
Method Article
Polarisation des cellules et analyses dérivées des monocytes humains M1 et M2 avec cytométrie du débit sur l’infection par mycobactérie tuberculeuse
Dans cet article
Erratum Notice
Résumé
Ce protocole fournit une méthode pour étudier l’infection de mycobactérie de tuberculose dans les macrophages humains M1- ou M2-polarisés basés sur la différenciation des monocytes périphériques de sang aux cellules macrophage-comme qui sont infectées par la souche virulente GFP-étiquetée H37Rv, et analysées avec la cytométrie de flux utilisant un panneau de 10 couleurs comprenant l’expression des marqueurs M1/M2 choisis.
Résumé
Les macrophages humains sont les principales cellules hôtes de l’infection intracellulaire de Mycobacterium tuberculosis (Mtb) et jouent ainsi un rôle central dans la lutte immunitaire contre la tuberculose. Nous avons établi un protocole expérimental pour suivre la polarisation immunitaire des cellules dérivées des myéloïdes en cellules macrophages M1 (classiquement activées) ou M2 (alternativement activées) par l’évaluation avec un panneau de cytométrie de flux de 10 couleurs qui permet la visualisation et la caractérisation profonde de la tonte vert fluorescent-protéine (GFP) étiquetée dans divers sous-ensembles de macrophages. Les monocytes obtenus auprès de donneurs de sang sains ont été polarisés en cellules M1 ou M2 en utilisant la différenciation avec le facteur macrophage-colonie-stimulant des granulocytes (GM-CSF) ou le facteur stimulant macrophage-colonie (M-CSF) suivi de la polarisation avec IFN-γ et lipopolysaccharide (LPS) ou IL-4, respectivement. Les cellules M1 et M2 entièrement polarisées ont été infectées par le Mtb-GFP pendant 4 heures avant que les macrophages détachés infectés par le vtt ne soient tachés de cytométrie d’écoulement à 4 ou 24 heures après l’infection. L’acquisition de l’échantillon a été effectuée avec la cytométrie du débit et les données ont été analysées à l’aide d’un logiciel d’analyse de cytométrie de flux. Le gating manuel aussi bien que la réduction de dimensionnalité avec l’approximation et la projection multiples uniformes (UMAP) et l’analyse phénographique ont été exécutés. Ce protocole a eu comme conséquence la polarisation effective de M1/M2 caractérisée par des niveaux élevés de CD64, CD86, TLR2, HLA-DR et CCR7 sur les cellules M1 non infectées, alors que les cellules M2 non infectées ont montré une forte régulation des marqueurs phénotypes M2 CD163, CD200R, CD206 et CD80. Les cellules polarisées par M1 contenaient généralement moins de bactéries que les cellules polarisées par M2. Plusieurs marqueurs M1/M2 ont été régulés après l’infection à vtt, ce qui suggère que le vtt peut moduler la polarisation des macrophages. En outre, 24 groupes cellulaires différents de différentes tailles se sont révélés répartis de façon unique entre les cellules non infectées et infectées par le vtt M1 et M2 à 24 heures après l’infection. Ce protocole de cytométrie de flux M1/M2 pourrait être utilisé comme colonne vertébrale dans la recherche sur le mtb-macrophage et être adopté pour des besoins spéciaux dans différents domaines de recherche.
Introduction
Les macrophages sont des cellules immunitaires qui contribuent de manière significative à la régulation de l’homéostasie tissulaire, de l’inflammation et des pathologies de la maladie. Étant une composante essentielle de l’immunité innée, la lignée monocyte-macrophage des cellules exprime des phénotypes hétérogènes en réponse à des indices environnementaux modifiés, qui reflètent leur plasticité et leur adaptation à différents endroits anatomiques et immunologiques1. Selon les facteurs de croissance, cytokines et autres médiateurs présents dans le microenvironnement, les macrophages ont été classés en deux populations réversibles majeures, chacune ayant un rôle différent dans le contrôle bactérien et ledégagement 2: les macrophages polarisés M1 pro-inflammatoires et classiquement activés et les macrophages polarisés M2 anti-inflammatoires, alternativement activés, qui ont été initialement nommés pour imiter la nomenclature cellulaire T helper (Th)3. Ce regroupement de macrophages polarisés immunitaires est souvent considéré comme simpliste, car l’activation et la différenciation des macrophages ne sont pas linéaires, mais plus exactement illustrées comme un continuum où chaque population a des caractéristiques et des rôles fonctionnels différents dans les résultats du développement et de la progressionde la maladie 4,5,6,7. Néanmoins, il existe de nombreux avantages expérimentaux avec le modèle macrophage M1/M2 qui peuvent être utilisés dans plusieurs domaines de recherche différents.
Mycobacterium tuberculosis (Mtb) est l’agent causal de la tuberculose et a été estimé à infecter une personne sur deux et est considéré comme l’agent infectieux le plus mortel au monde (Rapport mondial sur la tuberculose 2019). Puisque les voies respiratoires sont la voie principale de l’infection de vtt, les macrophages alvéolaires sont les cellules hôtes préférées pour être infectées par la tuberculose et représentent à la fois les barrières primaires et le réservoir infectieux pour la tuberculose dans les poumons. La polarisation des macrophages en réponse à différents stimuli a été largement étudiée au fildes ans 7 et dans la plupart des travaux publiés, La polarisation M1 des cultures monocytes in vitro est induite par granulocyte-macrophage colony stimulating factor (GM-CSF) avec IFN-γ et LPS8,9, tandis que la polarisation M2 est induite par macrophage Colony Stimulating Factor (M-CSF) et IL-410,11. Les macrophages M1 sont des cellules effectrices puissantes qui médiation les réponses antimicrobiennes contre les pathogènes intracellulaires et ont un rôle essentiel dans l’immunité antitumorale12. Les macrophages M2, d’autre part, ont une fonction anti-inflammatoire, une capacité phagocytique élevée et sont principalement impliqués dans la cicatrisation des plaies et la réparation des tissus ainsi que dans les infections parasitaires12. Par conséquent, les macrophages M1 sont considérés comme plus efficaces dans le contrôle intracellulaire de la tonte par rapport aux macrophages M213. Cependant, les bactéries de Mtb ont également le potentiel de moduler la polarisation de macrophage pour subvertir l’immunité innée14,15,16,17.
Bien qu’il soit courant de générer des macrophages à partir de la différenciation des monocytes obtenus à partirdu sang périphérique 18, les macrophages pourraient également être générés à partir de cellules souches pluripotentes induites (IPSCs)19 ou de macrophages dérivés de la moelleosseuse à partir de souris 20,21. Ce sont des techniques réalisables pour étudier les cellules macrophages primaires obtenues à partir de progéniteurs monocytes/macrophages qui prolifèrent et se différencieront en une population homogène de cellules macrophages matures. Cependant, ces protocoles fournissent rarement la connaissance approfondie sur le phénotype et la fonction des cellules obtenues ni expliquent l’hétérogénéité normale observée parmi les macrophages obtenus in vivo. Comme le vtt est un agent pathogène humain strict, il y a aussi un avantage à étudier le vtt dans les systèmes modèles humanisés. La cytométrie de flux est une technologie puissante qui offre la possibilité d’évaluer les caractéristiques phénotypiques et fonctionnelles multiples des cellules simples dans la suspension22, quelquechose qui pourrait être assez difficile avec les cellules adhérentes telles que les macrophages qui sont également connus pour être autofluorescents23,24. En plus du détachement chimique des macrophages fermement adhérents, l’infection de vtt peut poser un facteur de stress significatif aux cellules qui ajoute un autre niveau de complexité dans les analyses cytométriques de flux des macrophages Mtb-infectés.
Dans ce protocole expérimental, nous avons employé un modèle précédemment établi d’infection humaine de macrophage basé sur la polarisation immunisée des cellules périphériques-sang-monocyte-dérivées primaires qui sont infectées par la souche virulente de Mtb de laboratoire H37Rv, et analysées avec la cytométrie de flux utilisant un panneau de 10 couleurs comprenant l’expression des marqueurs M1 et M2choisis 25. Ce protocole fournit une méthode efficace et reproductible pour étudier les réponses à l’infection à vtt dans les macrophages polarisés dérivés des monocytes M1 ou M2. En outre, l’utilisation de la cytométrie du débit sur les macrophages infectés par les vtt adhérents nous permet d’étudier une variété de marqueurs de surface associés aux macrophages conventionnels M1 et M2 et leur réponse longitudinale à l’infection par le vtt. Fait important, ce protocole peut facilement être adopté pour des études d’infections avec d’autres agents pathogènes, dans des études antitumorales ou dans des études de conditions inflammatoires, pour le dépistage des médicaments, etc. et pourrait également être exploité pour l’évaluation de la polarisation macrophage M1/M2 dans des échantillons cliniques humains.
Protocole
Le sang périphérique humain des donneurs anonymes sains de sang a été obtenu de la banque de sang à l’hôpital universitaire de Karolinska, Huddinge, Suède (approbation éthique Dnr 2010/603-31/4). Toutes les étapes expérimentales impliquant la tonte virulente vivante ont été exécutées au laboratoire de niveau de biosécurité-3 (BSL-3) à l’Agence suédoise de santé publique (FOHM), Solna, Suède.
1. Préparation des médias, des tampons et des cultures bactériennes
REMARQUE : Des détails sur tous les reagents et consommables sont fournis dans le Tableau des matériaux.
- RPMI milieu complet : Supplément RPMI 1640 avec pyruvate de sodium de 1 mM, 2 mM L-glutamine, 10 mM HEPES, et 10% sérum bovin foetal inactivé par la chaleur (FBS). Évitez les antibiotiques dans le milieu de culture cellulaire lorsque vous travaillez avec l’infection à vtt.
- Milieu RPMI sans sérum : Supplément RPMI 1640 avec pyruvate de sodium de 1 mM, 2 mM L-glutamine et 10 mM HEPES.
- Tampon de lavage : Préparer la solution saline tampon de phosphate (PBS) contenant 0,05 % (v/v) Tween-80.
- Tampon FACS : Préparez pbs contenant 2,5% (v/v) FBS et 0,5 mM EDTA.
- Tampon de fixation : Préparer le PBS contenant 4 % de formaldéhyde à PBS. Assurez-vous qu’il est fraîchement préparé avant utilisation, par exemple, mélangé à partir d’une solution de stock de formaldéhyde à 37 %.
- Tampon de perméabilisation : Ajouter 0,1 % de citrate de sodium et 0,1 % de Triton X-100 à l’eau déionisée.
- Tampon de lavage (pour l’immunofluorescence) : Préparez le PBS contenant 0,1 % de BSA et 0,1 % de Tween-20.
- Tampon de blocage : Préparer le PBS contenant 0,1 % de BSA et 10 % de sérum de chèvre normal (NGS) à PBS.
- Tampon de coloration (pour l’immunofluorescence) : Préparer le PBS contenant 0,1 % de BSA à PBS.
- Tb milieu complet: Supplément Middle Brook 7H9 bouillon avec 0,05% (v/v) Tween-80, 0,5% (v/v) glycérol, kanamycine (20 μg/mL), 10% (v/v) Acide oléique Middlebrook, albumine, dextrose et enrichissement catalase (Middlebrook OADC Enrichment).
- Cultures bactériennes : Utiliser la souche de laboratoire virulente standard de vtt, H37Rv, exprimant constitutivement la protéine fluorescente verte (GFP), pour l’infection des cellules dérivées des monocytes. Cette souche de vtt est porteuse d’un plasmide pFPV2 qui contient un gène codant GFP, ainsi qu’un gène de résistance à la kanamycine. La résistance aux antibiotiques permet une sélection continue des bactéries exprimant le plasmide dans les cultures contenant de la kanamycine. Conserver les bactéries dans la tuberculose complète moyenne et 70% glycérol (1:1 dilution) à -80 °C.
2. Isolement périphérique de cellules mononucléaires de sang des couches buffy
REMARQUE : Effectuez tout le travail avec du sang humain (potentiellement contagieux) à l’intérieur d’une armoire de biosécurité de classe II. Inactiver les produits sanguins résiduels avec des désinfectants pendant 15 minutes avant de les jeter. Le sang a été obtenu des volontaires en bonne santé dans ce cas. Ce protocole de différenciation macrophage in vitro a été mis en place pour inclure 10 x10 6 PBMCs/donneur/puits isolés. De chaque donneur, une couche buffy contient environ 50 mL d’une suspension concentrée de leucocyte provenant du sang entier, qui fournit normalement 500-800 x 106 PBMCs à partir de laquelle approximativement 10% ou 50-80 x 106 monocytes peuvent être récupérés.
- Chargez 15 mL de sang bouffi sur 15 mL de dénivelé moyen préparé dans un tube de 50 mL. Superposez lentement le sang au-dessus de la couche de gradient de densité en appuyant la pointe de pipette sur la paroi du tube.
- Faites tourner les tubes à 600 x g pendant 25 min à température ambiante (RT) avec 0 accélération et 0 décélération.
REMARQUE : Fermez soigneusement les couvercles avant la centrifugation et vérifiez toujours les porte-tubes pour qu’ils se déversent après la centrifugation. - Retirez la couche plasmatique supérieure à l’aide d’une pipette Pasteur stérile et recueillez ensuite soigneusement la couche cellulaire mononucléaire dans un nouveau tube de 50 mL à l’aide d’une pipette Pasteur stérile. Ajouter le milieu RPMI sans sérum à la pastille PBMC pour obtenir un volume final de 50 mL. Mélanger soigneusement en inversant le tube à quelques reprises avant la centrifugation à 500 x g pendant 5 min à RT.
- Jetez soigneusement le supernatant et résuspendez la pastille cellulaire en renversant le fond du tube dans les doigts.
- Pour éliminer la contamination moyenne du gradient de densité des PBMCs, lavez les cellules 2 à 3 fois avec un RPMI sans sérum pour obtenir un volume final de 50 mL. Centrifugeuse à 500 x g pendant 5 min à RT. Laver jusqu’à ce que le supernatant cellulaire devienne transparent.
- Jetez le supernatant et resuspendez les cellules dans 20 mL de milieu RPMI sans sérum.
- Comptez les cellules en testant la coloration bleue, manuellement à l’aide d’un hémocytomètre ou à l’aide d’un compteur de cellules automatisé. Diluer la suspension cellulaire en bleu trypan en 1:2 ou 1:10 dilution en mélangeant l’échantillon bleu trypan cellulaire dans une plaque de puits de 96 par exemple, 50 μL + 50 μL (pour le comptage de l’hémocytomètre) ou 10 μL + 10 μL (pour le comptage automatisé des cellules) et compter les cellules pour obtenir le nombre de cellules vivantes / mL.
ATTENTION: Trypan bleu est toxique et doit être jeté dans un déchet chimique distinct.
3. Différenciation et polarisation des cellules dérivées des monocytes
REMARQUE : Pour la différenciation et la polarisation des cellules dérivées des monocytes, un protocole que nous avons précédemment établi pour les cellules M0, M1 et M2 ainsi que pour les cellules polarisées entièrement M1 et M2 a étésuivi 25. Par souci de simplicité, seuls les macrophages M1 et M2 entièrement polarisés sont décrits ici.
- Utilisez l’adhérence plastique pour l’isolement des monocytes. En bref, semencer des PBMCs fraîchement isolés dans une plaque de culture de 6 puits à une concentration appropriée, par exemple, 10 x 106 PBMCs/puits dans un milieu RPMI sans sérum de 2 mL et incuber à 37 °C et 5 % de CO2.
- Après 2-3 h, retirer les cellules non adhérentes avec une pipette et laver les puits 3 fois avec un milieu sans sérum de 1 mL. Les cellules attachées sont des monocytes et représentent environ 10% du total des PBMCs ajoutés au puits, soit 106 monocytes récupérés à partir de 10 x 106 PPC ajoutés par puits.
- Pour la différenciation des macrophages, préparez une solution de travail contenant respectivement 50 ng/mL GM-CSF ou M-CSF pour la polarisation macrophage M1 et M2, ajoutée en 2 mL de milieu complet RPMI par puits. Culture des cellules dans un incubateur de CO2 à 5% en 37 °C pendant 3 jours.
- Le jour 3, retirez soigneusement 1 mL du milieu de culture cellulaire de la couche supérieure de chaque puits et complétez les cultures cellulaires avec 1 mL de milieu complet RPMI frais contenant la double concentration de M-CSF ou gm-CSF pour obtenir une concentration finale de 50 ng/mL dans les puits. Ajoutez les facteurs de croissance dans une solution de travail pré-faite de 100 ng/mL/well.
- Le jour 6, ajouter différents stimuli pour les 18-20 dernières heures de différenciation cellulaire pour obtenir m1 entièrement polarisé et mature (interféron-γ; IFN-γ, et lipopolysaccharide; LPS (E. coli O55:B5)) ou M2 (interleukine 4; IL-4) macrophages. Pour la polarisation M1, préparez IFN-γ et LPS dans rpmi milieu complet et ajouter 50 μL par puits pour obtenir une concentration finale de 50 ng/mL IFN-γ et 10 ng/mL LPS dans les cultures cellulaires. Pour la polarisation M2, préparer l’IL-4 dans le milieu complet du RPMI et ajouter 50 μL par puits pour obtenir une concentration finale de 20 ng/mL dans les cultures cellulaires.
- Pour la différenciation des macrophages polarisés M0, stimulez les cellules avec M-CSF seulement, sans cytokines supplémentaires (fournissant un phénotype M2-like)25.
- Vérifiez régulièrement la morphologie des cultures cellulaires dérivées des monocytes à l’aide d’une microscopie légère pour vous assurer que les monocytes plus petits sont différenciés en cellules macrophages plus grandes. En outre, surveiller les différences morphologiques potentielles entre la polarisation M1 et M2, c’est-à-dire les cellules M1 allongées et étirées par rapport aux cellules M2 avec une formeplus arrondie 25.
- Le jour 7, transférer les plaques avec des cellules dérivées des monocytes à un laboratoire BSL-3 pour l’infection par la tonte virulente.
4. Préparation des cultures vtt
REMARQUE : Les étapes suivantes doivent être effectuées dans une installation BSL-3. Pour tous les travaux avec la tuberculose virulente, utilisez des vêtements de protection, une protection respiratoire et des gants résistants à l’éthanol.
- Décongeler un flacon avec 1 mL d’aliquot bactérien et mélanger avec 9 mL de toy milieu complet (1:10 dilution) dans un tube de capuchon filtré de 50 mL. Culture de la suspension dans un incubateur à 37 °C et 5% de CO2.
- Après 24 h, faire tourner la suspension bactérienne à 2 300 x g pendant 10 min et verser délicatement le milieu. Resuspendez la pastille bactérienne avec 15-20 mL de tomodenne fraîche moyenne complète dans un nouveau tube de culture de bouchon filtré de 50 mL et incuber à 37 °C et 5% CO2. Mélanger les bactéries sédentaires dans le tube tous les 2-3 jours pour maintenir un approvisionnement homogène en nutriments pour toutes les cellules bactériennes.
- Après 7-10 jours, mélanger la suspension bactérienne correctement en pipetting de haut en bas avant de transférer à un tube de bouchon de vis de 50 mL.
- Ajouter 35 à 40 mL de tampon de lavage stérile au tube de 50 mL et faire tourner la suspension bactérienne à 2 300 x g pendant 10 min. Répétez les étapes de lavage une fois. Resuspendez la pastille bactérienne dans 1 mL de milieu RPMI sans sérum par pipetting avec une micropipette.
- Ajouter un autre 9 mL de milieu RPMI sans sérum et sonifier la suspension bactérienne à l’intérieur d’un cabinet de biosécurité de classe II pendant 5 min à 37 °C, pour perturber les touffes bactériennes. Tremper le tube à plusieurs reprises (3-4 fois) dans le sonicateur du bain d’eau pour assurer une perturbation maximale des touffes bactériennes. Mesurer la densité optique (OD) de 1 mL de suspension bactérienne à une longueur d’onde de 600 nm à l’aide d’un spectrophotomètre placé à l’intérieur de l’armoire de biosécurité. Utilisez un support RPMI sans sérum pour définir la référence.
- Calculer le nombre d’unités de formation de colonies (CFU) à l’aide de la formule : (OD+0,155)/0,161 = Y, et Y x 107= Y x 106 CFU/mL, par exemple, une valeur od 0,32 fournit une concentration bactérienne de (0,32 + 0,155)/0,161 = 2,95, 2,95 x 107= 29,5 x 106 CFU/mL.
5. Infection à la tonte des cellules dérivées des monocytes
REMARQUE : Les étapes suivantes doivent être effectuées dans une installation BSL-3.
- Resuspendez la pastille bactérienne dans un milieu RPMI sans sérum dans un nouveau tube stérile de 50 mL et ajustez la concentration bactérienne finale à environ 5 x 106 CFU/mL.
- Retirez le milieu de culture cellulaire des 6 plaques de puits contenant des cellules dérivées des monocytes. Ajouter 1 mL de milieu RPMI sans sérum à chaque puits. Ajouter 1 mL de suspension bactérienne par puits pour obtenir une multiplicité d’infection (MOI) 5:1, c’est-à-dire 5 x 106 CFU par 106 macrophages en 2 mL/puits et incuber les plaques pendant 4 h en 37 °C et 5 % de CO2.
- Après l’infection, laver les cellules 3 fois avec 1 mL de tampon de lavage stérile pour éliminer les bactéries extracellulaires. Inclinez la plaque et retirez soigneusement l’ensemble du tampon de lavage des coins. Resuspendez les cellules dérivées de monocytes infectées par la tonte dans 2 mL de RPMI milieu complet sans antibiotiques et procéder à couler la cytométrie souiller ou incuber les cellules pendant un autre 24 h (ou d’autres points de temps) avant la cytométrie de flux.
6. Coloration de cytométrie d’écoulement des cellules monocyte-dérivées Mtb-infectées
REMARQUE : Les étapes suivantes doivent être effectuées dans une installation BSL-3. La coloration de cytométrie d’écoulement pourrait être exécutée dans une plaque de 96 puits au lieu des tubes.
- Détacher les cellules infectées par la touffe (et les témoins non infectés) des puits de la plaque de 6 puits par incubation avec 1 mL de tampon FACS par puits pendant au moins 30 min à 37 °C et 5 % de CO2.
- Pipette doucement de haut en bas à quelques reprises pour s’assurer que les cellules sont détachées. Si possible, confirmer le décollement cellulaire par microscopie. Transférer la suspension cellulaire de chaque puits dans un tube de microcentrifugeuse plafonné à vis et faire tourner les tubes à 200 x g pendant 5 min. Jetez soigneusement le supernatant par pipetting.
- Lavez la pastille cellulaire dans chaque tube deux fois avec le tampon FACS et faites tourner les cellules à 200 x g pendant 5 min.
- Tacher les cellules (environ 0,5 x 106 à 1 x 106 cellules/tube) avec environ 50 cocktails μL d’anticorps anti-humains conjugués au fluorochrome, y compris TLR2 (AF647), CD206 (APC-Cy7), CD163 (BV605), CD80 (1 BV650), CCR7 (BV711), CD86 (BV786), CD200R (PE), CD64 (PE-Dazzle 594), HLA-DR (PE-Cy5) (Tableau 1) en combinaison avec colorant de viabilité Zombie-UV pendant 30 min à 4 °C (réfrigérateur) dans l’obscurité.
- Lavez les cellules tachées deux fois avec 400 μL de tampon FACS et faites tourner les cellules à 200 x g pendant 5 min.
- Fixer les cellules tachées avec 200 μL de tampon de fixation (fraîchement préparé) pendant 30 min à RT dans l’obscurité pour assurer l’inactivation complète des mycobactéries.
- Lavez les cellules deux fois avec 400 μL de tampon FACS et tournez à 200 x g pendant 5 min pour éliminer l’excès de tampon fixe.
- Resuspendez les cellules fixes dans 400 μL de tampon FACS et transférez les échantillons dans de nouveaux tubes de microcentrifugeuse de 1 mL avant de les sortir du laboratoire BSL-3 pour la cytométrie d’écoulement dans BSL-2. Conserver les cellules tachées en +4 °C jusqu’à l’acquisition de l’échantillon.
REMARQUE : Vaporisez les tubes avec 70 % d’éthanol avant de les sortir du laboratoire BSL-3. Le formaldéhyde est toxique (cancérigène) et doit être manipulé dans une armoire de biosécurité de classe II. Jeter les déchets de formaldéhyde dans un déchet chimique distinct.
7. Acquisition et analyse de données cytométriques de flux de cellules dérivées de monocytes infectées par la tonte
REMARQUE : Les étapes 7.1-7.2 doivent être exécutées avant la coloration de cytométrie d’écoulement décrite ci-dessus. Pour éviter les problèmes d’agglutination cellulaire et de dissociation des colorants tandem après fixation cellulaire, l’acquisition d’échantillons de cellules infectées par la touffe et non infectées est effectuée dans les 4-10 h après la coloration primaire des anticorps.
- Avant la coloration de cytométrie d’écoulement décrite ci-dessus, compensez le signal fluorescent pour chaque anticorps fluorochrome-conjugué énuméré dans le panneau de coloration (tableau 1) utilisant des perles de compensation (positives et négatives).
- Titrer la dilution des anticorps pour la coloration des macrophages humains pour obtenir le signal optimal pour chaque fluorochrome.
- Utilisez des cellules non tachées pour déterminer le niveau de fluorescence de fond nécessaire pour fixer la porte pour la population de cellules négatives permettant de visualiser les cellules tachées (les macrophages sont très auto fluorescents).
- Acquérir un minimum de 50 000 cellules/échantillon dans le cytomètre à flux à l’aide du logiciel recommandé pour l’acquisition de données.
- Exportez les fichiers d’acquisition à partir du cytomètre de flux dans le format standard de cytométrie de flux (FCS) 3.1.
- Analyser les fichiers FCS dans le logiciel d’analyse de cytométrie de flux.
- Macrophages de porte en fonction de leurs caractéristiques de dispersion avant et latérale (FSC et SSC) et exclure les cellules mortes par gating cellule vivante / morte en utilisant le colorant de viabilité Zombie-UV.
- Visualiser les macrophages infectés par H37Rv-GFP dans le canal FITC.
- Identifier la fréquence des cellules tachées positivement et l’intensité géométrique moyenne de fluorescence (IMF) pour tous les marqueurs (tableau 1).
8. Coloration par immunofluorescence des cellules dérivées des monocytes infectés par la tonte
REMARQUE : L’infection à vtt doit être effectuée dans un établissement BSL-3.
- Pour l’immunostaining, graine 2 x10 5 PBMCs/puits dans 500 μL de milieu RPMI sérique-libre dans 8 glissières de chambre de puits pour obtenir 2 x 104 monocytes/puits. Après la différenciation et la polarisation M1/M2 des monocytes, procéder à l’infection de vtt comme décrit ci-dessus. Fixer les glissières après 24 h d’infection à vtt avec tampon de fixation pendant 30 min. Les glissières fixes sont conservées au congélateur à -20 °C jusqu’à ce que d’autres analyses soient effectuées.
- Lavez les cellules dérivées des monocytes deux fois avec 200 μL de PBS pendant 10 min chacune.
- Perméabiliser les cellules avec 200 μL de tampon de perméabilisation pendant 5 min à RT.
- Lavez les cellules 3 fois avec 200 μL de PBS pendant 5 min chacune.
- Lavez les cellules deux fois avec 200 μL de tampon de lavage pendant 5 min chacun.
- Bloquez la liaison non spécifique avec 200 μL de tampon de blocage pendant 30 min à RT.
- Diluer les anticorps primaires 1:100 dans le tampon de coloration et incuber les cellules M1 avec un anticorps CD64 non poursuivi (Clone: 10.1) et les cellules M2 avec un anticorps CD163 non poursuivi (polyclonal) pendant 2 h à RT.
- Ensuite, lavez les cellules 3 fois avec 200 μL de tampon de lavage pendant 10 min chacun.
- Diluer les anticorps secondaires étiquetés fluorescents 1:1,000 dans le tampon de coloration et d’incuber les cellules M1 avec un anti-souris IgG-Alexa Fluor 594 et les cellules M2 avec un anti-lapin IgG-Alexa Fluor 594 pour 1 h à RT.
- Lavez les cellules 3 fois avec un tampon de lavage de 200 μL pendant 10 min chacun.
- Retirez la grille de chambre et ajoutez ~20 μL de milieu de montage DAPI dans chaque puits et mettez un coverslip de 1,5 mm sur chaque diapositive.
- Sceller le coverslip avec une couche de vernis à ongles.
- Acquérir des images à l’aide d’un microscope confocal avec des lasers émettant à 486 nm pour l’excitation de GFP (canal vert), 402 nm pour DAPI (bleu) et 560 nm pour les anticorps secondaires (rouge) respectivement.
Résultats
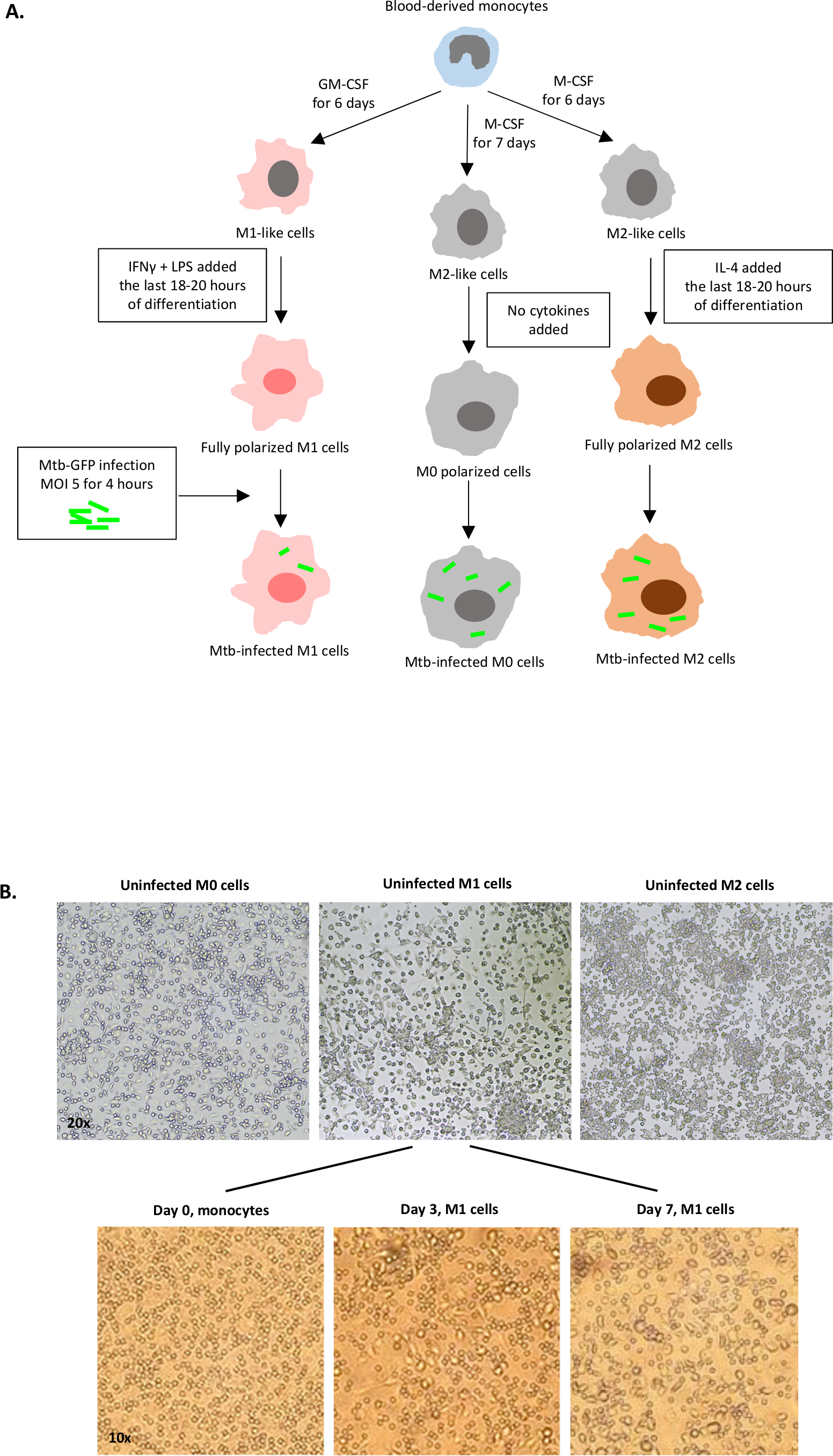
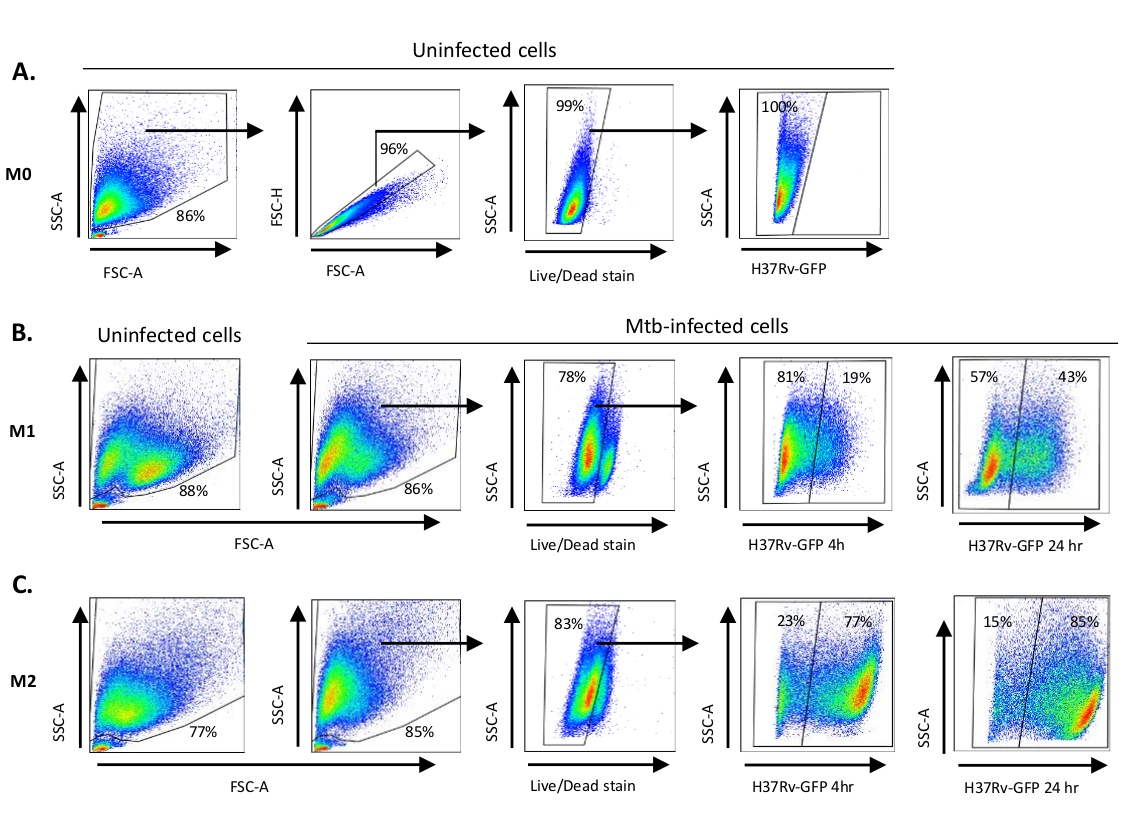
Une illustration schématique des stimulations cytokine utilisées pour la polarisation des cellules dérivées des monocytes à M0 (cellules m2), M1 (cellules M1 entièrement polarisées) et M2 (cellules M2 entièrement polarisées) est présentée à la figure 1A,tandis que des images représentatives des cultures cellulaires M0, M1 et M2 ainsi que des cultures M1 aux jours 0, 3 et 7, sont présentées à la figure 1B. Des cellules M0 non infectées ont été utilisées pour démontrer la stratégie de base de gating (figure 2A). Initialement, les cellules myéloïdes (~85%) ont été fermés en fonction de leurs propriétés de dispersion vers l’avant (FSC) et de dispersion latérale (SSC), y compris les plus grandes cellules à haute granularité et à l’exclusion des débris de petite taille avec un faible SSC et FSC qui se trouvent dans le coin inférieur gauche de la parcelle de points. Dans la deuxième parcelle, les doublets (c.-à-d. les touffes cellulaires) ont été définis comme ayant une surface accrue mais une hauteur similaire par rapport aux cellules individuelles et ont été exclus de l’analyse plus poussée. Par conséquent, seules les cellules proportionnées entre fsc-zone et FSC-Hauteur (cellules simples) ont été inclus à l’intérieur de la porte de forme inclinée. Ensuite, le colorant de viabilité Zombie-UV qui tache les protéines cytoplasmiques à l’intérieur des cellules mortes, a été utilisé pour exclure les cellules mortes de l’analyse ultérieure. Comme prévu, les cellules M0 non infectées viables étaient négatives pour l’expression Mtb-GFP visualisée dans le canal FITC.
Ensuite, nous avons appliqué la même stratégie de gating aux macrophages M1 et M2 non infectés ainsi qu’aux macrophages M1 et M2 non infectés à 4 heures après l’infection (figure 2B,C). Deux sous-populations ont été détectées dans la porte FCS/SSC des macrophages polarisés M1 non infectés; une population ayant une plus petite taille (FCS) et une granularité plus élevée (SSC) et l’autre population avec une plus grande taille et une granularité inférieure (figure 2B), tandis que la porte principale des cellules M2 non infectées semblait plus homogène (Figure 2C). Les cellules dérivées des monocytes M1 et M2 ont affiché un déplacement vertical vers une granularité plus élevée et une taille cellulaire réduite lors de l’infection à vtt, ce qui peut refléter une complexité accrue à l’intérieur des cellules causée par l’absorption de bactéries mtb intracellulaires(figure 2B,C). En outre, la tache de viabilité a indiqué une mort cellulaire accrue (17-22%) chez les cellules M1 et M2 infectées par la tonte à un MOI de 5, comparativement aux cellules M0 non infectées (99 %) (Figure 2A-C) ou cellules M1 et M2 non infectées (données non montrées). Les données représentatives ont montré que l’expression Mtb-GFP (c.-à-d. infectiosité de la tb) était sensiblement plus élevée chez les cellules M2 (77 % de cellules gfp-positives) que dans les cellules M1 (19 % de cellules gfp-positives) après 4 heures d’infection(figure 2B,C). Après 24 heures d’infection, l’expression de Mtb-GFP était 43% et 85% dans les cellules M1 et M2 respectivement, suggérant que les cellules M1 ont eu une augmentation relativement plus élevée de GFP-expression de 4-24 heures après infection de Mtb comparée aux cellules M2, 126% contre 10.4% augmentation de GFP-expression dans les cellules M1 et M2 de 4-24 heures, respectivement.
Pour caractériser l’efficacité de la polarisation M1/M2 dans les cellules non infectées dérivées des monocytes, des parcelles de points ont été utilisées pour identifier les cellules M1 doublement positives pour le CD64 et le CD86 (CD64+CD86+) et les cellules M2 qui étaient doublement positives pour le CD163 et le CD200R (CD163+CD200R+; Figure 3A,B). La sélection des marqueurs M1/M2 a été principalement faite sur la base des résultats de nostravaux précédents 25 mais aussi d’autresétudes 26,27,28,29. Les quadrants des cellules tachées ont été fixés à l’aide de barrières correspondantes pour les cellules M1/M2 non tachées (figure 3A). Aucun de ces marqueurs n’est exclusivement exprimé par les cellules M1 ou M2, mais la proportion de cellules positives ainsi que l’intensité de l’expression de surface est différente. Cela était particulièrement évident à partir de la tache M1 où environ 95% des cellules M1 et 79% des cellules M2 étaient CD64+CD86+, mais l’intensité de coloration était sensiblement plus élevé dans le sous-ensemble M1 (Figure 3A). Alors que 27% des cellules M1 étaient positives pour le CD200R à marqueur M2, seulement 1% étaient positives pour le CD163, fournissant 0,5% de CD163+CD200R+ cellules M1 contre 63% de CD163+CD200R+ cellules M2 (Figure 3A). Après 4 heures d’infection par la tonte, une augmentation de la fréquence des cellules CD200R+ a été observée dans les cellules polarisées M1 positives au Mtb-GFP (16 %), tandis que l’expression du CD163 a été réduite dans les cellules M2 (figure 3B). La carte thermique démontre une forte intensité d’expression GFP dans les cellules CD163+CD200R+ M2, mais aussi dans le sous-ensemble CD64+CD86+ M2 par rapport aux sous-ensembles de cellules M1 correspondants (Figure 3B). Dans l’ensemble, le changement d’expression des marqueurs M1 et M2 respectifs est également visualisé dans les histogrammes de la figure 3C. En outre, les bactéries Mtb-GFP ont également été visualisées dans les cellules CD64+ M1 et dans les cellules CD163+ M2 par microscopie confoccale, qui a soutenu une absorption intracellulaire accrue et/ou la croissance de la tome à l’intérieur de M2 par rapport aux cellules M1(figure 3D).
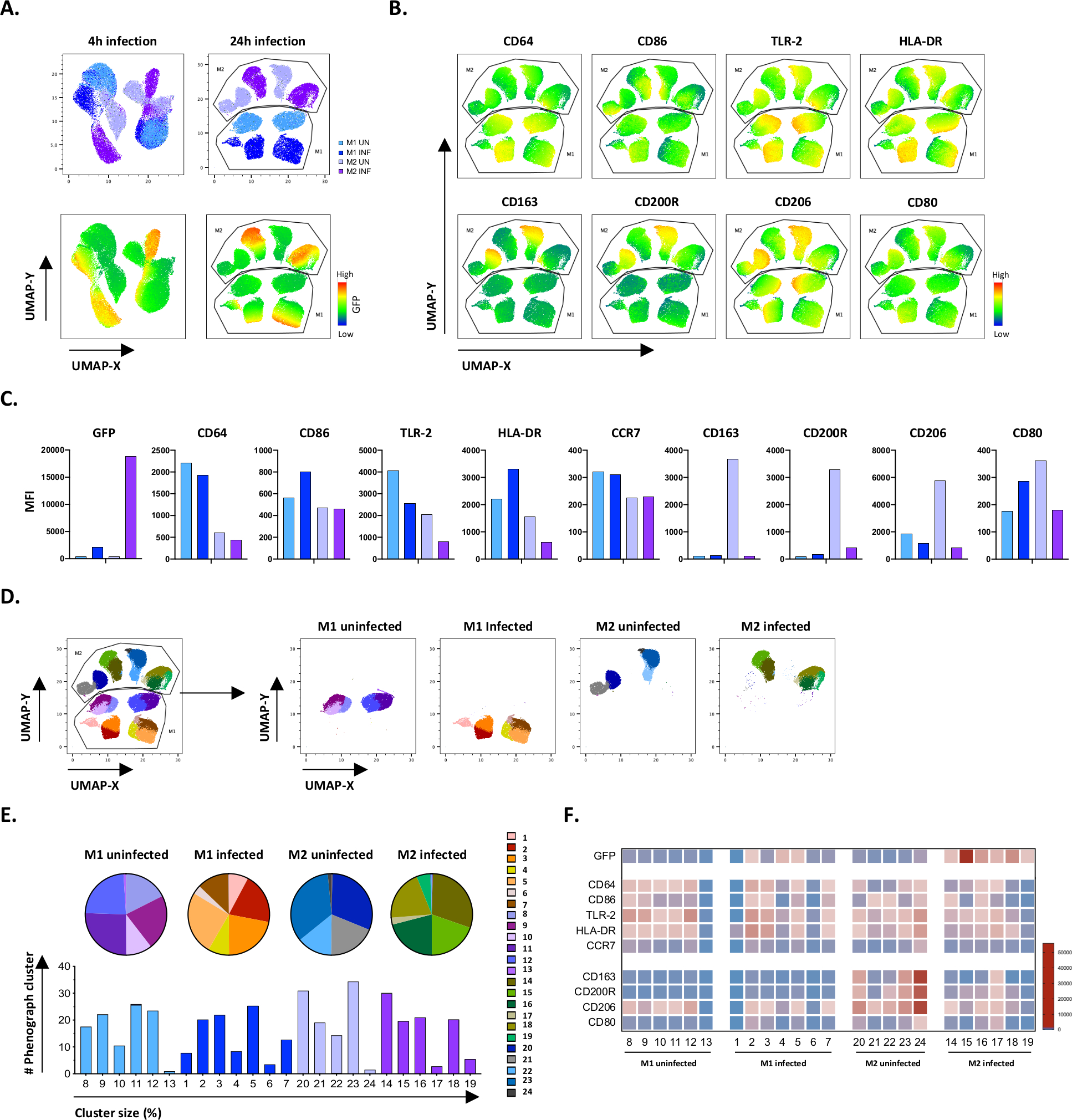
Pour vérifier les résultats du gating manuel, nous avons appliqué la réduction de dimensionnalité utilisant l’approximation et la projection uniformes multiples (UMAP). L’analyse umap a montré que l’infection à vtt pendant 4 heures n’était pas suffisante pour affecter la polarisation des macrophages, contrairement à 24 heures d’infection, ce qui a entraîné des grappes clairement séparées de cellules M1 et M2 non infectées et infectées (figure 4A). Les macrophages M1 non infectés affichaient une expression plus élevée des macrophages CD64, CD86, TLR2, HLA-DR et CCR7 par rapport aux macrophages M2, tandis que les cellules M2 non infectées présentaient une forte régulation des marqueurs phénotypeS M2 CD163, CD200R, CD206 et CD80(figure 4B,C). En accord avec le gating manuel, l’infection à vtt après 24 heures a provoqué une nette baisse de la réglementation des CD163, CD200R et CD206 sur les cellules M2 et une déréglementation du CD86 et du HLA-DR sur les cellules M1 (figure 4B,C), ce qui suggère que le VTT peut moduler la polarisation des macrophages. L’analyse phénographique subséquente (figure 4D-F) a permis d’identifier 24 grappes différentes de différentes tailles qui ont été réparties de façon unique entre les cellules non infectées et infectées par le vtt M1 et M2, comme l’illustrent les graphiques umap (figure 4D), les diagrammes à secteurs (figure 4E) et les cartes thermiques (figure 4F). Au total, ces résultats montrent l’efficacité prometteuse de ce protocole pour générer phénotypiquement et fonctionnellement diverses cellules polarisées M1 et M2 in vitro qui sont encore modulées par infection de vtt.

Figure 1 : Illustration schématique de la différenciation in vitro et de la polarisation des cellules myéloïdes-dérivées humaines. (A) M0 (M2-like), M1 (classiquement activé) et M2 (alternativement activé) cellules sont représentées. Les monocytes obtenus auprès de donneurs de sang sains ont été polarisés avec différentes cytokines comme décrit dans le protocole et infectés par la souche de vtt étiquetée GFP, H37Rv, pendant 4 heures avant l’analyse avec cytométrie de flux de 10 couleurs. Les cellules polarisées par M1 contiennent généralement moins de bactéries que les cellules polarisées par M2. (B) Images microscopiques de cellules M0, M1 et M2 entièrement polarisées et non infectées dans les plaques de 6 puits au jour 7, et images représentatives de la différenciation cellulaire M1 des monocytes aux jours 0, 3 et 7. Le grossissement est de 20x (panneau supérieur) et 10x (panneau inférieur). Notez que les cellules M1 sont plus allongées et étirées par rapport aux cellules M0 et M2 plus arrondies (panneau supérieur). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Stratégie de gating des cellules myéloïdes différenciées dérivées. Parcelles à points représentatives montrant (A) Les propriétés de dispersion vers l’avant (FSC) et de dispersion latérale (SSC) des macrophages M0 non infectés. La parcelle FSC-A/FSC-H montre le gating manuel des cellules simples proportionnées pour la zone et la hauteur. La porte cellulaire vivante excluait les cellules qui étaient positives pour zombie-UV (colorant de viabilité). Le vtt intracellulaire a été détecté par expression GFP dans les cellules vivantes observées dans le canal FITC. (B) Gating de M1 et (C) macrophages M2 montrant FCS / SSC parcelles de points à la fois dans les cellules non infectées et les cellules infectées par le vtt 4 h et 24 h après l’infection. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Efficacité du protocole de polarisation in vitro M1/M2. Parcelles de points représentatives et gating de quadrant montrant les fréquences sous-ensemble des cellules polarisées M1 et M2 à l’aide de cellules non infectées CD64 et CD86 (M1) ou CD163 et CD200R (M2) dans (A) cellules non tachées et tachées non infectées et (B) cellules tachées infectées par le VTT 4 h après l’infection. Les parcelles de points dans (B) illustre l’intensité de fluorescence de gfp-expression (carte thermique) dans M1- et M2-polarisé macrophages obtenus à partir de différentes sous-portes. (C) Moyenne géométrique de l’intensité de fluorescence (IMF) est montrée dans les histogrammes d’un donneur représentatif après 4 h d’infection de VTT. Les valeurs de l’IMF dans les cellules M1 (bleu clair) et M2 non infectées (violet clair) sont présentées dans le panneau supérieur et les cellules M1 (bleu profond) et M2 (violet profond) infectées par le VTT sont présentées dans le panneau inférieur. (D) Des images confocales représentatives de cellules polarisées M1 et M2 non infectées et infectées par le VTT sont montrées. Les cellules M1 et M2 ont été tachées pour l’expression CD64 et CD163, respectivement, utilisant l’immunofluorescence. La coloration positive de surface est montrée dans les bactéries intracellulaires rouges et GFP-exprimant est montrée en vert. Les noyaux tachés de DAPI sont représentés en couleur bleue. Échelle – 10 μm. Le grossissement des images vers la droite est de 350x. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Réduction de la dimensionnalité avec approximation et projection multiples uniformes (UMAP) et analyse phénographique des cellules M1 et M2 non infectées et infectées par le vtt. (A) UMAP, créé en concatenating 11000 cellules vivantes à partir de cultures cellulaires M1 et M2 non infectées et infectées par le VTT à partir de deux donneurs de sang représentatifs, 4 h (graphiques gauches) ou 24 h (graphiques de droite) après l’infection. La carte thermique pour l’expression GFP (panneau inférieur) indique les cellules non infectées et infectées par le vtt. (B-C) IMF des marqueurs exprimés dans les cellules M1 et M2 non infectées et infectées par le VTT 24 h après l’infection, montrées comme (B) heatmap ou (C) parcelles de barre. (D-F) L’analyse phénographique a permis d’identifier 24 grappes qui sont réparties différemment entre les cultures M1 et M2 non infectées et infectées par le vtt. Les grappes 8 à 13 sont uniques dans les cellules M1 non infectées, les grappes 1 à 7 sont uniques dans les cellules M1 infectées par le vtt, les grappes 20 à 24 sont uniques dans les cellules M2 non infectées et les grappes 14 à 19 sont uniques dans les cellules M2 infectées par le VTT. L’IMF de chaque marqueur de chaque groupe phénographique est affichée dans (F). Les données sont présentées sous forme de cellules M1 (bleu clair) et M2 non infectées (violet clair) et M1 (bleu profond) et M2 (violet foncé). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Tableau 1 : Liste des anticorps utilisés pour la cytométrie d’écoulement.
| laser | filtre | Fluorochrome | phénotype | fonction | clone | Catalogue no. | compagnie |
| 639 | 670/30 | AF647 (af647) | TLR2 (en) | Récepteur de reconnaissance des pathogènes | TL2.1 (en) | 309714 | BioLegend (BioLegend) |
| 639 | 780/60 | APC-Cy7 (en) | CD206 (EN) | Récepteur Mannose | 15-2 | 321120 | BioLegend (BioLegend) |
| 405 | 610/20 | BV605 (BV605) | CD163 (EN) | Récepteur charognard | IGS/61 | 333616 | BioLegend (BioLegend) |
| 405 | 670/30 | BV650 (BV650) | CD80 (en) | Molécule co-stimulante | 2D10 (2D10) | 305227 | BioLegend (BioLegend) |
| 405 | 710/50 | BV711 (en) | CCR7 (en) | Récepteur de chimiokine | G043H7 | 353228 | BioLegend (BioLegend) |
| 405 | 780/60 | BV785 (BV785) | CD86 (en) | Molécule co-stimulante | IT2.2 (en) | 305442 | BioLegend (BioLegend) |
| 488 | 530/30 | GFP (GFP) | VTT | Bactéries intracellulaires | |||
| 561 | 586/15 | Pe | CD200R | Récepteur inhibiteur | OX-108 (OX-108) | 329306 | BioLegend (BioLegend) |
| 561 | 620/14 | PE/ÉBLOUISSEMENT 594 | CD64 (en) | Fc gamma receptor-I d’IgG | 10.1 | 305032 | BioLegend (BioLegend) |
| 561 | 661/20 | PE-Cy5 (PC5) | HLA-DR | Molécule de classe II du MHC | L243 (L243) | 307608 | BioLegend (BioLegend) |
| 355 | 450/50 | BUV395 (BUV395) | Colorant de viabilité | Marqueur de cellules vivantes/mortes | Zombie UV | 423108 | Invitrogen (Invitrogen) |
Discussion
Ce protocole expérimental décrit la polarisation efficace des cellules dérivées des myéloïdes en phénotypes M1 ou M2, y compris l’évaluation avec un panneau de cytométrie de flux de 10 couleurs qui permet la visualisation et la caractérisation profonde de la tonte étiquetée GFP dans divers sous-ensembles de macrophages. Bien que la tuberculose soit une ancienne maladie humaine, il n’existe actuellement aucun modèle standard d’or pour étudier les interactions Mtb-macrophage, et la cytométrie du flux multicolore des macrophages pourrait être compliquée par rapport aux analyses des réponses des lymphocytes. Peu de protocoles disponibles pour la différenciation in vitro des monocytes humains aux macrophages présentent une connaissance approfondie du type de macrophages générés. Un protocole de base pour la polarisation des macrophages et l’évaluation cytométrique du flux de l’activation des macrophages à l’aide d’un solide panel de marqueurs peut probablement faciliter une telle caractérisation et offrir des possibilités d’explorer d’autres caractéristiques des cellules polarisées traitées dans différentes conditions. Cela comprend des analyses des cellules cultivés in vitro ainsi que des analyses de cellules in vivo dans des échantillons cliniques, c’est-à-dire des suspensions PBMC et unicell cellules à partir de liquides organiques (c.-à-d. lavage bronchoalveolar) ou de tissus homogénéisés. Par conséquent, la différenciation et/ou l’état d’activation des monocytes et des macrophages obtenus des patients pourraient être liés aux résultats de la maladie. L’expansion du CD16+CD163+ monocytes dans le sang périphérique ont été rapportées dans les patients pulmonaires deTB 30. Une fréquence accrue de CD163+ cellules a également été détectée dans la peau enflammée des patients atopiques de dermatite31. De même, il a été démontré que les macrophages semblables à cd206+ M2 inhibent la prolifération et la différenciation des cellules dans le microenvironnement du tissu adipocyte32 et qu’ils sont enrichis en échantillons de moelle osseuse chez des patients atteints de leucémie myéloïde aiguë (LAM)29. Un rapport élevé des cellules CD64 (M1) à CD163 (M2) dans le sang entier des patients présentant l’ostéoarthrite s’est trouvé pour être associé à la sévéritéde la maladie 33. Une autre étude a employé CD86 (M1) et CD163 (M2) pour démontrer que l’expression élevée de M1 dans le tissu a corrélé à de pires résultats dans un sous-groupe des tumeurs malignes decerveau 34.
Il y a plusieurs avantages significatifs de ce protocole expérimental de cytométrie d’écoulement de M1/M2. Ce modèle offre l’occasion d’étudier les réponses immunitaires innées à l’infection virulente à la tonte et peut être développé pour contenir des études sur les réponses immunitaires adaptatives en ajoutant des lymphocytes T autologues ainsi que des macrophages M1 ou M2 dans les réactions mixtes des lymphocytes (MLR). Le protocole convient également au dépistage et au dépistage des différents composés immunomodulateurs et antimicrobiens. Ici, nous avons précédemment étudié les effets de la vitamine D et de l’inhibiteur de l’histone deacetylase phénylbyrate sur les cellules dérivées des myéloïdes après l’infection àvtt 25,35. La cytométrie d’écoulement M1/M2 pourrait également être employée pour évaluer l’activation de macrophage après conditionnement avec des supernatants de culture cellulaire ou le plasma patient. Bien que les études in vivo sur la co-infection tuberculeuse par le VIH ou les helminthes ou la comorbidité tb-diabète puissent être difficiles, le modèle M1/M2 moins complexe peut faciliter les études sur les comorbidités in vitro. De même, le protocole pourrait être exploité pour des études de transmission afin d’examiner l’infectiosité des cellules mtb ou d’étudier la capacité de présentation phagocytique et antigène des cellules M1/M2 individuelles. La cytométrie d’écoulement M1/M2 est également attrayante pour une utilisation dans les études de biomarqueurs et de vaccins, pour suivre le pronostic de la maladie pendant le traitement et pour tester des thérapies ciblant les cellules dérivées des myéloïdes. Fait important, un certain nombre de méthodes différentes pourraient être appliquées parallèlement à la cytométrie du débit pour l’évaluation simultanée des phénotypes de polarisation des macrophages et des réponses fonctionnelles à l’aide de microscopie confocale (Figure 3D), PCR en temps réel, tache occidentale, analyses de multiplexe et ELISA des facteurs solubles dans le supernatant de culture aussi bien que l’évaluation de l’infectivité et de la croissance bactériennes intracellulaires utilisant GFP-expression (cytométrie de flux et microscopie confocale) et unités de formation de colonie (CFU). L’infection des cellules M1 ou M2 par la bactérie Mtb-GFP permet également de trier les cellules non infectées et infectées par la tuberculose à partir du même échantillon pour une seule analyse de séquençage de l’ARN cellulaire.
Le protocole décrit a également certaines limites, y compris les inconvénients techniques et scientifiques. L’inconvénient de l’utilisation de macrophages dérivés des monocytes provenant de donneurs de sang humains est que la variabilité du donneur est souvent élevée et le fait que les cellules ne sont pas polarisées dans l’environnement physiologique des tissus humains. Une grande variabilité de l’efficacité de la polarisation M1/M2 ou de l’infectiosité du vtt entre les donneurs peut entraîner des problèmes avec les variations intra-et interexpérimentales, une faible puissance statistique et la nécessité d’inclure de nombreux donneurs pour obtenir des résultats fiables. En outre, l’adhérence plastique des monocytes des PBMCs a comme conséquence un nombre donneur-dépendant des monocytes/puits qui peuvent par la suite fournir un MOI arbitraire qui pourrait avoir un impact sur la polarisation de macrophage et la viabilité cellulaire après l’infection de Mtb. Les étapes critiques du protocole impliquent un lavage approprié pour empêcher d’autres types de cellules de contaminer les cultures cellulaires qui pourraient également affecter la polarisation des macrophages. Bien qu’un MOI trop faible puisse imiter l’infection tuberculeuse latente, un MOI trop élevé tuera les cellules, soulignant l’importance d’utiliser un MOI approprié. En outre, il pourrait être difficile de récupérer des cellules fermement adhérentes lors du détachement, ce qui peut entraîner une représentation biaisée de certains sous-ensembles de macrophages utilisés pour les analyses de cytométrie du flux. Une étape cruciale dans l’analyse de cytométrie de flux implique l’utilisation appropriée de la matrice de compensation de perles et des contrôles négatifs tels que les cellules non tachées ou fmo (Fluorescence Minus One) contrôles pour assurer le gating manuel correct.
Une autre limitation implique la polarisation des monocytes dérivés du sang et non de l’environnement tissulaire local. La marque de fabrique de la tuberculose humaine est la formation de granulomes dans les tissus infectés par la tuberculose et, par conséquent, l’immunopathologie dans la tuberculose devrait être étudiée de préférence au site tissulaire local. Cependant, les monocytes sont recrutés au poumon à partir du sang périphérique lors de l’inflammation/ infection, où les cellules peuvent se différencier en macrophages en présence de cytokines inflammatoires tels que GM-CSF12. Fait important, dans le milieu physiologique des tissus in vivo, il y a probablement une grande hétérogénéité de polarisation des macrophages, y compris un mélange et différents ratios de diverses populations de macrophages de type M1 et M2 qui contribuent au sort de l’infection tuberculeuse36. Nous avons précédemment développé un modèle humain de tissu pulmonaire organotypique qui permet des études 3D de la formation de granulome macrophage-24 dans TB37. Il pourrait être intéressant d’exploiter le protocole actuel de polarisation M1/M2 en combinaison avec le modèle des tissus pulmonaires pour étudier plus avant la formation de granulomes tuberculeux, les fonctions effectrices et le rapport M1/M2 dans les tissus expérimentaux.
Ce protocole de flowcytometry M1/M2 pourrait facilement être adapté pour inclure un panneau étendu des marqueurs myéloïdes utiles pour l’évaluation des dispositifs liés aux réponses inhibitrices aussi bien qu’inflammatoires. Il y a un grand intérêt de recherche dans les molécules inhibitrices de point de contrôle immunitaire telles que PD-1, SIRP-α, IDO et arginases qui pourraient moduler des réponses de macrophage38. Dans ce contexte, la polarisation des cellules myéloïdes pourrait également impliquer d’autres stimuli qui favorisent les macrophages immunoréglementaires (Mreg) ou les cellules suppressrices dérivées des myéloïdes (MDSC) qui se sont manifestées comme impliquées dans plusieurs maladies dont la tuberculose38. Les panneaux de cytométrie d’écoulement plus avancés des sous-ensembles macrophages M1/M2/Mreg peuvent également inclure la coloration intracellulaire des cytokines/chimiokines IL-1β, TNF-α, IL-10 et MCP-1 ou d’autres facteurs solubles ou molécules effectrices telles que l’oxyde nitrique inductible (iNOS) et les peptides antimicrobiens. Ceci pourrait augmenter les possibilités d’étudier des réponses polyfonctionnelles de macrophage, semblables à ce qui a été abondamment décrit pour des cellules T39.
Actuellement, les panneaux de coloration de cytométrie d’écoulement peuvent inclure jusqu’à 30-40 couleurs, qui fournit la capacité d’immunophénotyper plusieurs sous-ensembles cellulaires et molécules simultanément. La mise en place expérimentale de base de ce protocole de cytométrie d’écoulement M1/M2 peut être utilisée comme une colonne vertébrale compatible avec la plupart des cytomètres anciens et nouveaux flux et peut être construite et adaptée aux besoins individuels, y compris les défis posés par le travail avec la tonte virulente dans un environnement BSL-3. Aujourd’hui, des techniques de réduction de la dimensionnalité telles que l’UMAP sont disponibles dans les nouvelles versions du logiciel de cytométrie de flux, qui permet l’analyse d’un grand nombre de paramètres générés dans les études unicelliques qui est essentiel pour améliorer la visualisation et l’interprétation des données de haute dimension40. Les améliorations technologiques constantes de la cytométrie du débit se poursuivront probablement au cours des prochaines années, y compris la combinaison du phénotypage multi-paramétrique ainsi que des capacités modernes de tri cellulaire, où ce protocole pourrait s’avérer utile dans plusieurs tests d’infection à vtt à base de macrophages.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions nos collègues de l’Agence suédoise de santé publique, Matilda Svensson et Solomon Ghebremichael, pour leur aide dans le laboratoire BSL-3.
Ces travaux ont été soutenus par des subventions de la Fondation suédoise du cœur et du poumon (HLF) (2019-0299 et 2019-0302 à SB), du Conseil suédois de la recherche (VR) (2014-02592, 2019-01744 et 2019-04720 à SB), la Fondation pour la prévention de la résistance aux antibiotiques (Resist), les Fondations Karolinska Institutet et KID to SB (financement partiel de l’enseignement doctoral pour Marco Loreti) de l’Institut Karolinska. ML a reçu le soutien de la Swedish Children’s Cancer Foundation (TJ2018-0128 et PR2019-0100).
matériels
| Name | Company | Catalog Number | Comments |
| 8-well chamber slides | Lab-Tek | 154534 | |
| BD Comp bead plus | BD | 560497 | |
| Bovine Serum Albumin | Sigma-Aldrich | A7906 | |
| DAPI Mounting media | Vector Laboratories | H-1200-10 | |
| EDTA (0.5 M) | Karolinska University hospital, Huddinge | N/A | |
| Falcon 6-well Flat Bottom plates | Corning Life Sciences | 353046 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich | F7524 | |
| Formaldehyde | Sigma-Aldrich | F8775 | |
| Glycerol (70%) | Karolinska University hospital, Huddinge | N/A | |
| GM-CSF | Peprotech | 300-03 | |
| Goat anti-mouse IgG Alexa Fluor 594 secondary antibody | Invitrogen | R37121 | Secondary antibody for CD64 |
| Goat anti-Rabbit IgG Alexa Fluor 594 secondary antibody | Invitrogen | A-11037 | Secondary antibody for CD163 |
| HEPES | GE Healthcare Life Sciences | SH30237.01 | |
| IFN-γ | Peprotech | 300-02 | |
| IL-4 | Peprotech | 200-04 | |
| L-Glutamine | GE Healthcare Life Sciences | SH30034.01 | |
| LPS (Escherichia coli O55:B5) | Sigma-Aldrich | L6529 | |
| Lymphoprep | Alere Technologies AS | 11508545 | |
| M-CSF | Peprotech | 300-25 | |
| Middle Brook 7H10 agar plates | Karolinska University hospital, Huddinge | N/A | |
| Middle Brook 7H9 media | Karolinska University hospital, Huddinge | N/A | |
| Mouse anti-human CD64 primary antibody | Bio-Rad | MCA756G | Clone: 10.1 |
| Na-pyruvate | GE Healthcare Life Sciences | SH300239.01 | |
| Normal goat serum | Jackson ImmunoResearch | 005-000-121 | |
| Rabbit anti-human CD163 primary antibody | GeneTex | GTX81526 | Polyclonal |
| RPMI 1640 | Life Technologies Corporation | SH30096.01 | |
| Triton X-100 | Sigma-Aldrich | X-100 | |
| TubeSpin bioreactor tubes | TPP Techno Plastic Products AG | 87050 | |
| Tween-20 | Sigma-Aldrich | P9416 | |
| Tween-80 | Sigma-Aldrich | P4780 |
Références
- Sica, A., Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation. 122 (3), 787-795 (2012).
- Cassetta, L., Cassol, E., Poli, G. Macrophage polarization in health and disease. Scientific World Journal. 11, 2391-2402 (2011).
- Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J., Hill, A. M. M-1/M-2 macrophages and the Th1/Th2 paradigm. Journal of Immunology. 164 (12), 6166-6173 (2000).
- Martinez, F. O., Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000 Prime Reports. 6, 13 (2014).
- Atri, C., Guerfali, F. Z., Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. International Journal of Molecular Sciences. 19 (6), (2018).
- Flynn, J. L., Gideon, H. P., Mattila, J. T., Lin, P. L. Immunology studies in non-human primate models of tuberculosis. Immunological Reviews. 264 (1), 60-73 (2015).
- Mosser, D. M., Edwards, J. P. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 8 (12), 958-969 (2008).
- Fleetwood, A. J., Lawrence, T., Hamilton, J. A., Cook, A. D. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. Journal of Immunology. 178 (8), 5245-5252 (2007).
- Nathan, C. F., Murray, H. W., Wiebe, M. E., Rubin, B. Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. Journal of Experimental Medicine. 158 (3), 670-689 (1983).
- Leidi, M., et al. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. Journal of Immunology. 182 (7), 4415-4422 (2009).
- Stein, M., Keshav, S., Harris, N., Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. Journal of Experimental Medicine. 176 (1), 287-292 (1992).
- Italiani, P., Boraschi, D. From Monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Frontiers in Immunology. 5, 514 (2014).
- Verreck, F. A., et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proceedings of the National Academy of Sciences. 101 (13), 4560-4565 (2004).
- Redente, E. F., et al. Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. Journal of Leukocyte Biology. 88 (1), 159-168 (2010).
- Refai, A., Gritli, S., Barbouche, M. R., Essafi, M. Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Frontiers in Cellular and Infection Microbiology. 8, 327 (2018).
- Kahnert, A., et al. Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. European Journal of Immunology. 36 (3), 631-647 (2006).
- Marino, S., et al. Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infection and Immunity. 83 (1), 324-338 (2015).
- Erbel, C., et al. An in vitro model to study heterogeneity of human macrophage differentiation and polarization. Journal of Visualized Experiments. (76), e50332 (2013).
- Lee, C. Z. W., Kozaki, T., Ginhoux, F. Publisher Correction: Studying tissue macrophages in vitro: are iPSC-derived cells the answer. Nature Reviews Immunology. 18 (11), 726 (2018).
- Ying, W., Cheruku, P. S., Bazer, F. W., Safe, S. H., Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. Journal of Visualized Experiments. (76), e50323 (2013).
- Van den Bossche, J., Baardman, J., de Winther, M. P. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. Journal of Visualized Experiments. (105), e53424 (2015).
- McKinnon, K. M. Flow Cytometry: An Overview. Current Protocols in Immunology. 120, 1-11 (2018).
- Njoroge, J. M., et al. Characterization of viable autofluorescent macrophages among cultured peripheral blood mononuclear cells. Cytometry. 44 (1), 38-44 (2001).
- Li, F., et al. Autofluorescence contributes to false-positive intracellular Foxp3 staining in macrophages: a lesson learned from flow cytometry. Journal of Immunological Methods. 386 (1-2), 101-107 (2012).
- Rao Muvva, J., Parasa, V. R., Lerm, M., Svensson, M., Brighenti, S. Polarization of human monocyte-derived cells with vitamin D promotes control of Mycobacterium tuberculosis infection. Frontiers in Immunology. 10, 3157 (2019).
- Tarique, A. A., et al. functional, and plasticity features of classical and alternatively activated human macrophages. American Journal of Respiratory Cell and Molecular Biology. 53 (5), 676-688 (2015).
- Hristodorov, D., et al. Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies. MAbs. 7 (5), 853-862 (2015).
- Jaguin, M., Houlbert, N., Fardel, O., Lecureur, V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cellular Immunology. 281 (1), 51-61 (2013).
- Xu, Z. J., et al. The M2 macrophage marker CD206: a novel prognostic indicator for acute myeloid leukemia. Oncoimmunology. 9 (1), 1683347 (2020).
- Liu, Q., et al. Differential expression and predictive value of monocyte scavenger receptor CD163 in populations with different tuberculosis infection statuses. BMC Infectious Diseases. 19 (1), 1006 (2019).
- Sugaya, M., et al. Association of the numbers of CD163(+) cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. Journal of Dermatological Science. 68 (1), 45-51 (2012).
- Nawaz, A., et al. CD206(+) M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nature Communications. 8 (1), 286 (2017).
- Liu, B., Zhang, M., Zhao, J., Zheng, M., Yang, H. Imbalance of M1/M2 macrophages is linked to severity level of knee osteoarthritis. Experimental and Therapeutic Medicine. 16 (6), 5009-5014 (2018).
- Lee, C., et al. M1 macrophage recruitment correlates with worse outcome in SHH Medulloblastomas. BMC Cancer. 18 (1), 535 (2018).
- Rekha, R. S., et al. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy. 11 (9), 1688-1699 (2015).
- Mattila, J. T., et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. Journal of Immunology. 191 (2), 773-784 (2013).
- Braian, C., Svensson, M., Brighenti, S., Lerm, M., Parasa, V. R. A 3D Human Lung Tissue Model for Functional Studies on Mycobacterium tuberculosis Infection. Journal of Visualized