecnica di microscopia descritti dettagliatamente di seguito.
Protocollo
1. Raccolta, fissaggio e conservazione dei campioni
NOTA: Le piante completamente MH si trovano di solito nel sottobosco della foresta oscura 12,13, principalmente in aree umide e abbondanti di rifiuti, mentre le piante parzialmente MH possono essere trovate nelle foreste più aperte12,13. Le piante MH di solito hanno organi sotterranei ben sviluppati in una varietà di forme e dimensioni.
- Quando raccogli le specie MH, esplora il terreno intorno alla base vegetale, facendo attenzione a non danneggiare gli organi sotterranei ed evita di estrarre le piante da terra per evitare di scollegare gli organi aerei da quelli sotterranei.
- Scava attentamente intorno alle strutture aeree usando una cazzuola da giardinaggio mentre esplori gli organi sotterranei come radici, steli, rizomi e organi di conservazione, senza danneggiare queste strutture.
- Rimuovere le particelle di terreno per preservare le strutture fragili e lavare delicatamente questi organi con acqua di rubinetto per sciacquare via le particelle di terreno rimanenti prima di fissare i campioni.
- Le piante MH associate alla lettiera di foglie richiedono un'attenzione particolare; Raccogliere con cura gli organi collegati al materiale in decomposizione attraverso le loro ife, evitare di estrarre questi organi dalle strutture collegate e raccoglierli con cura poiché queste parti sono molto delicate. Preservare le strutture con tali connessioni e raccogliere anche i rifiuti per l'analisi.
- Se si sceglie di analizzare materiale fresco utilizzando tecniche di imaging, mantenere i campioni in sacchetti di plastica chiusi con umidità adeguata, abbastanza acqua che evapora e idrata la pianta, evitando che l'acqua eccessiva venga a contatto con i campioni. Trasportarli immediatamente in laboratorio e analizzare i campioni lo stesso giorno in cui sono stati raccolti, prestando attenzione se i campioni sono ancora conservati durante l'analisi.
- Portare i fissativi al sito di raccolta in contenitori ben sigillati. Fissare rapidamente i campioni dopo la raccolta per microscopia ottica (LM) e microscopia elettronica a scansione (SEM) in uno dei seguenti fissativi: formalina tamponata neutra al 10% (NBF14, Tabella 1) o soluzione di Karnovsky (modificata15, Tabella 2). La soluzione di Karnovsky può essere preparata con tampone fosfato 0,2 M15, la cui ricetta è descritta nella Tabella 3.
- Per l'analisi mediante microscopia elettronica a trasmissione (TEM), sezionare i campioni con uno spessore di 4-3 mm all'interno di una goccia di tampone cacodilato glutaraldeide-sodico (modificato16, Tabella 4) in sezioni più piccole di 1-2 mm di spessore. Scartare i bordi tagliati all'esterno della goccia. Trasferire immediatamente le sezioni in una provetta di raccolta con un volume di fissativo più di 10 volte maggiore del volume dei campioni, in quanto è un fissativo additivo (cioè, le sue molecole vengono aggiunte chimicamente alle proteine fissate16).
ATTENZIONE: I tre fissativi descritti sono altamente tossici. Evitare l'inalazione, soprattutto durante il loro uso sul campo. Preparare tutti i fissativi in una cappa aspirante usando i guanti. Non mescolare cacodilato e acidi, poiché si può formare gas arsenico16. - Se i campioni fissi galleggiano nel fissativo, ciò indica la presenza di gas nei tessuti vegetali. L'aria e gli altri gas impediscono al fissativo di penetrare nell'intero campione2. Prelevare il gas dai tessuti ricampionandoli in parti più piccole e utilizzando una pompa per vuoto (pressione da -300 a -400 inHg) fino a quando tutti i campioni affondano sul fondo della soluzione17. Prestare attenzione poiché una pressione eccessiva esercitata dalla pompa può danneggiare i campioni.
- Dopo almeno 48 ore nella soluzione di Karnovsky o al 10% di NBF, lavare i campioni in 0,2 M PB (Tabella 3) e disidratarli con una serie di etanolo al 10%, 30%, 50% e infine 70%. Per campioni delicati, disidratare per 30 minuti in ogni concentrazione; Per campioni più grandi, disidratare per 1 ora o più.
NOTA: Una soluzione di etanolo al 70% è il mezzo ideale per la conservazione dei campioni. I campioni in etanolo al 70% possono essere conservati a temperatura ambiente per anni. Non conservare materiale vegetale per lunghi periodi nei fissativi, poiché la rimozione degli agenti fissanti è un passaggio essenziale dopo la fissazione2. - Conservare i campioni in glutaraldeide-cacodilato a 4 °C prima di procedere alla postfissazione (fase 9.1).
| 10% formalina tamponata neutra (NBF)14 | |
| Passo 1 | aggiungere 10 ml di soluzione di formaldeide al 37-40% in 80 ml di acqua distillata |
| Passo 2 | aggiungere 0,4 g di fosfato di sodio monobasico monoidrato (NaH2PO4· H2O) alla soluzione |
| Passo 3 | aggiungere 0,65 g di sodio fosfato bibasico, anidro (Na2HPO4) |
| Passo 4 | portare il volume a 100 mL |
Tabella 1: Ricetta di formalina tamponata neutra al 10% 14.
| Soluzione di Karnovsky (modificata15) | |
| Passo 1 | in 20 mL di acqua distillata a 60-70 °C |
| Passo 2 | aggiungere 0,8 g di paraformaldeide (per ottenere il 4% p/v), mescolando |
| Passo 3 | aggiungere 1-4 gocce di NaOH al 40% e mescolare fino a quando la soluzione diventa limpida |
| Passo 4 | raffreddare e aggiungere 30 ml di tampone fosfato 0,2 M pH 7,2 (Tabella 3) |
| Passo 5 | diluire il 25% di glutaraldeide in 0,1 M PB (pH 7,2) per ottenere l'1% di glutaraldeide (volume finale: ~60 ml) |
| Passo 6 | aggiungere l'1% di glutaraldeide (fase 5) alla soluzione ottenuta al punto 4 fino a ottenere fino a 100 ml di fissativo |
Tabella 2: Ricetta della soluzione di Karnovsky (modificata15).
| Tampone fosfato 0,2 M (PB) pH 7,2 | |
| Passo 1 | aggiungere 14,196 g di sodio fosfato bibasico, anidro (Na2HPO4) a 400 mL di acqua distillata |
| Passo 2 | aggiungere 13,8 g di fosfato di sodio monobasico monoidrato (NaH2PO4· H2O) |
| Passo 3 | Mescolare fino a quando la soluzione è limpida |
| Passo 4 | regolare il volume finale a 500 ml con acqua distillata |
| Passo 5 | regolare il pH a 7,2 |
| Passo 6 | per un PB da 0,1 M, diluire 1:1 |
Tabella 3: Ricetta tampone fosfato 0,2 M.
| 3% glutaraldeide 0,2 M tampone cacodilato di sodio (modificato16) | |
| Passo 1 | Tampone cacodilato 0,2 M: aggiungere 4,28 g di cacodilato di sodio triidrato in 100 ml di acqua distillata |
| Passo 2 | regolare il pH a 7,2 |
| Passo 3 | aggiungere 12 mL di glutaraldeide al 25% in 25 mL della soluzione nella fase 2 (tampone cacodilato 0,2 M pH 7,2) |
| Passo 4 | portare il volume a 100 ml con acqua distillata |
Tabella 4: 3% glutaraldeide 0,2 M di tampone di cacodilato di sodio ricetta (modificata16).
2. Analisi superficiale di organi in materiale fisso e non fisso
- Per analizzare le ife superficiali negli organi, soprattutto quelli sotterranei, e quelli a contatto con la lettiera di foglie, osservare materiale fisso o fresco in un microscopio da dissezione (stereomicroscopio) ad un ingrandimento di 7,5x o superiore, a seconda dei campioni analizzati.
- Visualizza i campioni immersi nel fissativo, etanolo al 70% (se conservato in esso) o acqua di rubinetto nel caso di materiale fresco. Evitare la luce diretta dal microscopio da dissezione in quanto può asciugare e danneggiare i campioni.
- Ricerca delle aree di interesse nei campioni, guidata dalle ife superficiali e dai rizomorfi. Selezionare campioni che contengono aree con rizomorfi superficiali in quanto questi possono essere sezionati per visualizzare pelotoni e bobine ife all'interno delle cellule corticali nelle radici e negli steli.
- Dopo la selezione, seguire i passaggi 1.6 e 1.9 se i campioni non sono ancora stati corretti. Se lo si desidera, fotografare campioni freschi utilizzando un microscopio ottico senza fissarli, come descritto nella sezione 3.
- Utilizzare la fotocamera accoppiata allo stereomicroscopio per raccogliere immagini dalle superfici degli organi, dai rizomorfi e da altre strutture osservate. In questi casi, disporre un colore di sfondo adeguato per contrastare bene con il materiale e, se possibile, scegliere un materiale di sfondo con una superficie meno ruvida (ad esempio, carta).
3. Sezioni a mano libera di organi vegetali
NOTA: le sezioni a mano libera degli organi vegetali possono essere impegnative, specialmente per le strutture piccole e sottili. Tuttavia, queste sezioni di tessuti con endofiti fungini possono, in alcuni casi, mostrare meglio ife e altre caratteristiche rispetto alle sezioni sottili.
- Sezionare campioni freschi o fissi con una lama affilata, tagliandoli il più sottile possibile e ponendoli immediatamente in una piccola capsula di Petri con acqua (se fresca) o etanolo al 70% (se fissa). Utilizzare un piccolo pennello per manipolare le sezioni senza danneggiarle.
- Per facilitare il sezionamento di materiali più difficili (cioè organi piccoli, sottili e flessibili), circondare il campione in una struttura, ad esempio polistirene o cecropia picciolo. Intagliare il supporto per ospitare il campione e creare una sezione sottile del campione e del supporto del tutto.
- Colorare e montare i campioni come descritto nella sezione 6.
4. Incorporazione di campioni di piante in resina e sezionamento
- Disidratare ulteriormente i campioni conservati in etanolo al 70% in 80%, 96% e 2x in etanolo al 100%, per 30 minuti a 2 ore a seconda delle dimensioni e della composizione dei campioni.
- Utilizzare un kit di resina glicole metacrilato (GMA) secondo le istruzioni del produttore. Controllare Gerrits e Horobin (1996)18 per ulteriori considerazioni. Seguire di conseguenza i passaggi di infiltrazione e incorporamento.
ATTENZIONE: La resina GMA è tossica, può causare reazioni allergiche e irritazione della pelle, degli occhi e delle mucose. Utilizzare i reagenti in una cappa aspirante e utilizzare i guanti. - Utilizzare vassoi di stampaggio in polietilene per l'incorporamento, selezionati in base alle dimensioni dei campioni (ad esempio, 13 mm x 19 mm x 5 mm per campioni più grandi, 6 mm x 8 mm x 5 mm per quelli più piccoli). Prestare attenzione all'orientamento del campione desiderato all'interno dello stampo e utilizzare un ago per orientarsi.
- Lasciare per la polimerizzazione, preferibilmente a temperatura ambiente, fino a completa solidificazione. Il processo di indurimento richiede solitamente alcune ore, anche se si consiglia di sformare i blocchi il giorno seguente. Dopo la polimerizzazione, staccare con cura i blocchi di resina dagli stampi e procedere ad attaccare i blocchi il prima possibile per evitare la curvatura dei blocchi.
- Carteggiare la faccia del blocco di resina che verrà attaccato, creando una superficie piana. Incollare il blocco di resina su un cuboide di legno (consigliato 2 cm x 2 cm x 3 cm) con un adesivo cianoacrilato liquido di media viscosità (vedi Tabella dei materiali). Assicurarsi che la resina sia completamente attaccata per evitare di compromettere il sezionamento.
- Eseguire il sezionamento in un microtomo rotante come descritto di seguito.
ATTENZIONE: Le lame dei coltelli microtomo sono molto affilate e possono causare incidenti. Assicurati di gestirli seguendo tutte le misure di sicurezza. Prima di avvicinarsi al coltello (ad esempio, per cambiare il blocco, per inumidire la resina), bloccare il volantino grossolano e posizionare il coperchio di sicurezza della lama. Conservare le lame monouso in casi appropriati. Prestare estrema attenzione quando si sostituiscono le lame.
NOTA: Diversi tipi di coltelli (ad esempio, monouso o fissi; vetro o acciaio) possono essere utilizzati per la sezione della resina GMA18. La qualità delle sezioni dipende da quanto è affilato il coltello. Assicurati che il coltello sia ben attaccato e non possa muoversi. Potrebbe essere necessario cambiare regolarmente i coltelli usa e getta per ottenere un migliore sezionamento.- Attaccare saldamente il cuboide di legno al supporto del blocco. Regolare l'orientamento del sezionamento utilizzando le viti di orientamento e garantire un angolo adeguato del coltello utilizzando l'inclinazione del coltello. Selezionare lo spessore delle sezioni; utilizzare un'impostazione più spessa per il taglio e un'impostazione più sottile per le sezioni selezionate, poiché le sezioni GMA aderiscono in modo più appropriato al vetrino quando sono più sottili; Lo spessore suggerito per i tessuti vegetali è 5-8 μm.
- Prima di iniziare, raffreddare la stanza, se necessario, poiché temperature più elevate peggiorano la qualità delle sezioni. Preparare quanto segue per il sezionamento: un becher con acqua distillata, una piastra calda, pennelli (almeno due), pinzette a punta fine, una pipetta Pasteur, vetrini, carta da filtro (o carta velina) e una matita (per identificare i campioni da sezionare).
- Regolare la piastra riscaldante a 50 °C e posizionare il becher su di essa. Prestare attenzione alle differenze di riscaldamento a seconda dell'area della piastra riscaldante (di solito, l'area centrale riscalda più dei bordi; riscaldare l'acqua nel mezzo, preferibilmente).
- Scegli un vetrino, identificalo con una matita e pipetta l'acqua distillata calda su tutta la superficie del vetrino. Se necessario, utilizzare una soluzione (ad esempio, detergente e acqua, o etanolo al 70%) per rompere la tensione tra l'acqua e il vetro, in modo che l'intero vetrino sia ugualmente coperto. Alcuni tipi di vetrini devono essere pre-puliti con etanolo al 70% per ottenere un'adeguata aderenza alla sezione.
- Inizia facendo avanzare gradualmente la faccia del blocco di resina verso la lama del coltello. Non provare a sezionare senza prima far avanzare il blocco, altrimenti l'attrezzatura e il blocco potrebbero essere danneggiati. Se necessario, tagliare il blocco utilizzando uno spessore maggiore (10 μm e oltre).
- Quando ci si avvicina a una sezione adatta, fare un movimento deciso a senso unico con il volantino, in modo che la sezione sia fatta contemporaneamente. Tenere sotto controllo l'umidità della resina. Durante il sezionamento, idratare regolarmente la faccia del blocco da tagliare usando un pennello immerso in acqua distillata se c'è un problema con le sezioni di arricciatura. Rimuovere l'acqua in eccesso con una carta velina.
- Con un paio di pinzette a punta fine, posizionare la sezione ottenuta nell'acqua sopra il vetrino. A contatto con l'acqua, la resina GMA si allunga. Se necessario, utilizzare un pennello per aprire delicatamente e allungare le sezioni. Utilizzare un altro pennello per mantenere costantemente la lama libera da detriti di resina. Non passare da un pennello all'altro per evitare di bagnare la lama.
- Dopo aver messo in coda tutte le sezioni desiderate nella diapositiva, asciugare la faccia inferiore della diapositiva e posizionarla sopra la piastra riscaldante. Rimuovere l'acqua in eccesso dalla parte superiore del vetrino tamponando delicatamente con una carta da filtro (opzionale). Le sezioni aderiranno quando l'acqua evapora dallo scivolo. Non lasciare le guide troppo a lungo per evitare che le sezioni vengano danneggiate dal calore eccessivo.
- Conservare i vetrini in una scatola di scorrimento, lontano dalla polvere e dal sole, e usarli per la colorazione e altre procedure. Le diapositive possono essere conservate per diversi anni.
5. Congelamento di campioni di piante e sezionamento con criostato
NOTA: La considerazione essenziale nella criosezione del tessuto biologico è quella di ridurre i danni dovuti alla formazione di cristalli di ghiaccio durante il congelamento dei campioni. La crioprotezione viene solitamente eseguita infondendo soluzioni chimicamente inerti come glicerolo o saccarosio 19,20.
- Un giorno prima del sezionamento del campione, attenersi alla seguente procedura.
- Diluire 100 mL di 0,2 M PB (Tabella 3) in 100 mL di acqua distillata per ottenere 200 mL di 0,1 M PB. Preparare soluzioni di saccarosio al 10%, 20% e 30% in 0,1 M PB (ad esempio, per una soluzione al 10%, aggiungere 2 g di saccarosio in 20 ml di tampone).
- Per i campioni freschi, lavarli in 0,1 M PB per 30 minuti. Per i campioni in un fissativo, lavarli nello stesso tampone utilizzato per preparare il fissativo per 30 minuti. Per i campioni in etanolo al 70%, idratarli in etanolo al 50% e al 30% e lavare in 0,1 M PB per 1 ora in ciascuna soluzione.
- Incubare i campioni per 2 ore in 10% di saccarosio, 2 ore in 20% di saccarosio e 2 ore in 30% di saccarosio, a temperatura ambiente. Successivamente, incubare per una notte in saccarosio al 30% a 4 °C (o almeno per 3 ore; il tempo massimo è di 48 ore).
- Il giorno del sezionamento, preparare il 40% e il 50% di saccarosio in 0,1 M PB; Non preparare soluzioni di saccarosio con più di 12 ore di anticipo. Incubare per 2 ore in ciascuna concentrazione di saccarosio, a 4 °C.
- Per l'incorporazione, in piccoli stampi, fare uno strato di composto OCT (mezzo di temperatura di taglio ottimale, utilizzato per l'incorporazione e il congelamento dei campioni) e mantenerlo a -20 °C per congelare. Gli stampi possono essere stampi istologici regolari, anche se per facilitare lo sformatura dei blocchi, quelli di carta o carta stagnola possono essere realizzati utilizzando un piccolo cuboide come cornice e nastro adesivo.
- Dopo che lo strato inferiore del composto OCT è congelato negli stampi, lavorare all'interno di una camera di criostato (ca. -27 °C). Posizionare i campioni incubati al 50% di saccarosio negli stampi, nell'orientamento in cui verranno sezionati. La faccia superiore di un blocco cuboide è di solito una faccia migliore del sezionamento. Segnare nello stampo dove sono posizionati i campioni, in modo che il blocco possa essere facilmente tagliato e mantenuto l'orientamento corretto.
- Circondare i campioni nel composto OCT e far scoppiare qualsiasi bolla d'aria che tocca i campioni. Congelarli a -20 °C. Con i blocchi completamente congelati, posizionarli all'interno della camera del criostato (ca. -27 °C). Sformare ciascuno solo prima dell'uso e prestare attenzione ai segni che indicano la posizione del campione all'interno del blocco.
- Poiché il composto OCT è facilmente tagliabile con una lama, tagliare opportunamente i blocchi prima di posizionarli sui mandrini. Inserire un composto OCT nel mandrino del criostato e posizionare il blocco in modo che la faccia superiore sia sezionata. Le facce con aree più piccole forniscono sezioni migliori. Attendere che il blocco sia ben attaccato al mandrino e testarlo prima di iniziare la sezione.
- Posizionare saldamente il mandrino nel supporto del mandrino. Regolate l'orientamento del sezionamento utilizzando le viti di orientamento. Regolare l'angolo del coltello usando l'inclinazione del coltello. Selezionate lo spessore delle sezioni. I campioni possono essere sezionati più spessi delle normali sezioni di resina. Le sezioni in un intervallo compreso tra 5-20 μm sono ottenute con successo, con sezioni più spesse più facili da realizzare (meno arricciature e meno danni alle strutture).
- Avanzare la faccia del blocco congelato verso la lama del coltello. Non provare a sezionare senza farlo, altrimenti il blocco può essere staccato dal mandrino e danneggiato. Se necessario, tagliare il blocco utilizzando uno spessore maggiore (10 μm e oltre).
- Quando ci si avvicina a una sezione adatta, posizionare la piastra antirollio (cioè una piastra trasparente che manterrà la sezione) sopra il coltello e fare un movimento deciso a senso unico con il volantino, in modo che la sezione sia fatta contemporaneamente. I problemi di arricciatura possono essere causati se la piastra antirollio deve essere regolata (di solito è regolabile in riferimento alla lama) o se ci sono detriti nella lama. Pulire costantemente la lama con un pennello per rimuovere i detriti.
- Utilizzare vetrini speciali in modo che le sezioni si attacchino facilmente, come vetrini silanizzati (commerciali o preparati con aminoalchilsilano al 2% in acetone 21), o vetrini preparati con 500 μg/ml di poli-L-lisina in acqua distillata 21 o gelatina allo 0,2% (vedi dettagli21). Mantenere i vetrini a temperatura ambiente.
- Per far aderire la sezione a una slitta, sollevare la piastra antirollio e far toccare rapidamente la sezione alla slitta. Poiché il vetrino è a temperatura ambiente, la sezione si scioglie immediatamente e aderisce al vetrino. Prestare attenzione a ruotare la faccia trattata della diapositiva verso la sezione, che potrebbe rimanere sopra il coltello o sulla faccia interna della piastra antirollio. Per evitare l'arricciamento delle sezioni, eseguire rapidamente questo passaggio non appena la piastra viene sollevata e fare attenzione a non contorcere la sezione.
- Lasciare il vetrino fuori dalla camera del criostato (a temperatura ambiente) se si devono aggiungere nuove sezioni. Dopo aver fatto aderire tutte le sezioni desiderate al vetrino, conservarlo all'interno della camera del criostato o nel congelatore (-20 °C o inferiore). Non esporre i vetrini all'umidità. Tienili in una casella di diapositive e ricorda di identificare le diapositive con una matita.
NOTA: i vetrini e i blocchi di OCT possono essere conservati a -20 °C, anche se non troppo a lungo. Per ottenere risultati migliori, utilizzare le diapositive e i blocchi entro pochi giorni.
6. Colorazione di sezioni vegetali ed endofiti per microscopia ottica
NOTA: Molti tipi di macchie possono essere utilizzati per sezioni di piante. È difficile colorare in modo differenziale i funghi endofiti e i tessuti vegetali. Sebbene non sia una procedura di colorazione, un metodo per marcare le strutture dei funghi è presentato nella sezione 7 (fluorescenza con un coniugato di agglutinina del germe di grano). Le sezioni a mano libera (spiegate nella sezione 3), le sezioni di resina (sezione 4) e le criosezioni (sezione 5) possono essere colorate, sebbene le macchie a base di fenolo e alcol siano difficili per questi campioni poiché la resina GMA e l'OCT perdono aderenza al vetrino in questi casi.
- Utilizzare uno o combinare i seguenti metodi di colorazione usuali per i campioni di piante.
- Blu di toluidina O22,23, un metodo ampiamente applicato per la colorazione generale delle sezioni vegetali. Preparare una soluzione di blu di toluidina O allo 0,05% in fosfato 0,1 M (pH 6,8) o tampone citrato 0,09 M (pH 4,5-4,8), a seconda della specie e dei tipi di tessuto. Incubare le sezioni di resina GMA per 2-10 minuti utilizzando un barattolo colorante per vetrini o posizionando alcune gocce sopra le sezioni se pochi vetrini sono macchiati. Lavare accuratamente con acqua distillata o tampone dopo l'incubazione e montare i vetrini con acqua o asciugarli su una piastra riscaldante per produrre vetrini permanenti come descritto al punto 6.3.
- Il reagente Lugol2 indica la presenza di amido. Preparare una soluzione di iodio al 5% (I2) e ioduro di potassio (KI) al 10% in acqua distillata. Colorare le sezioni per 2 minuti, aggiungendo alcune gocce sopra il vetrino, quindi lavare con acqua distillata. Questo test istochimico viene solitamente applicato ai vetrini temporanei.
- Sudan III, IV e colorazione nera B24,25 per diversi lipidi. Preparare una soluzione di Sudan allo 0,3% (III, IV o B nero) in etanolo al 70%, riscaldarla fino all'ebollizione e lasciare raffreddare. Utilizzare il surnatante, filtrarlo e incubare le sezioni per 15-30 minuti in una capsula di Petri chiusa. Lavare accuratamente le sezioni con etanolo al 70% e acqua distillata. Montare i vetrini con acqua (di solito applicata solo alle diapositive temporanee).
NOTA: Poiché il Sudan è un colorante a base alcolica, è più adatto per le sezioni a mano libera. Condurre accuratamente la colorazione della sezione resina GMA poiché di solito si staccano dal vetrino.
- Per le diapositive temporanee, montare le sezioni in acqua o glicerina e osservare successivamente. Sigillare la copertina con lo smalto per unghie per conservarle un po 'più a lungo.
- Per le guide permanenti, montare le sezioni con resine sintetiche (ad esempio, supporto di montaggio rapido, vedere Tabella dei materiali). Gocciolare alcune gocce del supporto di montaggio (può traboccare il coprivetrino), mettere il coprislip con attenzione per evitare bolle e utilizzare mollette per premere il vetrino contro il coprislip fino a completa asciugatura. Rimuovere l'eccesso di mezzo di montaggio asciutto con una lama di rasoio.
7. Applicazione di un fluorocromo coniugato all'agglutinina del germe di grano in fluorescenza e microscopia confocale
NOTA: Questo metodo può essere applicato a sezioni a mano libera (spiegate nella sezione 3), sezioni di resina (sezione 4) e criosezioni (sezione 5). Le criosezioni possono essere adeguate per scopi di microscopia confocale, poiché possono essere forniti campioni più spessi rispetto alle sezioni di resina, ma non così spessi come quelli a mano libera. Un fluorocromo coniugato all'agglutinina del germe di grano (WGA, vedi Tabella dei materiali) viene applicato all'imaging fungino nella microscopia a fluorescenza26. Un microscopio confocale non è essenziale, sebbene possa fornire chiare immagini tridimensionali delle strutture vegetali27.
- Preparare una soluzione di 0,2 mg/mL WGA-fluorocromo coniugato in 0,1 M PB28 (pH 7,2, controllare il punto 5.1.1 e la Tabella 3). Preparare una soluzione di Calcofluor White all'1% in 0,1 M PB (pH 7,2). Preparare piccole quantità di queste soluzioni, poiché le sezioni vengono direttamente incubate con esse.
- Incubare le sezioni nei vetrini per 30 minuti nella soluzione coniugata WGA-fluorocromo29, utilizzando un volume sufficiente a coprire le sezioni, quindi lavare in 0,1 M PB.
- Incubare le sezioni nella soluzione di calcofluor, utilizzando un volume sufficiente come mezzo di montaggio. La soluzione può essere mantenuta durante il periodo di osservazione.
- Mettere dei vetrini sui vetrini e osservare in un microscopio confocale o in un microscopio a luce a fluorescenza usando i seguenti filtri: TC / GFP (eccitazione: 470-440, emissione: 525-550, per WGA-fluorocromo nella tabella dei materiali - la parete cellulare fungina fluoresce verde sotto questo filtro29) e DAPI (eccitazione: 358, emissione: 463, per Calcofluor White) 30.
NOTA: Le immagini tridimensionali possono essere ottenute utilizzando la funzione serie Z nel microscopio confocale27.
8. Microscopia elettronica a scansione di organi vegetali
- Dopo aver fissato i campioni, eseguito la disidratazione e conservato in etanolo al 70% (sezione 1), una possibilità è quella di tagliare i campioni per esporre qualsiasi superficie desiderata per l'analisi SEM, se necessario (ad esempio, tessuti interni, struttura ovarica). Utilizzare una lama di rasoio affilata e nuova ed effettuare tagli con un movimento unidirezionale, evitando un aspetto danneggiato di queste aree in SEM. Se necessario, utilizzare uno stereomicroscopio per selezionare i campioni e considerare l'area degli stub metallici per determinare le dimensioni del campione.
- Ulteriori campioni disidratati per SEM in una serie etanolica: 80%, 96% e 2 volte in alcool etilico assoluto (≥99,8%). Mantenere campioni piccoli e delicati per 30 minuti in ogni concentrazione e campioni più grandi e più densi per 1 ora.
- Piegare piccole buste usando carta velina per organizzare i campioni per i passaggi successivi, i campioni più grandi possono essere gestiti senza busta. Identifica le buste con una matita scrivendo una lettera o un numero e tieni un registro di tutti i campioni in ciascuna. Conservare i campioni in etanolo assoluto, anche se non a lungo, e procedere al passaggio 8.4 il prima possibile.
- Procedere per l'asciugatura del punto critico (CP). Utilizzare un essiccatore CP secondo le procedure operative standard. Posizionare i campioni in etanolo assoluto (fluido intermedio) in una camera a pressione. Nel punto critico di CO2 (31 °C, 7,3 x 106 Pa) il fluido intermedio si dissolve nel fluido di transizione (anidride carbonica liquida) e i campioni vengono essiccati31.
- Dopo l'essiccazione CP, conservare i campioni il prima possibile in un contenitore di essiccazione, ad esempio un pallone sigillato contenente gel di silice. L'umidità atmosferica può distruggere i campioni se riassorbita31.
- Utilizzare mozziconi metallici per montare i campioni. Prima di montare, indossare i guanti per manipolare i mozziconi, immergerli nell'acetone per 5 minuti per eliminare il grasso e lasciarli asciugare. Utilizzare un nastro biadesivo conduttivo in carbonio per fissare i campioni sul tronco e uno stereomicroscopio per aiutare a posizionare i campioni, tenendo presente che la vista dall'alto è l'unica prospettiva possibile nelle immagini SEM.
- Manipolare i campioni con pinzette a punta fine, facendo attenzione poiché la parte del campione toccata dalle pinzette è solitamente danneggiata, quindi prova a toccare le parti posizionate lontano dalle aree di interesse (ad esempio, le aree a contatto con il nastro). Mantenere i mozziconi con campioni in una capsula di Petri sigillata con gel di silice. Procedere al passaggio 8.8 il prima possibile.
- Utilizzare uno sputter coater per depositare uno strato di metallo, di solito oro o platino, sulla superficie dei campioni in un'atmosfera a bassa pressione di un gas inerte, spesso argon31. Seguire le procedure operative standard quando si utilizza un rivestimento sputter. Lo spessore del rivestimento dipende dalla topografia dei campioni, di solito tra 15-40 nm32.
- Mantenere i mozziconi rivestiti in una capsula di Petri sigillata con gel di silice e, a condizione che il gel di silice trattenga l'umidità, i campioni possono essere conservati in questo modo per settimane. Utilizzare un microscopio elettronico a scansione per analizzare i campioni. I campioni nel vuoto sono colpiti da un fascio di elettroni e l'emissione di segnali da tale interazione è interpretata come immagini31. Per i dettagli sul funzionamento di un microscopio elettronico a scansione, leggere Jeffree e Read (1991)31 e Bozzola e Russell (1999)32.
- Per riutilizzare i mozziconi, tirare il nastro adesivo e strofinarli con lana metallica. Lavare in acqua di rubinetto, immergerli in etanolo assoluto e asciugarli adeguatamente, evitando l'ossidazione del metallo costituente.
9. Microscopia elettronica a trasmissione
- Campioni di prefisso con tampone glutaraldeide-cacodilato come spiegato nei punti 1.7 e 1.10. Dopo 12-24 ore di prefissazione, lavare i campioni 3 volte in tampone cacodilato 0,2 M (pH 7,25) per 10 minuti. Eseguire la postfissazione con tetrossido di osmio all'1% (OsO4) in tampone cacodilato da 0,2 M, per 12 ore al buio, a temperatura ambiente. Lavare 3 volte con acqua distillata per 5 min.
ATTENZIONE: Il cacodilato e il tetrossido di osmio sono altamente tossici e non devono essere inalati. Utilizzarli nelle cappe aspiranti, seguendo le rispettive schede di sicurezza. - Disidratare i campioni con etanolo al 30%, 50%, 70% e 96%, 2x in ciascuna concentrazione, per 10 minuti. Quindi, disidratare 3 volte in alcool assoluto, per 15 minuti ogni volta.
- Infiltrare i campioni in resine acriliche idrofile (vedi Tabella dei materiali), una volta con resina 1:1 + etanolo assoluto e 3x con resina pura per 8-12 ore ciascuna. Condurre la polimerizzazione in capsule di gelatina a 60 °C fino a completa solidificazione (massimo 12 ore17).
- Valutare attentamente l'orientamento dei campioni all'interno del blocco di resina; Tagliare la parte superiore del blocco, con una lama di rasoio, creando una forma piramidale che concentra il campione nella zona di sezionamento. Ricavare sezioni semisottili (250-500 nm)33 in un ultramicrotomo con un coltello diamantato e disporre su vetrini in poche gocce d'acqua.
- Conservare i vetrini su una piastra riscaldante a 60 °C. Colorare le sezioni con toluidina blu O come al punto 6.1.1 e lasciare asciugare completamente la macchia. Lavare accuratamente con acqua di rubinetto. Valutare la sezione ottenuta disegnando quattro quadranti e selezionando il quadrante più adatto per l'analisi.
- Taglia il blocco in modo che la forma piramidale concentri il quadrante scelto sulla faccia superiore del blocco. Produrre sezioni ultrasottili (50-100 nm)33,34. Lo spessore viene valutato in base al colore di interferenza delle sezioni: le sezioni con circa 70 nm appaiono argento-oro, con circa 100 nm appaiono oro, e con circa 200 nm appaiono blu34.
- Raccogliere le sezioni ultrasottili dall'acqua utilizzando griglie di rame e procedere al metodo di colorazione a contrasto con acetato di uranile e citrato di piombo, come descritto di seguito.
- Preparare una soluzione di citrato di piombo (Tabella 5) e congelare la soluzione finale in provette da microcentrifuga con 1 mL di soluzione in ciascuna, scongelandola solo subito prima dell'uso.
- Preparare una soluzione di acetato di uranile: sciogliere 0,625 g di acetato di uranile [UO 2(CH3COO)2] in 25 ml di acqua distillata recentemente bollita e raffreddata. Conservare in un matraccio scuro nel congelatore.
ATTENZIONE: Il nitrato di piombo è tossico se ingerito. 1 N NaOH è altamente corrosivo. L'acetato di uranile è radioattivo e tossico. Non deve essere ingerito, inalato o venire a contatto con la pelle. - Durante la colorazione, mettere entrambi i reagenti preparati in siringhe separate da 3 ml con unità filtranti (poro 0,22 μm, vedere Tabella dei materiali). Preparare una capsula di Petri capovolta con una pellicola termoplastica sigillante sopra di essa (vedi Tabella dei materiali) e all'interno di una capsula più ampia, con pellet di NaOH sui bordi come trappola per CO 2 32 (vedi Figura 2).
- Scartare la prima goccia e sopra il film posizionare una goccia di acetato di uranile e tre gocce di acqua distillata per ogni griglia macchiata. Fai lo stesso con il citrato di piombo e aggiungi altre tre gocce di acqua distillata.
- Incubare la griglia (con il lato opaco verso il basso, dove si trovano le sezioni) in uranile per 30 minuti (tempo variabile). Lavare 3 volte nelle gocce d'acqua distillata, asciugando delicatamente la griglia ogni volta con carta da filtro sul lato brillante. Ripetere con la goccia di citrato di piombo (30 min) e lavare.
- Dopo almeno 4 ore, analizzare le griglie in un microscopio elettronico a trasmissione. In questo microscopio, un fascio di elettroni passa attraverso le sezioni nel vuoto e l'immagine viene proiettata su uno schermo fluorescente. Per i dettagli sul funzionamento di un microscopio elettronico a trasmissione, leggere Bozzola e Russell (1999)32.
| soluzione di citrato di piombo (per colorazione con contrasto TEM) | |
| Passo 1 | Circondare un becher con carta stagnola |
| Passo 2 | sciogliere 0,266 g di nitrato di piombo [Pb(NO3)2] in 6 mL di acqua distillata recentemente bollita e raffreddata |
| Passo 3 | agitare per 2 min |
| Passo 4 | aggiungere 0,352 g di citrato trisodico [Na3(C6H5O7).2H2O] (la soluzione deve assumere un aspetto lattiginoso) |
| Passo 5 | agitare per 15 minuti, sigillare il becher con carta stagnola e trasferire la soluzione in un becher da 10 mL |
| Passo 6 | aggiungere 1,6 mL di 1N NaOH e 2,4 mL di acqua distillata (la soluzione deve essere traslucida) |
| Passo 7 | se necessario, regolare il pH vicino a 12 |
Tabella 5: Ricetta della soluzione di citrato di piombo.

Figura 2: Schema di colorazione a contrasto con soluzioni di citrato di piombo e acetato di uranile . (A) Preparare le piastre di Petri, una capovolta (al centro) con pellicola termoplastica in modo che le gocce possano essere posizionate sopra di esso, all'interno di una più ampia. I pellet NaOH sono posti intorno al piatto centrale. (B) Le gocce di acetato di uranile sono poste nei cerchi con la lettera U, e le gocce di citrato di piombo nei cerchi contrassegnati con L. DW indicano gocce di acqua distillata. Le griglie sono colorate in sequenza nella colonna, quindi cinque griglie possono essere colorate contemporaneamente come rappresentate. Fare clic qui per visualizzare una versione ingrandita di questa figura.
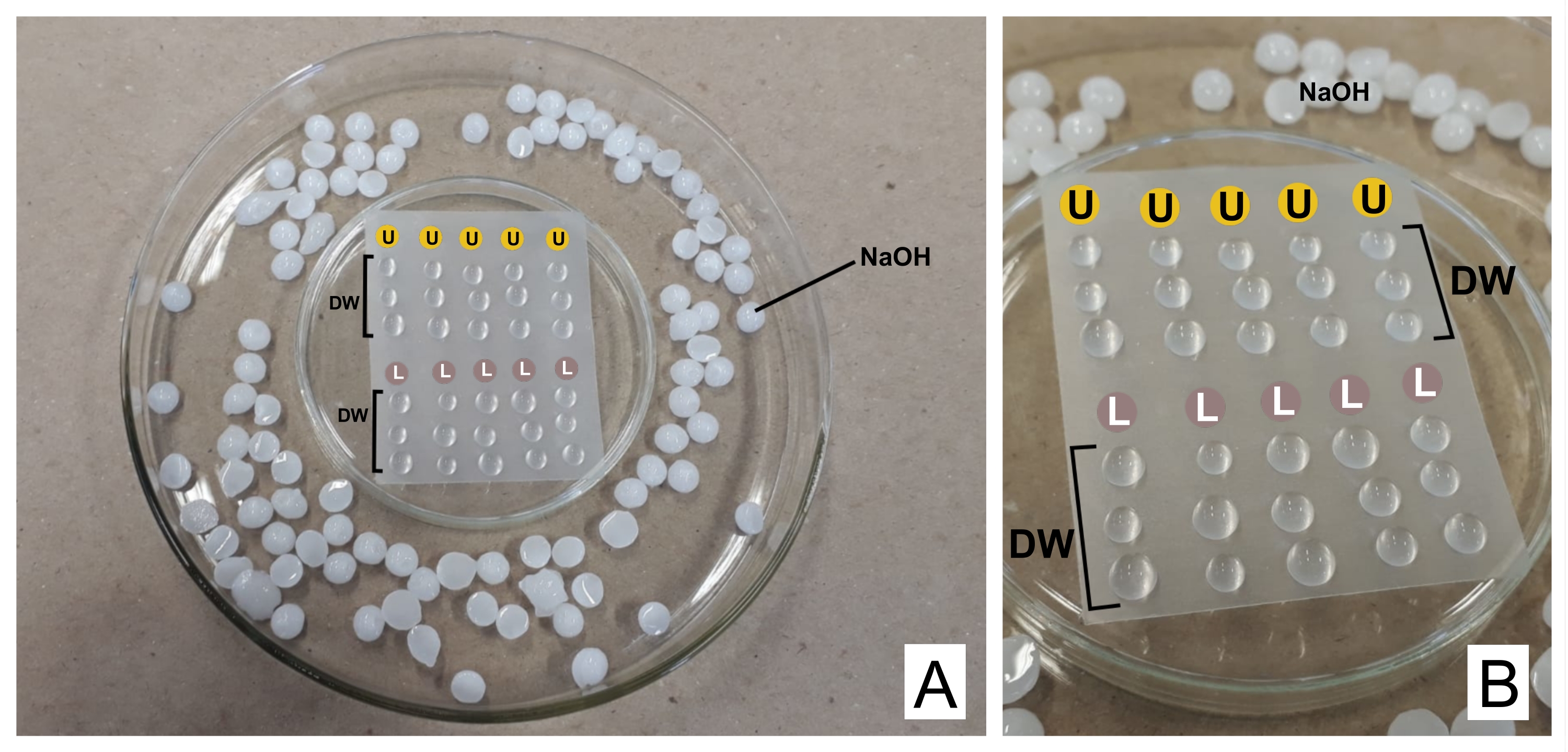{kind=link}
10. Germinazione simbiotica dei semi di orchidea
- Assicurarsi che le soluzioni e tutti i materiali utilizzati nella germinazione simbiotica dei semi siano sterili per evitare la contaminazione. Iniziare con l'autoclave per 20 minuti a 121 °C. Le fasi di germinazione simbiotica sono riassunte nella Figura 3.
- Disinfettare superficialmente frutti e semi immergendoli in una soluzione di ipoclorito di sodio contenente il 2% di cloro attivo per 10-15 minuti per i frutti e 7-10 minuti per i semi, considerando la rigidità e lo spessore del rivestimento del seme9. I semi sottili e fragili possono essere immersi in una soluzione di ipoclorito di sodio diluito 1: 1. Successivamente, lavare 3 volte in acqua distillata autoclavata per rimuovere la soluzione di ipoclorito.
- Recuperare i semi filtrando in tessuto serigrafico e utilizzare i semi per procedere con i test di germinazione (preferibilmente). Se necessario, conservarli in buste di carta da filtro all'interno di palloni di vetro con gel di silice, a 4 °C, chiudere ermeticamente i palloni e sigillarli con pellicola trasparente. Trasferire alcune gocce d'acqua dall'ultimo lavaggio all'agar destrosio di patate (PDA, 39 g/L), per valutare l'efficacia del processo di lavaggio.
- Prima di seminare i semi nel terreno di coltura, valutarne la vitalità attraverso il test del tetrazolio (facoltativo) come descritto di seguito35.
- Incubare circa 10 mg di semi in una provetta da microcentrifuga con 1 mL di saccarosio al 10% in acqua distillata, per 24 ore a temperatura ambiente (ca. 25 °C), in luce.
- Rimuovere la soluzione di saccarosio con una micropipetta e aggiungere 1 mL di soluzione di tetrazolio all'1% (trifeniltetrazolio cloruro) in acqua distillata. Incubare a 40 °C in un blocco termico per 24 ore al buio.
- Rimuovere la soluzione di tetrazolio con una micropipetta e lavare i semi con acqua distillata 2x o fino a quando la soluzione non viene rimossa. Rimuovere tutti i liquidi. Se necessario, i semi possono essere conservati nel congelatore per un massimo di una settimana (come nel passaggio 10.3) prima di essere analizzati.
- Risospendere i semi in acqua distillata e analizzarli al microscopio ottico. I semi vitali acquisiscono un colore da rosso chiaro a rosso scuro, mentre i semi non vitali mantengono il loro colore naturale.
- Eseguire il seguente protocollo adattato9 per la germinazione simbiotica dei semi di orchidea.
- Incubare i semi su dischi di carta da filtro autoclavati (1-2 cm di diametro) posti in piastre di Petri con terreno di coltura di farina d'avena agar (OMA) (2,5 g / L di fiocchi d'avena e 7 g / L di agar, pH 6).
- Al centro della capsula di Petri, inoculare un frammento di terreno di coltura (ca. 1 cm2) contenente micelio dal fungo isolato scelto per la procedura di germinazione. Sigillare le piastre di Petri con pellicola trasparente e incubarle al buio a circa 25 °C o a temperatura ambiente, poiché è più adatto alla crescita fungina.
- Preparare alcuni piatti con semi e senza inoculazione fungina, come controllo negativo per il test di germinazione.
- Analizzare settimanalmente i risultati della germinazione, raccogliendo dati quantitativi e qualitativi e fotografando protocormi e piantine. L'osservazione di semi e protocormi dovrebbe essere eseguita utilizzando uno stereomicroscopio per una valutazione più accurata della germinazione. Utilizzare una fonte di luce proveniente dal basso, in quanto consente un maggiore contrasto, rendendo possibile discriminare più facilmente il micelio fungino dai protocormi.
- Raccogliere campioni in diversi stadi di sviluppo e fissare per analisi anatomiche (sezione 1). Applicare tutte le analisi delle immagini precedentemente descritte per studiare gli endofiti fungini in semi, protocormi e piantine durante la germinazione (microscopia ottica - sezioni 4, 5 e 6; confocale e fluorescenza - sezione 7; SEM e TEM - sezioni 8 e 9).
- Generare risultati quantitativi seguendo la classificazione delle fasi secondo la tabella 6. Le fasi descrivono il solito sviluppo dei semi delle orchidee micoeterotrofiche. Raccogli dati settimanalmente e tabella con le date iniziali di ogni fase osservata.
- Inoltre, raccogliere dati quantitativi stimando la percentuale e il tasso di germinazione. Conta almeno 100 semi o definisci i campi di conteggio35. Delimitare tre o più campi di conteggio per capsula di Petri, costituiti da regioni fisse con un'area standardizzata, e valutare settimanalmente. Calcola i dati raccolti in base all'equazione dell'indice di crescita (IG):

dove N 0 è il numero di semi contati nella fase0 , N 1 si riferisce alla fase1 e segue fino alla fase 6 (registrata come N6)36.

Figura 3: Riassunto schematico della metodologia della germinazione simbiotica dei semi. Gli schemi forniscono indicazioni sui passaggi dettagliati del protocollo. Abbreviazioni: OMA = agar farina d'avena, PDA = agar destrosio di patate. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
| Fase di germinazione | Descrizione |
| 0 | Nessuna germinazione |
| 1 | Gonfiore dell'embrione |
| 2 | Rottura della testa |
| 3 | I peli assorbenti si sviluppano |
| 4 | Si sviluppa la proiezione dello stelo |
| 5 | Sviluppo di scaglie protettive (brattee) |
| 6 | Le prime radici si sviluppano |
Tabella 6: Descrizione degli stadi di sviluppo del protocormo applicati alle analisi periodiche dei test di germinazione. Modificato dagli stadi descritti in Otero et al.36.
Risultati
Seguendo le fasi essenziali di fissaggio del tessuto vegetale si ottengono strutture cellulari il più possibile simili allo stato vivente, considerando la morfologia, il volume e l'organizzazione spaziale dei componenti cellulari e dei tessuti16. Osservare tali tratti nei campioni dopo la fissazione chimica (Figura 4). La figura 4C-F rappresenta campioni adeguatamente fissati al microscopio ottico. Segui...
Discussione
Le analisi delle immagini nell'anatomia e morfologia delle piante hanno un importante potenziale per raggiungere gli obiettivi e aiutare a comprendere le relazioni tra le piante micoeterotrofe e i loro indispensabili endofiti fungini, come dimostrato dagli studi sugli organi sotterranei6,40, dalle analisi strutturali della germinazione simbiotica dei semi39 e dalle strutture aeree e riproduttive 41 . La botanica str...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Gli autori ringraziano i finanziamenti di FAEPEX e FAPESP (2015/26479-6). MPP ringrazia Capes per la sua borsa di studio di master (processo 88887.600591/2021-00) e CNPq. JLSM ringrazia CNPq per le sovvenzioni di produttività (303664/2020-7). Gli autori ringraziano anche l'accesso alle attrezzature e all'assistenza fornita da LME (Laboratorio di Microscopia Elettronica - IB/Unicamp), INFABiC (Istituto Nazionale di Scienza e Tecnologia sulla Fotonica Applicata alla Biologia Cellulare - Unicamp), e LaBiVasc (Laboratorio di Biologia Vascolare - DBEF/IB/Unicamp); LAMEB (UFSC) e Eliana de Medeiros Oliveira (UFSC) per i contributi al protocollo di crioprotezione; LME per contributi al protocollo TEM.
Materiali
| Name | Company | Catalog Number | Comments |
| Acetone | Sigma-Aldrich | 179124 | (for SEM stubs mounting) |
| Agar-agar (AA) | Sigma-Aldrich | A1296 | (for seeds germination tests) |
| Calcofluor White Stain | Sigma-Aldrich | 18909 | fluorescent dye (detects cellulose) |
| Citrate Buffer Solution, 0.09M pH 4.8 | Sigma-Aldrich | C2488 | (for toluidine blue O staining) |
| Conductive Double-Sided Carbon Tape | Fisher Scientific | 50-285-81 | (for SEM) |
| Confocal Microscope | Zeiss | (any model) | |
| Copper Grids | Sigma-Aldrich | G4776 | (for TEM) |
| Critical-point dryer | Balzers | (any model) | |
| Cryostat | Leica Biosystems | (any model) | |
| Dissecting microscope | Leica Biosystems | (= stereomicroscope, any model) | |
| Entellan | Sigma-Aldrich | 107960 | rapid mounting medium for microscopy |
| Ethyl alcohol, pure (≥99.5%) | Sigma-Aldrich | 459836 | (= ethanol, for dehydration processes) |
| Formaldehyde solution, 37% | Sigma-Aldrich | 252549 | (for NBF solution preparation) |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128 | histological tissue fixative |
| Gelatin capsules for TEM | Fisher Scientific | 50-248-71 | (for resin polymerisation in TEM) |
| Gelatin solution, 2% in H2O | Sigma-Aldrich | G1393 | (dilute for slides preparation - OCT adherence) |
| Glutaraldehyde solution, 25% | Sigma-Aldrich | G6257 | (for Karnovsky’s solution preparation) |
| HistoResin | Leica Biosystems | 14702231731 | glycol methacrylate (GMA) embedding kit |
| Iodine | Sigma-Aldrich | 207772 | (for Lugol solution preparation) |
| Lead(II) nitrate | Sigma-Aldrich | 228621 | Pb(NO3)2 (for TEM contrast staining) |
| Light Microscope | Olympus | (any model) | |
| LR White acrylic resin | Sigma-Aldrich | L9774 | hydrophilic acrylic resin for TEM |
| Lugol solution | Sigma-Aldrich | 62650 | (for staining) |
| Metal stubs for specimen mounts | Rave Scientific | (for SEM, different models) | |
| Microtome | Leica Biosystems | manual rotary microtome or other model | |
| Oatmeal agar (OMA) | Millipore | O3506 | (for seeds germination tests) |
| OCT Compound, Tissue-Tek | Sakura Finetek USA | 4583 | embedding medium for frozen tissues |
| Osmium tetroxide | Sigma-Aldrich | 201030 | OsO4 (for TEM postfixation) |
| Parafilm M | Sigma-Aldrich | P7793 | sealing thermoplastic film |
| Paraformaldehyde | Sigma-Aldrich | 158127 | (for Karnovsky’s solution preparation) |
| Poly-L-lysine solution, 0.1% in H2O | Sigma-Aldrich | P8920 | (for slides preparation - OCT adherence) |
| Poly-Prep Slides | Sigma-Aldrich | P0425 | poly-L-lysine coated glass slides |
| Polyethylene Molding Cup Trays | Polysciences | 17177A-3 | (6x8x5 mm, for embbeding samples in GMA resin) |
| Polyethylene Molding Cup Trays | Polysciences | 17177C-3 | (13x19x5 mm, for embbeding samples in GMA resin) |
| Potassium iodide | Sigma-Aldrich | 221945 | (for Lugol solution preparation) |
| Potato Dextrose Agar (PDA) | Millipore | 70139 | (for seeds germination tests) |
| Scanning Electron Microscope | Jeol | (any model) | |
| Silane [(3-Aminopropyl)triethoxysilane] | Sigma-Aldrich | A3648 | (for slides preparation - OCT adherence) |
| Silane-Prep Slides | Sigma-Aldrich | S4651 | glass slides coated with silane |
| Silica gel orange, granular | Supelco | 10087 | (for dessicating processes) |
| Sodium cacodylate trihydrate | Sigma-Aldrich | C0250 | (for glutaraldehyde-sodium cacodylate buffer) |
| Sodium hydroxide | Sigma-Aldrich | S5881 | NaOH (for Karnovsky’s solution preparation and TEM contrast staining) |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044 | NaClO (for seeds surface disinfection) |
| Sodium phosphate dibasic, anhydrous | Sigma-Aldrich | 71640 | Na2HPO4 (for NBF solution and PB preparation) |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638 | NaH2PO4·H2O (for NBF and PB) |
| Sputter coater | Balzers | (any model) | |
| Sucrose | Sigma-Aldrich | S0389 | C12H22O11 (for cryoprotection and germination test) |
| Sudan III | Sigma-Aldrich | S4131 | (for staining) |
| Sudan IV | Sigma-Aldrich | 198102 | (for staining) |
| Sudan Black B | Sigma-Aldrich | 199664 | (for staining) |
| Syringe | (3 mL, any brand, for TEM contrast staining) | ||
| Syringe Filter Unit, Millex-GV 0.22 µm | Millipore | SLGV033R | PVDF, 33 mm, gamma sterilized (for TEM contrast staining) |
| Tek Bond Super Glue 793 | Tek Bond Saint-Gobain | 78072720018 | liquid cyanoacrylate adhesive, medium viscosity |
| Toluidine Blue O | Sigma-Aldrich | T3260 | (for staining) |
| Transmission Electron Microscope | Jeol | (any model) | |
| Triphenyltetrazolium chloride | Sigma-Aldrich | T8877 | (for the tetrazolium test in seeds germination) |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804 | Na3(C6H5O7)·2H2O (for TEM contrast staining) |
| Ultramicrotome | Leica Biosystems | (any model) | |
| Uranyl acetate | Fisher Scientific | 18-607-645 | UO2(CH3COO)2 (for TEM contrast staining) |
| Vacuum pump | (any model) | ||
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | TermoFisher Scientific | W11261 | fluorescent dye-conjugated lectin (detects sialic acid and N-acetylglucosaminyl residues) |
Riferimenti
- Evert, R. F. . Esau’s Plant Anatomy: Meristems, Cells, and Tissues of the Plant Body: Their Structure, Function, and Development. , (2006).
- Yeung, E. C. T., Stasolla, C., Sumner, M. J., Huang, B. Q. . Plant Microtechniques and Protocols. , (2015).
- Sokoloff, D. D., Jura-Morawiec, J., Zoric, L., Fay, M. F. Plant anatomy: at the heart of modern botany. Botanical Journal of the Linnean Society. 195 (3), 249-253 (2021).
- Leake, J. R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytologist. 127 (2), 171-216 (1994).
- Bidartondo, M. I. The evolutionary ecology of myco-heterotrophy. New Phytologist. 167 (2), 335-352 (2005).
- Imhof, S., Massicotte, H. B., Melville, L. H., Peterson, R. L. Subterranean morphology and mycorrhizal structures. Mycoheterotrophy. , 157-214 (2013).
- Rasmussen, H. N., Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos. 118 (3), 334-345 (2009).
- Rasmussen, H. N., Dixon, K. W., Jersáková, J., Těšitelová, T. Germination and seedling establishment in orchids: a complex of requirements. Annals of Botany. 116 (3), 391-402 (2015).
- Zettler, L. W. Terrestrial orchid conservation by symbiotic seed germination: techniques and perspectives. Selbyana. 18 (2), 188-194 (1997).
- Stewart, S. L., Kane, M. E. Symbiotic seed germination and evidence for in vitro mycobiont specificity in Spiranthes brevilabris (Orchidaceae) and its implications for species-level conservation. In Vitro Cellular & Developmental Biology - Plant. 43 (3), 178-186 (2007).
- Zhao, D. -. K., et al. Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Frontiers in Plant Science. 12, 701152 (2021).
- Selosse, M. A., Roy, M. Green plants that feed on fungi: facts and questions about mixotrophy. Trends in Plant Science. 14 (2), 64-70 (2009).
- Merckx, V. S. F. T., Mennes, C. B., Peay, K. G., Geml, J. Evolution and diversification. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 215-244 (2013).
- Boon, M. E., Drijver, J. Routine Cytological Staining Techniques: Theoretical Background and Practice. Macmillan International Higher Education. , (1986).
- Karnovsky, M. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. Journal of Cell Biology. 27 (2), 137-138 (1964).
- Hayat, M. . Fixation for Electron Microscopy. , (1981).
- Roland, J. C., Vian, B. General preparation and staining of thin sections. Electron Microscopy of Plant Cells. 1, 675 (1991).
- Gerrits, P. O., Horobin, R. W. Glycol methacrylate embedding for light microscopy: basic principles and trouble-shooting. Journal of Histotechnology. 19 (4), 297-311 (1996).
- Zhang, Z., Niu, L., Chen, X., Xu, X., Ru, Z. Improvement of plant cryosection. Frontiers in Biology. 7 (4), 374-377 (2012).
- BeneŠ, K. On the media improving freeze-sectioning of plant material. Biologia Plantarum. 15 (1), 50-56 (1973).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. Cold Spring Harbor Protocols. 2008 (5), (2008).
- Sakai, W. S. Simple method for differential staining of paraffin embedded plant material using toluidine blue O. Stain Technology. 48 (5), 247-249 (1973).
- O’Brien, T., Feder, N., McCully, M. E. Polychromatic staining of plant cell walls by toluidine blue O. Protoplasma. 59 (2), 368-373 (1964).
- Ventrella, M. C., Almeida, A. L., Nery, L. A., Coelho, V. P. d. e. M. Métodos Histoquímicos Aplicados às Sementes. Universidade Federal de Viçosa. , (2013).
- Pearse, A. G. E. . Histochemistry, Theoretical and Applied. , (1960).
- Andrade-Linares, D. R., Franken, P. Fungal endophytes in plant roots: taxonomy, colonization patterns, and functions. Symbiotic Endophytes. , 311-334 (2013).
- Wymer, C. L., Beven, A. F., Boudonck, K., Lloyd, C. W. Confocal microscopy of plant cells. Confocal Microscopy Methods and Protocols. , 103-130 (1999).
- Marques, J. P. R., Soares, M. K. M. Manual de Técnicas Aplicadas à Histopatologia Vegetal. FEALQ. , (2021).
- Navarro, B. L., Marques, J. P. R., Appezzato-da-Glória, B., Spósito, M. B. Histopathology of Phakopsora euvitis on Vitis vinifera. European Journal of Plant Pathology. 154 (4), 1185-1193 (2019).
- Marques, J. P. R., et al. Sugarcane cell wall-associated defense responses to infection by Sporisorium scitamineum. Frontiers in Plant Science. 9, 698 (2018).
- Jeffree, C. E., Read, N. D. Ambient-and low-temperature scanning electron microscopy. Electron Microscopy of Plant Cells. , 313-413 (1991).
- Bozzola, J. J., Russell, L. D. . Electron Microscopy: Principles and Techniques for Biologists. , (1999).
- Murray, S. Basic transmission and scanning electron microscopy. Introduction to electron Microscopy for Biologists. , 3-18 (2008).
- . Glossary of TEM terms Available from: https://www.jeol.co.jp/en/words/emterms/ (2021)
- Seaton, P. T., et al. Orchid seed and pollen: a toolkit for long-term storage, viability assessment and conservation. Orchid Propagation: From Laboratories to Greenhouses—Methods and Protocols. , 71-98 (2018).
- Otero, J. T., Ackerman, J. D., Bayman, P. Differences in mycorrhizal preferences between two tropical orchids. Molecular Ecology. 13 (8), 2393-2404 (2004).
- Koch, R. A., et al. Marasmioid rhizomorphs in bird nests: Species diversity, functional specificity, and new species from the tropics. Mycologia. 112 (6), 1086-1103 (2020).
- Webster, J., Weber, R. . Introduction to Fungi. , (2007).
- Sisti, L. S., et al. The role of non-mycorrhizal fungi in germination of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. Frontiers in Plant Science. 10, 1589 (2019).
- Martos, F., et al. Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytologist. 184 (3), 668-681 (2009).
- Alves, M. F., et al. Reproductive development and genetic structure of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. BMC Plant Biology. 21 (1), 332 (2021).
- Merckx, V. S. F. T. Mycoheterotrophy: an introduction. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 1-17 (2013).
- Hall, J. L., Hawes, C. . Electron Microscopy of Plant Cells. , (1991).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati