
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
菌従属栄養植物組織における真菌のコロニー形成と種子の共生発芽を解釈するための顕微鏡技術
要約
このプロトコルは、菌根菌で発芽した植物組織および種子における真菌コロニー形成を研究するために、走査型および透過型電子顕微鏡、光学顕微鏡、共焦点顕微鏡、蛍光顕微鏡などのさまざまな顕微鏡技術を適用して、マイコ従属栄養植物サンプルを収集、固定、および維持するための詳細な手順を提供することを目的としています。
要約
構造植物学は、植物の生態、生理、発達、進化を完全に理解するために不可欠な視点です。菌従属栄養植物(菌類から炭素を得る植物)を研究する場合、その構造適応、菌類による組織コロニー形成のパターン、地下器官の形態解剖学的構造などの顕著な側面は、その発生戦略と栄養源である菌糸との関係を解明することができます。共生菌のもう一つの重要な役割は、ランの種子の発芽に関連しています。すべてのラン科種は、発芽および実生段階(初期マイコヘテロトロフィー)の間に菌従属栄養性であり、成虫段階で光合成するものでさえあります。ランの種子には栄養貯蔵が不足しているため、真菌共生生物は基質を提供し、発芽を可能にするために不可欠です。構造的な観点から発芽段階を分析することは、真菌と種子との相互作用に関する重要な質問に答えることもできます。この記事で提案されているように、植物組織中の真菌内生植物を明らかにするために、さまざまなイメージング技術を適用できます。植物器官のフリーハンドおよび薄い切片を染色し、光学顕微鏡を使用して観察することができます。小麦胚芽凝集素に結合した蛍光色素を真菌に適用し、Calcofluor Whiteと共生して、共焦点顕微鏡で植物細胞壁を強調することができます。さらに、菌従属栄養ランの走査型および透過型電子顕微鏡の方法論が詳述されており、そのようなプロトコルを関連植物に適用する可能性が探求されています。ラン種子の共生発芽(すなわち、菌根菌の存在下で)は、光、共焦点、および電子顕微鏡による分析のために発芽の異なる段階から得られた構造を調製する可能性とともに、プロトコルに詳細に記載されている。
概要
植物の形態学と解剖学を網羅する植物学の構造研究は、生物全体を理解する上で基礎であり1,2、植物の生態・生理・発生・進化に関する知識を統合し、貢献するために不可欠な視点を提供します3。植物の形態学と解剖学の方法は、現在、最近および1世紀以上前に開発されたプロトコル、機器、および知識で構成されています2。古典的な方法(光学顕微鏡など)の継続的な実行と適応は、より最近の技術(共焦点顕微鏡、X線マイクロトモグラフィーなど)と同じ本質的な基礎を持っています:方法論の開発を可能にする理論的知識。
植物の解剖学と形態学の主なツールは画像です。このような分析は単純な観察であるという誤解にもかかわらず、主観的な解釈にスペースを与えます2、この領域の画像を分析および理解するには、適用される方法(機器、分析の種類、方法論的手順)、細胞成分、組織化学、および植物体(組織の組織と機能、個体発生、形態学的適応)。さまざまな 方法で得られた画像を 解釈することで、形態と機能の相関、構造の化学組成の解読、分類群の記述の裏付け、植物病原体による感染の理解などにつながります。
菌従属栄養植物(菌根菌から炭素を得る非光合成植物4,5)を研究する場合、その構造適応、真菌による組織コロニー形成のパターン、および地下器官の形態解剖学的構造の顕著な側面は、それらの発生戦略と栄養素の源である菌糸との関係を啓発することができます。MH植物の地下器官は通常、土壌真菌との関連に関連する重要な適応を示すため、これらの解剖学的および形態学的調査を行うことが不可欠です6。MH種の空中器官は、菌根菌でなくても、これらの組織にも内生菌が存在する可能性があるため、無視してはなりません(個人的な観察、まだ公開されていません)。
菌根菌とMH種との関連がライフサイクル全体にわたって確立された必須性に加えて7、すべてのラン種は、独立栄養種でさえ、自然環境において最初の絶対的な菌従属栄養段階を持っています。これは、ランの胚が未分化であり、胚乳や子葉を欠いているため、真菌パートナーの栄養サポートなしに自然環境で発達および確立することができないために発生します4,8。それを考慮すると、共生発芽プロトコルはMH種だけでなく、光合成ランにも適用でき、発芽と原始球の発生におけるラン菌特異性を調査することを目的としており、絶滅危惧種の保全イニシアチブに広く適用されている方法論です9,10,11。
この方法の組み立てでは、解剖学的研究のためのMH植物サンプルの収集、固定、および保管(セクション1)、表面分析とサンプル選択(セクション2)、セクショニング方法(フリーハンド:セクション3、ミクロトミー:セクション4、クライオミクロトミー:セクション5)、染色とマウント(セクション6)、真菌内生植物の蛍光および共焦点顕微鏡(セクション7)、走査型電子顕微鏡(セクション8)、 透過型電子顕微鏡(セクション9)。さらに、ラン種子の共生発芽法(MHと独立栄養、セクション10)について述べ、前述のイメージング法は、発芽過程における種子、原球菌、および苗の真菌コロニー形成の分析にうまく適用できるためです。

図1:イメージング方法の概略図。回路図は、それらが詳述されているプロトコルステップの指示を提供します。略語:GMA =グリコールメタクリレート、OCT =最適な切断温度化合物、SEM =走査型電子顕微鏡。この図の拡大版を表示するには、ここをクリックしてください。
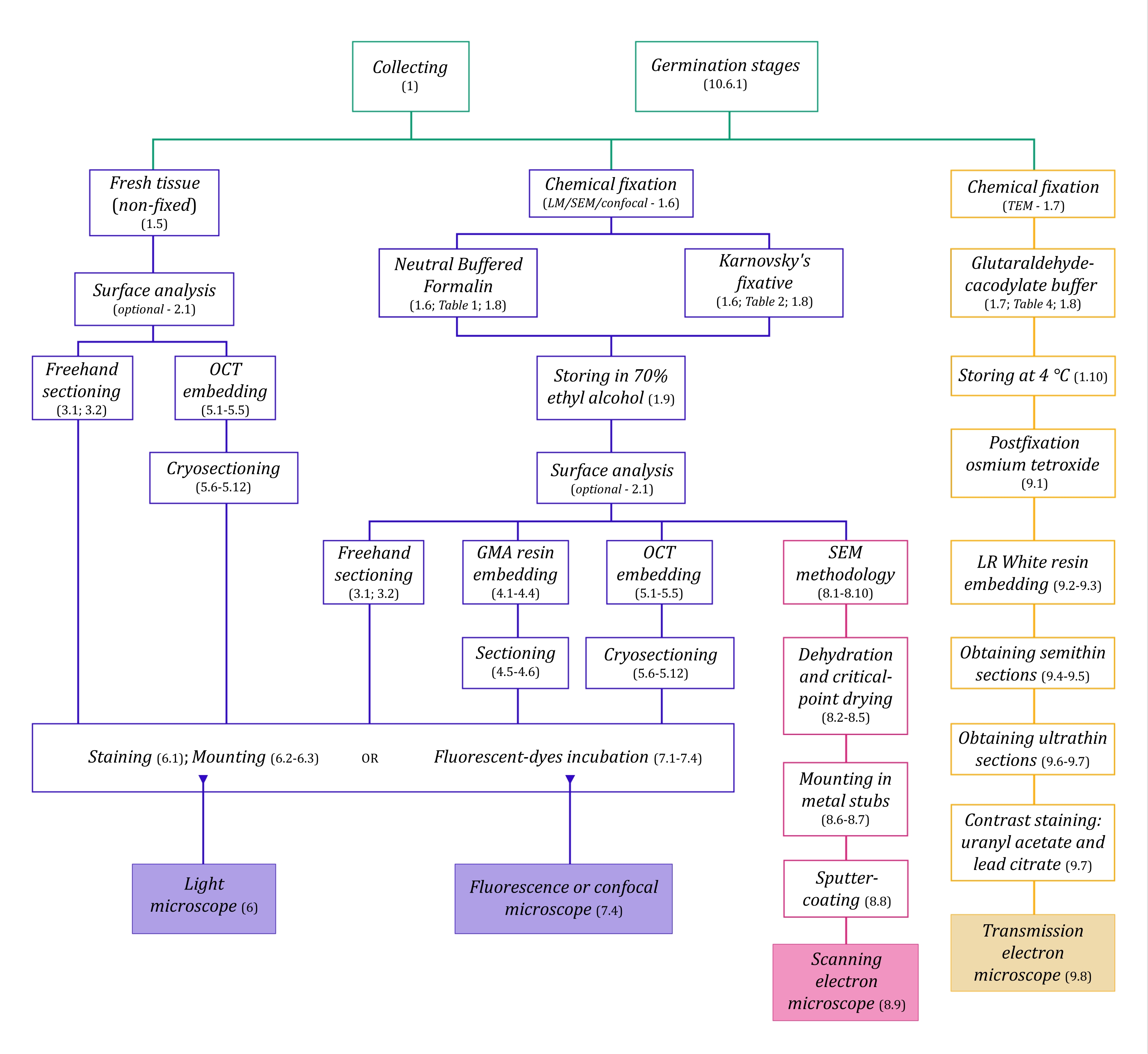{kind=link}
ここで詳細に説明する顕微鏡技術(図1)の前には、サンプルの収集、固定、脱水、埋め込み、および切片作成という重要なステップがあります。手順は選択した技術に応じて可変であるため(図1)、準備して収集サイトに輸送する固定液、固定前のサンプルの準備方法、使用する脱水プロセス(セクション1)、およびさまざまな埋め込みの可能性とセクショニング方法(セクション4、5、 および9)。 図1 は、以下に徹底的に説明する各顕微鏡技術に必要なすべてのステップを順番にまとめたものです。
プロトコル
1.サンプルの収集、修正、および保守
注:完全にMHの植物は通常、主に湿気の多いゴミの多い地域の暗い森の下層12,13に見られますが、部分的にMHの植物はより開放的な森林で見つけることができます12,13。MH植物は通常、さまざまな形や大きさのよく発達した地下器官を持っています。
- MH種を採取するときは、地下の臓器を傷つけないように注意しながら植物基盤周辺の土壌を探索し、地上の臓器と地上の臓器が外れないように地面から植物を引っ張らないようにします。
- ガーデニングこてを使用して空中構造物を注意深く掘り起こし、根、茎、根茎、貯蔵器官などの地下器官を探索しながら、これらの構造を損傷することなく探索します。
- 壊れやすい構造を保存するために土壌粒子を取り除き、サンプルを固定する前に、これらの臓器を水道水で繊細に洗って残りの土壌粒子を洗い流します。
- 落葉に関連するMH植物は特別な注意を必要とします。菌糸を通して分解物質につながっている臓器を丁寧に集め、つながった構造物から引っ張らないようにし、デリケートな部分なので丁寧に集めてください。そのようなつながりを持つ構造物を保存し、分析のためにゴミも集めます。
- イメージング技術を使用して新鮮な材料を分析することを選択した場合は、十分な水分、十分な水分が植物を蒸発および保湿する密閉ビニール袋にサンプルを維持し、過剰な水がサンプルと接触しないようにします。すぐに実験室に輸送し、サンプルを採取したその日に分析し、分析時にサンプルがまだ保存されているかどうかに注意してください。
- 固定剤は、密閉容器に入れて収集場所に運びます。光学顕微鏡(LM)および走査型電子顕微鏡(SEM)のために採取後すぐにサンプルを次の固定液のいずれかに固定します:10%中性緩衝ホルマリン(NBF14、表 1)またはカルノフスキー溶液(修正15、 表2)。カルノフスキーの溶液は、0.2 Mリン酸緩衝液15で調製することができ、そのレシピは 表3に記載されています。
- 透過型電子顕微鏡(TEM)による分析のために、グルタルアルデヒド-カコジル酸ナトリウム緩衝液(修飾16、表4)の滴の中に厚さ 4〜3 mmのサンプルを1〜2 mmの厚さのより小さな切片に分割します。ドロップの外側にカットされたエッジを破棄します。添加剤の固定液であるため、サンプルの体積の10倍を超える固定液の量を有する収集チューブに切片を直ちに移す(すなわち、その分子は固定されたタンパク質に化学的に添加される16)。
注意: 記載されている3つの固定剤は非常に有毒です。特に現場での使用中は吸入を避けてください。手袋を使用して、ヒュームフード内のすべての固定剤を準備します。ヒ素ガスが形成される可能性があるため、カコジル酸塩と酸を混合しないでください16。 - 固定サンプルが固定液に浮遊している場合、これは植物組織にガスが存在することを示しています。空気やその他のガスは、固定液がサンプル2全体に浸透するのを防ぎます。すべてのサンプルが溶液17の底に沈むまで、組織をより小さな部分に再サンプリングし、真空ポンプ(-300〜-400 inHg圧力)を使用して、組織からガスを除去します。ポンプによって過度の圧力がかかると、サンプルが損傷する可能性があるため、注意してください。
- カルノフスキー溶液または10%NBFで少なくとも48時間後、サンプルを0.2 M PB(表3)で洗浄し、一連の10%、30%、50%、最後に70%エタノールで脱水します。デリケートなサンプルの場合は、各濃度で30分間脱水します。サンプルが大きい場合は、1時間以上脱水します。
注:70%エタノール溶液は、サンプルを保存するための理想的な媒体です。70%エタノール中のサンプルは、室温で何年も保存できます。固定剤の除去は固定後の必須ステップであるため、植物材料を固定液に長期間保管しないでください2。 - ポストフィックスに進む前に、サンプルをグルタルアルデヒド-カコジル酸塩で4°Cで保存します(ステップ9.1)。
| 10% 中性緩衝ホルマリン(NBF)14 | |
| ステップ1 | 80 mLの蒸留水に10 mLの37-40%ホルムアルデヒド溶液を加えます |
| ステップ 2 | 0.4gのリン酸ナトリウム一塩基性一水和物(NaH2PO4·H2O)を溶液にする |
| ステップ 3 | 0.65gのリン酸二ナトリウム、無水(Na2HPO4)を加える |
| ステップ 4 | 容量を100mLに構成します |
表1:10%中性緩衝ホルマリンレシピ14。
| カルノフスキーの解決策 (修正15) | |
| ステップ1 | 60〜70°Cの蒸留水20mL中 |
| ステップ 2 | パラホルムアルデヒド0.8gを加え(4%w/vを得るため)、攪拌しながら |
| ステップ 3 | 40%NaOHを1〜4滴加え、溶液が透明になるまで攪拌します |
| ステップ 4 | 冷却し、pH 7.2の0.2 Mリン酸緩衝液30 mLを加えます(表3)。 |
| ステップ 5 | 25%グルタルアルデヒドを0.1 M PB(pH 7.2)で希釈し、1%グルタルアルデヒドを得る(最終容量:~60 mL) |
| ステップ 6 | ステップ4で得られた溶液に1%グルタルアルデヒドを加え(ステップ5)、最大100 mLの固定液を作ります |
表2:カルノフスキーの溶液レシピ (修正15)。
| 0.2 M リン酸緩衝液 (PB) pH 7.2 | |
| ステップ1 | 14.196gのリン酸二ナトリウム、無水(Na2HPO4)を400mLの蒸留水に加える |
| ステップ 2 | 13.8gのリン酸ナトリウム一塩基性一水和物(NaH2PO4·H2O) |
| ステップ 3 | 溶液が透明になるまでかき混ぜる |
| ステップ 4 | 蒸留水で最終容量を500mLに調整します |
| ステップ 5 | pHを7.2に調整します |
| ステップ 6 | 0.1 M PBの場合は、1:1に希釈します |
表3:0.2 Mリン酸緩衝液のレシピ。
| 3%グルタルアルデヒド0.2 Mカコジル酸ナトリウム緩衝液 (修飾16) | |
| ステップ1 | 0.2 Mカコジル酸緩衝液:100 mLの蒸留水に4.28 gのカコジル酸ナトリウム三水和物を加える |
| ステップ 2 | pHを7.2に調整します |
| ステップ 3 | ステップ2の溶液25 mLに12 mLの25%グルタルアルデヒドを加えます(0.2 Mカコジル酸緩衝液pH 7.2)。 |
| ステップ 4 | 蒸留水で容量を100 mLに構成します |
表4:3%グルタルアルデヒド0.2Mカコジル酸ナトリウム緩衝液レシピ (修正16)。
2. 固定・非固定材料中の臓器の表面分析
- 臓器、特に地下の臓器や落葉と接触する臓器の表在菌糸を分析するには、分析するサンプルに応じて、7.5倍以上の倍率で解剖顕微鏡(実体顕微鏡)で固定または新鮮な物質を観察します。
- 固定液、70%エタノール(保存されている場合)、または新鮮な材料の場合は水道水に浸したサンプルを視覚化します。解剖顕微鏡はサンプルを乾燥させて損傷する可能性があるため、直接光を避けてください。
- 表在性菌糸と根茎によって導かれるサンプル内の関心のある領域を検索します。表面的な根粒菌を含む領域を含むサンプルを選択して、根と茎の皮質細胞内のペロトンと菌糸コイルを視覚化することができます。
- 選択後、サンプルがまだ固定されていない場合は、手順1.6および1.9に従います。必要に応じて、セクション3で説明したように、固定せずに光学顕微鏡を使用して新鮮なサンプルを撮影します。
- 実体顕微鏡に結合されたカメラを使用して、臓器表面、根粒形、および観察された他の構造から画像を収集します。このような場合は、素材とのコントラストが合うように適切な背景色を配置し、可能であれば、表面が粗くない背景素材(紙など)を選択してください。
3.植物器官のフリーハンドセクション
注意: 植物器官のフリーハンドセクションは、特に小さくて薄い構造の場合、困難な場合があります。しかしながら、真菌性内生植物を有する組織のこれらの切片は、場合によっては、薄い切片と比較して菌糸および他の特徴をよりよく明らかにすることができる。
- 新鮮なサンプルまたは固定されたサンプルを鋭利な刃で切断し、できるだけ薄く切断し、水(新鮮な場合)または70%エタノール(固定されている場合)を入れた小さなペトリ皿にすぐに置きます。小さな絵筆を使用して、セクションを損傷することなく操作します。
- より困難な材料(すなわち、小さく、薄く、柔軟な器官)の切片化を容易にするために、サンプルをポリスチレンや セクロピア 葉柄などの構造で囲みます。サンプルを収容するためにサポートを彫り、サンプルとサポートの薄いセクションを作成します。
- セクション6の説明に従ってサンプルを染色してマウントします。
4.植物サンプルの樹脂への埋め込みと切片作成
- さらに、70%エタノールに保存されたサンプルを80%、96%、および100%エタノール中の2xで、サンプルのサイズと組成に応じて30分から2時間脱水します。
- グリコールメタクリレート(GMA)樹脂キットは、製造元の指示に従って使用してください。さらなる考察については、Gerrits and Horobin (1996)18 を参照のこと。それに応じて、侵入と埋め込みの手順に従います。
注意:GMA樹脂は有毒であり、アレルギー反応や皮膚、目、粘膜の炎症を引き起こす可能性があります。ヒュームフード内の試薬を使用し、手袋を使用してください。 - 埋め込みには、サンプルサイズによって選択されたポリエチレン成形トレイを使用します(例:大きなサンプルの場合は13 mm x 19 mm x 5 mm、小さいサンプルの場合は6 mm x 8 mm x 5 mm)。金型内の目的のサンプルの向きに注意を払い、針を使用して方向付けに役立てます。
- 完全に固化するまで、好ましくは室温で重合のために放置する。硬化プロセスには通常数時間かかりますが、翌日にブロックの型を外すことをお勧めします。重合後、樹脂ブロックを金型から慎重に取り外し、ブロックの湾曲を避けるためにできるだけ早くブロックの取り付けに進みます。
- 取り付ける樹脂ブロックの面を研磨し、平らな面を作ります。樹脂ブロックを中粘度の液体シアノアクリレート接着剤で木製の直方体(2 cm x 2 cm x 3 cmを推奨)に接着します( 材料の表を参照)。切片化が損なわれないように、レジンが完全に付着していることを確認してください。
- 以下に説明するように、回転式ミクロトームで切片化を実行します。
注意: ミクロトームナイフの刃は非常に鋭利で、事故を引き起こす可能性があります。すべての安全対策に従って取り扱ってください。ナイフに近づく前に(ブロックを交換する、樹脂を湿らせるなど)、粗いハンドホイールをロックし、ブレードの安全カバーを取り付けます。使い捨てブレードは適切な場合に保管してください。ブレードを交換するときは細心の注意を払ってください。
注:GMA樹脂18のセクションには、さまざまな種類のナイフ(使い捨てまたは固定、ガラスまたは鋼など)を使用できます。セクションの品質は、ナイフの鋭さによって異なります。ナイフがしっかりと取り付けられていて、動かないことを確認してください。使い捨てナイフは、より良い切片化を達成するために定期的に交換する必要がある場合があります。- 木製の直方体をブロックホルダーにしっかりと取り付けます。オリエンテーションネジを使用してセクショニングの向きを調整し、ナイフチルトを使用してナイフの適切な角度を確保します。セクションの厚さを選択します。GMAセクションは薄くなるとスライドガラスにより適切に接着するため、トリミングには厚い設定を使用し、選択したセクションには薄い設定を使用します。植物組織の推奨厚さは5〜8μmです。
- 開始する前に、温度が高くなるとセクションの品質が低下するため、必要に応じて部屋を冷やしてください。セクショニング用に次のものを準備します:蒸留水を入れたビーカー、ホットプレート、絵筆(少なくとも2つ)、ファインポイントピンセット、パスツールピペット、スライドガラス、ろ紙(またはティッシュペーパー)、および鉛筆(切片化されるサンプルを識別するため)。
- ホットプレートを50°Cに調整し、ビーカーをその上に置きます。ホットプレートの面積による加熱の違いに注意してください(通常、中央の領域は端よりも加熱され、できれば中央の水を加熱します)。
- スライドガラスを選び、鉛筆で識別し、スライド表面全体に温かい蒸留水をピペットで貼り付けます。必要に応じて、溶液(洗剤と水、または70%エタノールなど)を使用して水とガラスの間の張力を解き、スライド全体が均等に覆われるようにします。一部のスライドタイプは、セクションの適切な接着性を得るために、70%エタノールで事前に洗浄する必要があります。
- まず、樹脂ブロックの面をナイフの刃に向かって徐々に進めます。最初にブロックを進めずにセクショニングを試みないでください、さもなければ機器とブロックが損傷する可能性があります。必要に応じて、より高い厚さ(10 μm以上)を使用してブロックをトリミングします。
- 適切なセクションに近づくときは、ハンドホイールでしっかりと一方通行の動きをして、セクションがすぐに作られるようにします。樹脂の水分を確認してください。切片作成中、カール部分に問題がある場合は、蒸留水に浸した絵筆を使用して、切断するブロックの表面を定期的に保湿します。ティッシュペーパーで余分な水分を取り除きます。
- 一対の細かいピンセットで、得られた部分をスライドの上の水に入れます。水と接触すると、GMA樹脂が伸びます。必要に応じて、絵筆を使用してセクションをそっと広げて伸ばします。別の絵筆を使用して、ブレードに樹脂の破片が常にないようにしてください。ブレードを濡らさないように、絵筆を切り替えないでください。
- スライド内のすべての目的のセクションをキューに入れた後、スライドの底面を乾燥させ、ホットプレートの上に置きます。ろ紙で軽くたたくことで、スライドの上部から余分な水分を取り除きます(オプション)。水がスライドから蒸発すると、セクションが付着します。過度の熱によるセクションの損傷を防ぐために、スライドを長く放置しないでください。
- スライドは、ほこりや太陽を避けてスライドボックスに保管し、染色やその他の手順に使用してください。スライドは数年間保存できます。
5.植物サンプルの凍結とクライオスタットによる切片作成
注:生体組織の凍結切片化における重要な考慮事項は、サンプルを凍結する際の氷晶形成による損傷を減らすことです。凍結保護は通常、グリセロールやスクロース19,20などの化学的に不活性な溶液を注入することによって行われます。
- サンプル切片作成の 1 日前に、次の手順を実行します。
- 100 mLの0.2 M PB(表3)を100 mLの蒸留水で希釈し、200 mLの0.1 M PBを得ました。0.1 M PB中の10%、20%、および30%のスクロース溶液を調製します(たとえば、10%溶液の場合は、20 mLのバッファーに2 gのスクロースを追加します)。
- 新鮮なサンプルの場合は、0.1 M PBで30分間洗浄します。固定液中のサンプルについては、固定液の調製に使用したのと同じバッファーで30分間洗浄します。70%エタノール中のサンプルの場合は、50%および30%エタノールで水和し、各溶液で0.1 M PBで1時間洗浄します。
- サンプルを室温で10%スクロース中で2時間、20%スクロース中で2時間、30%スクロース中で2時間インキュベートします。その後、30%スクロース中で4°Cで一晩インキュベートします(または少なくとも3時間、最大時間は48時間です)。
- 切片化の日に、0.1M PB中に40%および50%スクロースを調製する。12時間以上前にスクロース溶液を調製しないでください。各スクロース濃度で4°Cで2時間インキュベートします。
- 埋め込むには、小さな金型にOCTコンパウンド(サンプルの埋め込みと凍結に使用される最適な切断温度媒体)の層を作り、-20°Cで保持して凍結します。型は通常の組織学的型にすることができるが、ブロックの型抜きを容易にするために、紙または錫箔のものは、フレームおよび粘着テープとして小さな直方体を使用して作ることができる。
- OCTコンパウンドの最下層を金型内で凍結させた後、クライオスタットチャンバー(約-27°C)内で作業します。50%スクロースでインキュベートしたサンプルを、切片化する方向に型に入れます。直方体ブロックの上面は、通常、セクショニングのより良い面です。サンプルが配置されている金型にマークを付けて、ブロックを簡単にトリミングし、正しい向きを維持できるようにします。
- サンプルをOCTコンパウンドで囲み、サンプルに触れる気泡を破裂させます。-20°Cで凍結します。 ブロックを完全に凍結させた状態で、クライオスタットチャンバー(約-27°C)に入れます。使用する前にのみそれぞれを型を外し、ブロック内のサンプルの位置を示すマークに注意してください。
- OCTコンパウンドはブレードで簡単に切断できるため、ブロックを適切にトリミングしてからチャックに配置してください。クライオスタットチャックにOCTコンパウンドを入れ、上面が切片になるようにブロックを配置します。面積が小さい面は、より良い断面を提供します。ブロックがチャックにしっかりと取り付けられるまで待ち、セクションを開始する前にテストします。
- チャックをチャックホルダーにしっかりと入れます。方向ネジを使用して、断面の方向を調整します。ナイフの傾きを使用してナイフの角度を調整します。断面の厚さを選択します。サンプルは、通常の樹脂セクションよりも厚く切断することができます。5〜20μmの範囲の切片が正常に得られ、厚い切片が作りやすくなります(カールが少なく、構造の損傷が少なくなります)。
- 凍結したブロックの面をナイフの刃に向かって進めます。そうせずにセクショニングを試みないでください、さもなければブロックはチャックから取り外されて損傷する可能性があります。必要に応じて、より高い厚さ(10 μm以上)を使用してブロックをトリミングします。
- 適切なセクションに近づくときは、アンチロールプレート(つまり、セクションを保持する透明なプレート)をナイフの上に置き、ハンドホイールでしっかりと一方向に動かすようにして、セクションがすぐに作成されます。カールの問題は、アンチロールプレートを調整する必要がある場合(通常はブレードに対して調整可能)、またはブレードに破片がある場合に発生する可能性があります。破片を取り除くために、絵筆で常に刃を掃除してください。
- シラン化スライド(市販またはアセトン21中の2%アミノアルキルシランで調製)や、蒸留水21または0.2%ゼラチン中の500 μg/mLポリL-リジンで調製されたスライド(詳細21を参照)のように、セクションが簡単に取り付けられるように特別なスライドを使用してください。スライドを室温に保ちます。
- セクションをスライドに接着するには、アンチロールプレートを持ち上げて、スライドをセクションにすばやく接触させます。スライドは室温であるため、切片はすぐに溶けてスライドに付着します。スライドの処理面を、ナイフの上またはアンチロールプレートの内面に残っている可能性のあるセクションに向けるように注意してください。セクションのカールを避けるために、プレートが持ち上げられたらすぐにこの手順を実行し、セクションをゆがめないように注意してください。
- スライドをクライオスタットチャンバーの外側(室温)に残して、新しいセクションを追加します。必要な切片をすべてスライドに接着した後、クライオスタットチャンバー内または冷凍庫(-20°C以下)に保管してください。スライドを湿気にさらさないでください。それらをスライドボックスに保管し、鉛筆でスライドを識別することを忘れないでください。
注:OCTのスライドとブロックは、-20°Cで保存できますが、長すぎません。より良い結果を得るには、数日以内にスライドとブロックを使用してください。
6. 光学顕微鏡のための植物切片および内生植物の染色
注:植物のセクションには多くの種類の汚れを使用できます。内生菌や植物組織を鑑別染色することは困難です。染色手順ではありませんが、真菌構造をマーキングする方法はセクション7(小麦胚芽凝集素コンジュゲートによる蛍光)に示されています。フリーハンド切片(セクション3で説明)、樹脂切片(セクション4)、および凍結切片(セクション5)を染色できますが、GMA樹脂とOCTがスライドへの接着を失うため、フェノールおよびアルコールベースの染色はこれらのサンプルでは困難です。
- 植物サンプルには、以下の通常の染色方法を使用するか、組み合わせます。
- トルイジンブルーO22,23は、植物切片の一般的な染色に広く適用された方法である。組織の種類および種類に応じて、0.1 Mリン酸(pH 6.8)または0.09 Mクエン酸緩衝液(pH 4.5〜4.8)中の0.05%トルイジンブルーOの溶液を調製する。スライド染色ジャーを使用するか、スライド染色ジャーを使用して、またはスライドがほとんど染色されていない場合はセクションの上に数滴を置いて、GMA樹脂切片を2〜10分間インキュベートします。インキュベーション後に蒸留水またはバッファーで注意深く洗浄し、スライドを水でマウントするか、ホットプレートで乾燥させて、手順6.3で説明されているように永久スライドを作成します。
- ルゴール試薬2 はデンプンの存在を示す。蒸留水中で5%ヨウ素(I2)および10%ヨウ化カリウム(KI)溶液を調製する。スライドの上に数滴を加えて2分間切片を染色し、蒸留水で洗います。この組織化学的検査は通常、一時的なスライドに適用されます。
- スーダンIII、IV、および黒B24,25は、異なる脂質について染色します。70%エタノール中の0.3%スーダン(III、IV、または黒B)の溶液を調製し、沸騰するまで温めて冷まします。上清を使用し、それを濾過し、そして閉じたペトリ皿中で15〜30分間切片をインキュベートする。切片を70%エタノールと蒸留水で注意深く洗浄します。スライドを水で取り付けます(通常は一時的なスライドにのみ適用されます)。
注:スーダンはアルコールベースの染料であるため、フリーハンドセクションに適しています。GMA樹脂セクションの染色は、通常スライドから外れるため、慎重に行ってください。
- 一時的なスライドの場合は、切片を水またはグリセリンに取り付け、その後観察します。カバーガラスをマニキュアで密封して、もう少し長く保存します。
- 永久スライドの場合は、合成樹脂(ラピッドマウントメディアなど、 材料表を参照)でセクションを取り付けます。封入剤を数滴滴下し(カバーガラスがオーバーフローする可能性があります)、泡を避けるためにカバースリップを注意深く置き、衣服ペグを使用してスライドをカバースリップに押し付けます。乾燥した余分な封入剤をかみそりの刃で取り除きます。
7. 小麦胚芽凝集素に結合した蛍光色素の蛍光および共焦点顕微鏡への応用
メモ: この方法は、フリーハンドセクション(セクション3で説明)、レジンセクション(セクション4)、および凍結セクション(セクション5)に適用できます。凍結切片は、樹脂切片と比較して厚いサンプルを提供できるため、共焦点顕微鏡の目的には十分ですが、フリーハンドのものほど厚くはありません。小麦胚芽凝集素に結合した蛍光色素(WGA、 材料の表を参照)を蛍光顕微鏡26における真菌イメージングに適用する。共焦点顕微鏡は必須ではありませんが、植物構造の鮮明な3次元画像を提供できます27。
- 0.1 M PB28 中の0.2 mg/mL WGA-蛍光色素コンジュゲートの溶液を調製します(pH 7.2、ステップ5.1.1および 表3を確認してください)。0.1 M PB(pH 7.2)中の1%カルコフルオールホワイトの溶液を調製します。切片はそれらと直接インキュベートされるので、少量のこれらの溶液を調製する。
- スライドガラス中の切片をWGA-蛍光色素コンジュゲート溶液29中で30分間インキュベートし、切片を覆うのに十分な容量を使用して、次いで0.1 M PBで洗浄する。
- カルコフルーア溶液中の切片を、封入剤として十分な量を使用してインキュベートします。溶液は観察期間中維持することができる。
- スライドにカバーガラスを置き、次のフィルターを使用して共焦点顕微鏡または蛍光光学顕微鏡で観察します:TC / GFP(励起:470-440、発光:525-550、 材料表のWGA-蛍光色素の場合-真菌細胞壁はこのフィルター29の下で緑色に蛍光を発します)およびDAPI(励起:358、発光:463、カルコフルオールホワイトの場合)30。
注:3次元画像は、共焦点顕微鏡27のZシリーズ機能を使用して取得できます。
8. 植物器官の走査型電子顕微鏡
- サンプルを固定し、脱水を行い、70%エタノール中に保存した後(セクション1)、1つの可能性は、必要に応じてSEM分析のために任意の所望の表面(例えば、内部組織、卵巣構造)を露出させるためにサンプルを切断することである。鋭くて新しいかみそりの刃を使用し、一方向の動きで切り込みを入れ、SEMでこれらの領域の外観が損傷しないようにします。必要に応じて、実体顕微鏡を使用してサンプルを選択し、金属スタブ領域を考慮してサンプルサイズを決定します。
- エタノール系列のSEM用サンプルをさらに脱水します:無水エチルアルコール(≥99.8%)で80%、96%、および2倍。小さくて繊細なサンプルを各濃度で30分間維持し、大きくて密度の高いサンプルを1時間維持します。
- ティッシュペーパーを使用して小さな封筒を折りたたんでサンプルを整理します 次のステップでは、大きなサンプルは封筒なしで処理できます。文字または数字を書いて鉛筆で封筒を識別し、それぞれのサンプルのすべてのログを保持します。サンプルを無水エタノールに保管しますが、長くはしませんが、できるだけ早くステップ8.4に進みます。
- 臨界点(CP)乾燥に進みます。CPドライヤーは、標準的な操作手順に従って操作してください。圧力チャンバー内の絶対エタノール(中間液)にサンプルを入れます。CO2臨界点(31°C、7.3 x 106 Pa)で、中間流体は遷移流体(液体二酸化炭素)に溶解し、サンプルは乾燥されます31。
- CP乾燥後、できるだけ早くサンプルを乾燥容器、例えばシリカゲルを含む密閉フラスコに保管してください。大気中の湿度は、再吸収された場合、サンプルを破壊する可能性があります31。
- 金属スタブを使用してサンプルをマウントします。取り付ける前に、手袋をはめてスタブを操作し、アセトンに5分間浸して脂肪を取り除き、乾燥させます。導電性両面カーボン粘着テープを使用してサンプルをスタブに固定し、実体顕微鏡を使用してサンプルの位置決めを支援し、上からの視界がSEM画像で唯一の可能な遠近法であることを念頭に置いてください。
- ピンセットでサンプルを操作すると、ピンセットが触れるサンプル部分が損傷することが多いので注意が必要ですので、対象領域から離れた位置にある部品(テープと接触する領域など)に触れてみてください。シリカゲルを含む密封されたペトリ皿にサンプルを含むスタブを維持します。できるだけ早く手順 8.8 に進みます。
- スパッタコーターを使用して、不活性ガス(多くの場合アルゴン31)の低圧雰囲気でサンプルの表面に金属層(通常は金または白金)を堆積させます。スパッタコーターを使用する場合は、標準的な操作手順に従ってください。コーティングの厚さは、サンプルのトポグラフィーに依存し、通常は15〜40nm32の間です。
- コーティングされたスタブをシリカゲルで密封されたペトリ皿に維持し、シリカゲルが湿度を保持している場合、サンプルはこの方法で数週間保存できます。走査型電子顕微鏡を使用してサンプルを分析します。 真空中の 試料は電子ビームによって打たれ、そのような相互作用からの信号の放出は画像31として解釈される。走査型電子顕微鏡の操作の詳細については、Jeffree and Read (1991)31 および Bozzola and Russell (1999)32 をお読みください。
- スタブを再利用するには、粘着テープを引っ張り、ワイヤーウールでこすります。水道水で洗い、無水エタノールに浸し、十分に乾燥させることで、構成金属の酸化を防ぎます。
9. 透過型電子顕微鏡
- ステップ1.7および1.10で説明されているように、サンプルにグルタルアルデヒド-カコジル酸バッファーを接頭辞として付けます。12〜24時間のプレフィクション後、サンプルを0.2 Mカコジル酸バッファー(pH 7.25)で10分間3回洗浄します。0.2 Mカコジル酸バッファー中の1%四酸化オスミウム(OsO4)を暗所で室温で12時間後固定します。蒸留水で3回5分間洗浄します。
注意: カコジル酸塩と四酸化オスミウムは非常に有毒であるため、吸入しないでください。.それぞれの安全データシートに従って、ヒュームフードで使用してください。 - サンプルを30%、50%、70%、および96%のエタノールで各濃度で2倍、10分間脱水します。次に、無水アルコール中で3回、毎回15分間脱水します。
- サンプルを親水性アクリル樹脂( 材料表を参照)に1:1樹脂+無水エタノールで1回、純樹脂で3倍ずつ8〜12時間浸します。ゼラチンカプセル中で、完全に固化するまで60°Cで重合を行う(12時間最大17時間)。
- 樹脂ブロック内のサンプルの向きを綿密に評価します。かみそりの刃でブロックの上部を切り取り、サンプルを切片領域に集中させるピラミッド型にします。ダイヤモンドナイフを使用してウルトラミクロトームで半薄切片(250-500 nm)33 を取得し、数滴の水を入れてスライドガラスの上に置きます。
- スライドを60°Cのホットプレートに保管してください。 手順6.1.1のように切片をトルイジンブルーOで染色し、汚れを完全に乾かします。水道水でよく洗ってください。得られた断面を4つの象限を描き、分析に最も適した象限を選択して評価します。
- ピラミッド形状が選択した象限をブロックの上面に集中するようにブロックをトリムします。超薄切片(50-100 nm)33,34を作成します。厚さは、切片の干渉色に従って評価される:約70nmの切片は銀金色に見え、約100nmの切片は金色に見え、約200nmの切片は青く見える34。
- 銅グリッドを用いて水から極薄切片を採取し、下記のように酢酸ウラニルとクエン酸鉛による造影染色法に進みます。
- クエン酸鉛溶液(表5)を調製し、それぞれ1 mLの溶液を入れたマイクロ遠心チューブで最終溶液を凍結し、使用直前に解凍します。
- 酢酸ウラニル溶液を調製する:0.625 gの酢酸ウラニル[UO 2(CH3COO)2]を最近沸騰および冷却した蒸留水25 mLに溶解します。冷凍庫の暗いフラスコに保管してください。
注意: 硝酸鉛は摂取すると有毒です。1 N NaOHは非常に腐食性があります。酢酸ウラニルは放射性で有毒です。摂取、吸入、または皮膚に触れないでください。. - 染色する場合は、調製した両方の試薬をフィルターユニット(0.22 μmの細孔、材料表を参照)を備えた別々の3 mLシリンジに入れます。シーリング熱可塑性フィルムを逆さまにして逆さまにし(材料の表を参照)、より広い皿の中に、CO 232のトラップとしてNaOHペレットを端に置いたペトリ皿を準備します(図2を参照)。
- 最初の一滴を捨て、フィルムの上に一滴の酢酸ウラニルと3滴の蒸留水を各格子染色のために置く。クエン酸鉛でも同じことを行い、さらに3滴の蒸留水を加えます。
- グリッドを(不透明な面を下にして、セクションがある場所)ウラニルで30分間(可変時間)インキュベートします。蒸留水滴で3回洗浄し、鮮やかな面のろ紙で毎回グリッドを穏やかに乾燥させます。クエン酸鉛滴(30分)で繰り返し、洗浄します。
- 少なくとも4時間後、透過型電子顕微鏡でグリッドを分析します。この顕微鏡では、電子ビームが 真空中の 切片を通過し、画像が蛍光スクリーンに投影されます。透過型電子顕微鏡の操作の詳細については、Bozzola and Russell (1999)32をお読みください。
| クエン酸鉛溶液 (TEM造影染色用) | |
| ステップ1 | ビーカーをティンフォイルで囲む |
| ステップ 2 | 0.266 gの硝酸鉛[Pb(NO3)2]を最近沸騰して冷却した蒸留水6 mLに溶解します。 |
| ステップ 3 | 2分間攪拌します |
| ステップ 4 | 0.352 gのクエン酸三ナトリウム[Na3(C6H5O7).2H2O]を加える(溶液は乳白色の外観を獲得しなければならない) |
| ステップ 5 | 15分間攪拌し、ビーカーをティンフォイルで密封し、溶液を10 mLビーカーに移します |
| ステップ 6 | 1.6 mLの1N NaOHと2.4 mLの蒸留水を追加します(溶液は半透明でなければなりません) |
| ステップ 7 | 必要に応じて、pHを12に近づけます |
表5:クエン酸鉛溶液のレシピ。

図2:クエン酸鉛および酢酸ウラニル溶液によるコントラスト染色スキーム 。 (A)ペトリ皿を準備し、1つは熱可塑性フィルムで逆さま(中央)にして、滴をその上、幅の広いものの内側に置くことができるようにします。NaOHペレットは中央の皿の周りの場所です。(B)酢酸ウラニルの滴は文字Uで円の中に置かれ、クエン酸鉛の滴はL.DWとマークされた円の中に置かれます。グリッドはカラム内で順次染色されるため、表されているように5つのグリッドを同時に染色できます。 この図の拡大版を表示するには、ここをクリックしてください。
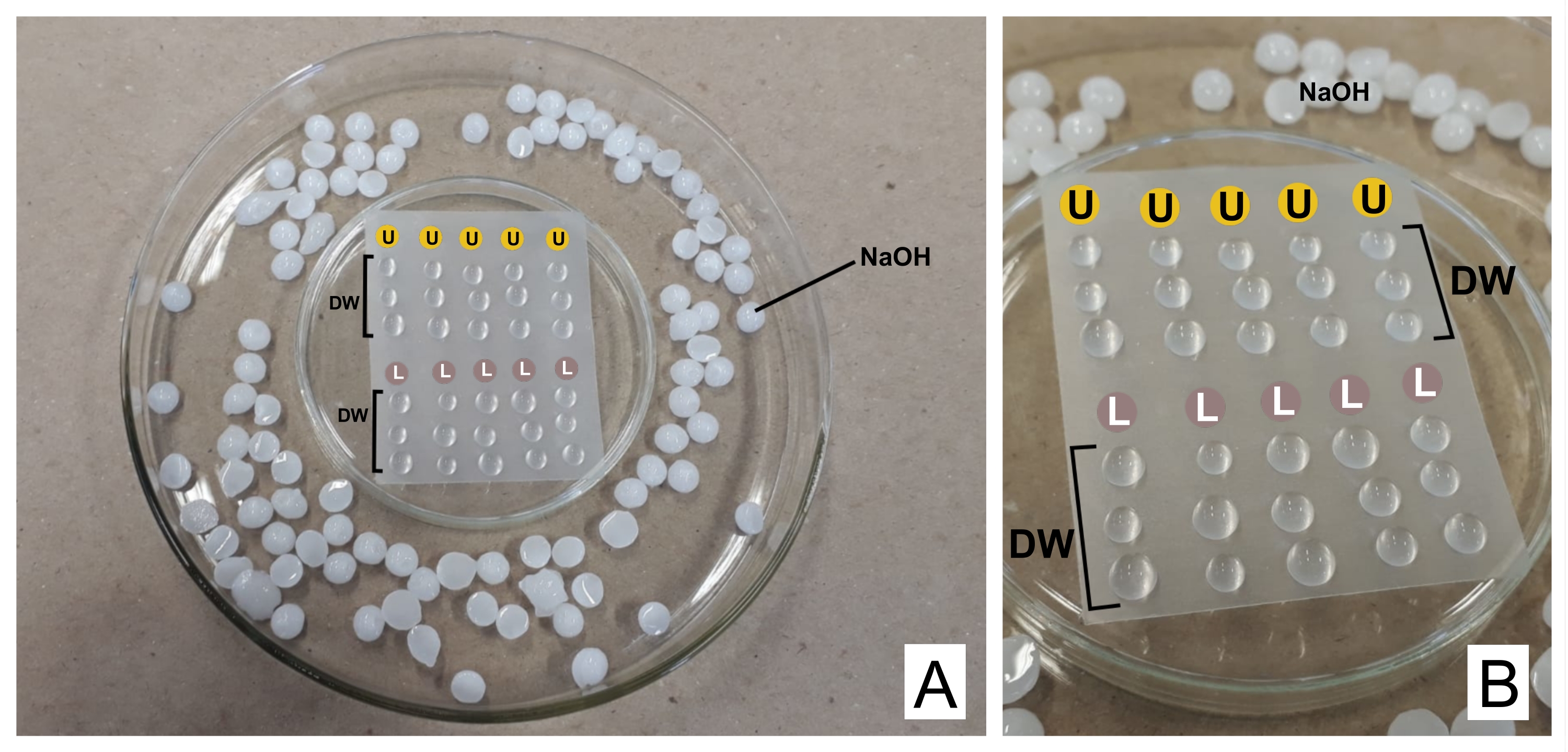{kind=link}
10.ラン種子の共生発芽
- 汚染を避けるために、種子の共生発芽に使用される溶液とすべての材料が無菌であることを確認してください。まず、121°Cで20分間オートクレーブします。 共生発芽ステップを 図3にまとめる。
- 種子皮の剛性と厚さを考慮して、果物の場合は2%活性塩素を含む次亜塩素酸ナトリウム溶液に10〜15分間、種子の場合は7〜10分間浸漬することにより、果物と種子を表面的に消毒します9。スリムで壊れやすい種子は、1:1の希釈次亜塩素酸ナトリウム溶液に浸すことができます。その後、オートクレーブ滅菌した蒸留水で3回洗浄し、次亜塩素酸塩溶液を除去します。
- セリグラフィ布で濾過することによって種子を回収し、種子を使用して発芽試験を進行させる(好ましくは)。必要に応じて、シリカゲルを含むガラスフラスコ内の濾紙封筒に4°Cで保管し、フラスコを密閉し、粘着フィルムで密封します。最後の洗浄から数滴の水をポテトデキストロース寒天培地(PDA、39 g / L)に移し、洗浄プロセスの有効性を評価します。
- 培養液に種を蒔く前に、後述するようにテトラゾリウム試験(任意)を通じてそれらの生存率を評価する35。
- 約10 mgの種子を微量遠心チューブに入れ、蒸留水中の10%スクロース1 mLを入れ、室温(約25°C)で光の中で24時間インキュベートします。
- マイクロピペットでスクロース溶液を取り除き、蒸留水中の1%テトラゾリウム溶液(塩化トリフェニルテトラゾリウム)1 mLを加えます。サーモブロック中で40°Cで暗所で24時間インキュベートします。
- マイクロピペットでテトラゾリウム溶液を取り除き、種子を蒸留水で2回または溶液が除去されるまで洗浄する。すべての液体を取り除きます。必要に応じて、種子は分析される前に(ステップ10.3のように)最大1週間冷凍庫に保管することができます。
- 種子を蒸留水に再懸濁し、光学顕微鏡で分析します。生存可能な種子は明るい色から濃い赤色を獲得しますが、生存不可能な種子は自然な色を保持します。
- ラン種子の共生発芽のために以下の適合9 プロトコルを実行する。
- オートクレーブ処理したろ紙ディスク(直径1〜2 cm)でシードをオートミール寒天(OMA)培地(2.5 g / Lのオーツ麦フレークと7 g / Lの寒天、pH 6)と一緒にペトリ皿に入れます。
- ペトリ皿の中央に、発芽手順のために選択された単離された真菌からの菌糸体を含む培養液の断片(約1 cm2)を接種します。ペトリ皿を粘着フィルムで密封し、真菌の増殖に適しているため、約25°Cまたは室温の暗所でインキュベートします。
- 発芽試験の陰性対照として、種子と真菌接種なしでいくつかの皿を準備します。
- 定量的および定性的なデータを収集し、原球茎と苗を撮影することにより、発芽結果を毎週分析します。種子および原球の観察は、発芽をより正確に評価するために実体顕微鏡を使用して実行する必要があります。下からの光源を使用すると、コントラストが高くなり、真菌菌糸体と原球菌をより簡単に区別できるようになります。
- さまざまな発達段階でサンプルを収集し、解剖学的分析のために修正します(セクション1)。発芽中の種子、原球菌、および苗の真菌内生植物を調べるために、前述のすべての画像分析を適用します(光学顕微鏡-セクション4、5、および6、共焦点および蛍光-セクション7;SEMおよびTEM - セクション8および9)。
- 表6に従ってステージの分類に続いて定量結果を生成します。段階は、菌従属栄養ランからの種子の通常の発達を説明する。毎週データを収集し、観察された各段階の最初の日付を表にします。
- さらに、発芽率と発芽率を推定する定量データを収集します。少なくとも 100 個のシードをカウントするか、カウント フィールド35 を定義します。標準化された領域を持つ固定領域で構成されるペトリ皿ごとに3つ以上のカウントフィールドを区切り、毎週評価します。成長指数(GI)の式に従って収集されたデータを計算します。

ここで、N 0はステージ0 でカウントされたシードの数であり、N 1はステージ1 を指し、ステージ6(N6として登録)36まで続きます。

図3:種子の共生発芽方法論の概略要約。 回路図は、プロトコルの詳細な手順を示しています。略語:OMA =オートミール寒天培地、PDA =ポテトデキストロース寒天培地。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| 発芽段階 | 形容 |
| 0 | 発芽なし |
| 1 | 胚の腫れ |
| 2 | テスタ破裂 |
| 3 | 吸収毛が発達する |
| 4 | 茎の投影が発達する |
| 5 | 保護スケール(苞葉)が発達する |
| 6 | 最初の根が発達する |
表6:発芽試験の定期的な分析に適用される原始球発育段階の説明。 Otero et al.36に記載されている段階から変更されました。
結果
植物組織を固定する重要な段階に続いて、細胞成分および組織の形態、体積、および空間的組織を考慮して、生きている状態に可能な限り類似した細胞構造をもたらす16。化学的固定後のサンプルでそのような特性を観察します(図4)。図4C-Fは、光学顕微鏡下で適切に固定されたサンプルを表しています。...
ディスカッション
植物の解剖学と形態学における画像解析は、地下器官の研究6,40、種子の共生発芽の構造解析39、および空中および生殖構造41によって実証されるように、目的を達成し、マイコ従属栄養植物とその不可欠な真菌内生植物の関係を理解するのに役立つ重要な可能性を秘めています。.構造植物学は、過去10年間で植物...
開示事項
著者は開示するものは何もありません。
謝辞
著者らは、FAEPEXおよびFAPESP(2015/26479-6)からの資金提供に感謝する。MPPは、修士号奨学金(プロセス88887.600591 / 2021-00)とCNPqに感謝します。 JLSMは、生産性助成金(303664 / 2020-7)に対してCNPqに感謝します。著者らはまた、LME(電子顕微鏡研究所-IB /ユニキャンプ)、INFABiC(細胞生物学に適用されるフォトニクスに関する国立科学技術研究所-ユニキャンプ)、およびLaBiVasc(血管生物学研究所-DBEF / IB /ユニキャンプ)によって提供される機器と支援へのアクセスに感謝します。LAMEB(UFSC)およびエリアナ・デ・メデイロス・オリベイラ(UFSC)は、凍結保護プロトコルへの貢献に対して。TEMプロトコルへの貢献のためのLME。
資料
| Name | Company | Catalog Number | Comments |
| Acetone | Sigma-Aldrich | 179124 | (for SEM stubs mounting) |
| Agar-agar (AA) | Sigma-Aldrich | A1296 | (for seeds germination tests) |
| Calcofluor White Stain | Sigma-Aldrich | 18909 | fluorescent dye (detects cellulose) |
| Citrate Buffer Solution, 0.09M pH 4.8 | Sigma-Aldrich | C2488 | (for toluidine blue O staining) |
| Conductive Double-Sided Carbon Tape | Fisher Scientific | 50-285-81 | (for SEM) |
| Confocal Microscope | Zeiss | (any model) | |
| Copper Grids | Sigma-Aldrich | G4776 | (for TEM) |
| Critical-point dryer | Balzers | (any model) | |
| Cryostat | Leica Biosystems | (any model) | |
| Dissecting microscope | Leica Biosystems | (= stereomicroscope, any model) | |
| Entellan | Sigma-Aldrich | 107960 | rapid mounting medium for microscopy |
| Ethyl alcohol, pure (≥99.5%) | Sigma-Aldrich | 459836 | (= ethanol, for dehydration processes) |
| Formaldehyde solution, 37% | Sigma-Aldrich | 252549 | (for NBF solution preparation) |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128 | histological tissue fixative |
| Gelatin capsules for TEM | Fisher Scientific | 50-248-71 | (for resin polymerisation in TEM) |
| Gelatin solution, 2% in H2O | Sigma-Aldrich | G1393 | (dilute for slides preparation - OCT adherence) |
| Glutaraldehyde solution, 25% | Sigma-Aldrich | G6257 | (for Karnovsky’s solution preparation) |
| HistoResin | Leica Biosystems | 14702231731 | glycol methacrylate (GMA) embedding kit |
| Iodine | Sigma-Aldrich | 207772 | (for Lugol solution preparation) |
| Lead(II) nitrate | Sigma-Aldrich | 228621 | Pb(NO3)2 (for TEM contrast staining) |
| Light Microscope | Olympus | (any model) | |
| LR White acrylic resin | Sigma-Aldrich | L9774 | hydrophilic acrylic resin for TEM |
| Lugol solution | Sigma-Aldrich | 62650 | (for staining) |
| Metal stubs for specimen mounts | Rave Scientific | (for SEM, different models) | |
| Microtome | Leica Biosystems | manual rotary microtome or other model | |
| Oatmeal agar (OMA) | Millipore | O3506 | (for seeds germination tests) |
| OCT Compound, Tissue-Tek | Sakura Finetek USA | 4583 | embedding medium for frozen tissues |
| Osmium tetroxide | Sigma-Aldrich | 201030 | OsO4 (for TEM postfixation) |
| Parafilm M | Sigma-Aldrich | P7793 | sealing thermoplastic film |
| Paraformaldehyde | Sigma-Aldrich | 158127 | (for Karnovsky’s solution preparation) |
| Poly-L-lysine solution, 0.1% in H2O | Sigma-Aldrich | P8920 | (for slides preparation - OCT adherence) |
| Poly-Prep Slides | Sigma-Aldrich | P0425 | poly-L-lysine coated glass slides |
| Polyethylene Molding Cup Trays | Polysciences | 17177A-3 | (6x8x5 mm, for embbeding samples in GMA resin) |
| Polyethylene Molding Cup Trays | Polysciences | 17177C-3 | (13x19x5 mm, for embbeding samples in GMA resin) |
| Potassium iodide | Sigma-Aldrich | 221945 | (for Lugol solution preparation) |
| Potato Dextrose Agar (PDA) | Millipore | 70139 | (for seeds germination tests) |
| Scanning Electron Microscope | Jeol | (any model) | |
| Silane [(3-Aminopropyl)triethoxysilane] | Sigma-Aldrich | A3648 | (for slides preparation - OCT adherence) |
| Silane-Prep Slides | Sigma-Aldrich | S4651 | glass slides coated with silane |
| Silica gel orange, granular | Supelco | 10087 | (for dessicating processes) |
| Sodium cacodylate trihydrate | Sigma-Aldrich | C0250 | (for glutaraldehyde-sodium cacodylate buffer) |
| Sodium hydroxide | Sigma-Aldrich | S5881 | NaOH (for Karnovsky’s solution preparation and TEM contrast staining) |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044 | NaClO (for seeds surface disinfection) |
| Sodium phosphate dibasic, anhydrous | Sigma-Aldrich | 71640 | Na2HPO4 (for NBF solution and PB preparation) |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638 | NaH2PO4·H2O (for NBF and PB) |
| Sputter coater | Balzers | (any model) | |
| Sucrose | Sigma-Aldrich | S0389 | C12H22O11 (for cryoprotection and germination test) |
| Sudan III | Sigma-Aldrich | S4131 | (for staining) |
| Sudan IV | Sigma-Aldrich | 198102 | (for staining) |
| Sudan Black B | Sigma-Aldrich | 199664 | (for staining) |
| Syringe | (3 mL, any brand, for TEM contrast staining) | ||
| Syringe Filter Unit, Millex-GV 0.22 µm | Millipore | SLGV033R | PVDF, 33 mm, gamma sterilized (for TEM contrast staining) |
| Tek Bond Super Glue 793 | Tek Bond Saint-Gobain | 78072720018 | liquid cyanoacrylate adhesive, medium viscosity |
| Toluidine Blue O | Sigma-Aldrich | T3260 | (for staining) |
| Transmission Electron Microscope | Jeol | (any model) | |
| Triphenyltetrazolium chloride | Sigma-Aldrich | T8877 | (for the tetrazolium test in seeds germination) |
| Trisodium citrate dihydrate | Sigma-Aldrich | S1804 | Na3(C6H5O7)·2H2O (for TEM contrast staining) |
| Ultramicrotome | Leica Biosystems | (any model) | |
| Uranyl acetate | Fisher Scientific | 18-607-645 | UO2(CH3COO)2 (for TEM contrast staining) |
| Vacuum pump | (any model) | ||
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | TermoFisher Scientific | W11261 | fluorescent dye-conjugated lectin (detects sialic acid and N-acetylglucosaminyl residues) |
参考文献
- Evert, R. F. . Esau’s Plant Anatomy: Meristems, Cells, and Tissues of the Plant Body: Their Structure, Function, and Development. , (2006).
- Yeung, E. C. T., Stasolla, C., Sumner, M. J., Huang, B. Q. . Plant Microtechniques and Protocols. , (2015).
- Sokoloff, D. D., Jura-Morawiec, J., Zoric, L., Fay, M. F. Plant anatomy: at the heart of modern botany. Botanical Journal of the Linnean Society. 195 (3), 249-253 (2021).
- Leake, J. R. The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytologist. 127 (2), 171-216 (1994).
- Bidartondo, M. I. The evolutionary ecology of myco-heterotrophy. New Phytologist. 167 (2), 335-352 (2005).
- Imhof, S., Massicotte, H. B., Melville, L. H., Peterson, R. L. Subterranean morphology and mycorrhizal structures. Mycoheterotrophy. , 157-214 (2013).
- Rasmussen, H. N., Rasmussen, F. N. Orchid mycorrhiza: implications of a mycophagous life style. Oikos. 118 (3), 334-345 (2009).
- Rasmussen, H. N., Dixon, K. W., Jersáková, J., Těšitelová, T. Germination and seedling establishment in orchids: a complex of requirements. Annals of Botany. 116 (3), 391-402 (2015).
- Zettler, L. W. Terrestrial orchid conservation by symbiotic seed germination: techniques and perspectives. Selbyana. 18 (2), 188-194 (1997).
- Stewart, S. L., Kane, M. E. Symbiotic seed germination and evidence for in vitro mycobiont specificity in Spiranthes brevilabris (Orchidaceae) and its implications for species-level conservation. In Vitro Cellular & Developmental Biology - Plant. 43 (3), 178-186 (2007).
- Zhao, D. -. K., et al. Orchid reintroduction based on seed germination-promoting mycorrhizal fungi derived from protocorms or seedlings. Frontiers in Plant Science. 12, 701152 (2021).
- Selosse, M. A., Roy, M. Green plants that feed on fungi: facts and questions about mixotrophy. Trends in Plant Science. 14 (2), 64-70 (2009).
- Merckx, V. S. F. T., Mennes, C. B., Peay, K. G., Geml, J. Evolution and diversification. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 215-244 (2013).
- Boon, M. E., Drijver, J. Routine Cytological Staining Techniques: Theoretical Background and Practice. Macmillan International Higher Education. , (1986).
- Karnovsky, M. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. Journal of Cell Biology. 27 (2), 137-138 (1964).
- Hayat, M. . Fixation for Electron Microscopy. , (1981).
- Roland, J. C., Vian, B. General preparation and staining of thin sections. Electron Microscopy of Plant Cells. 1, 675 (1991).
- Gerrits, P. O., Horobin, R. W. Glycol methacrylate embedding for light microscopy: basic principles and trouble-shooting. Journal of Histotechnology. 19 (4), 297-311 (1996).
- Zhang, Z., Niu, L., Chen, X., Xu, X., Ru, Z. Improvement of plant cryosection. Frontiers in Biology. 7 (4), 374-377 (2012).
- BeneŠ, K. On the media improving freeze-sectioning of plant material. Biologia Plantarum. 15 (1), 50-56 (1973).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. Cold Spring Harbor Protocols. 2008 (5), (2008).
- Sakai, W. S. Simple method for differential staining of paraffin embedded plant material using toluidine blue O. Stain Technology. 48 (5), 247-249 (1973).
- O’Brien, T., Feder, N., McCully, M. E. Polychromatic staining of plant cell walls by toluidine blue O. Protoplasma. 59 (2), 368-373 (1964).
- Ventrella, M. C., Almeida, A. L., Nery, L. A., Coelho, V. P. d. e. M. Métodos Histoquímicos Aplicados às Sementes. Universidade Federal de Viçosa. , (2013).
- Pearse, A. G. E. . Histochemistry, Theoretical and Applied. , (1960).
- Andrade-Linares, D. R., Franken, P. Fungal endophytes in plant roots: taxonomy, colonization patterns, and functions. Symbiotic Endophytes. , 311-334 (2013).
- Wymer, C. L., Beven, A. F., Boudonck, K., Lloyd, C. W. Confocal microscopy of plant cells. Confocal Microscopy Methods and Protocols. , 103-130 (1999).
- Marques, J. P. R., Soares, M. K. M. Manual de Técnicas Aplicadas à Histopatologia Vegetal. FEALQ. , (2021).
- Navarro, B. L., Marques, J. P. R., Appezzato-da-Glória, B., Spósito, M. B. Histopathology of Phakopsora euvitis on Vitis vinifera. European Journal of Plant Pathology. 154 (4), 1185-1193 (2019).
- Marques, J. P. R., et al. Sugarcane cell wall-associated defense responses to infection by Sporisorium scitamineum. Frontiers in Plant Science. 9, 698 (2018).
- Jeffree, C. E., Read, N. D. Ambient-and low-temperature scanning electron microscopy. Electron Microscopy of Plant Cells. , 313-413 (1991).
- Bozzola, J. J., Russell, L. D. . Electron Microscopy: Principles and Techniques for Biologists. , (1999).
- Murray, S. Basic transmission and scanning electron microscopy. Introduction to electron Microscopy for Biologists. , 3-18 (2008).
- . Glossary of TEM terms Available from: https://www.jeol.co.jp/en/words/emterms/ (2021)
- Seaton, P. T., et al. Orchid seed and pollen: a toolkit for long-term storage, viability assessment and conservation. Orchid Propagation: From Laboratories to Greenhouses—Methods and Protocols. , 71-98 (2018).
- Otero, J. T., Ackerman, J. D., Bayman, P. Differences in mycorrhizal preferences between two tropical orchids. Molecular Ecology. 13 (8), 2393-2404 (2004).
- Koch, R. A., et al. Marasmioid rhizomorphs in bird nests: Species diversity, functional specificity, and new species from the tropics. Mycologia. 112 (6), 1086-1103 (2020).
- Webster, J., Weber, R. . Introduction to Fungi. , (2007).
- Sisti, L. S., et al. The role of non-mycorrhizal fungi in germination of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. Frontiers in Plant Science. 10, 1589 (2019).
- Martos, F., et al. Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytologist. 184 (3), 668-681 (2009).
- Alves, M. F., et al. Reproductive development and genetic structure of the mycoheterotrophic orchid Pogoniopsis schenckii Cogn. BMC Plant Biology. 21 (1), 332 (2021).
- Merckx, V. S. F. T. Mycoheterotrophy: an introduction. Mycoheterotrophy: The Biology of Plants Living on Fungi. , 1-17 (2013).
- Hall, J. L., Hawes, C. . Electron Microscopy of Plant Cells. , (1991).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved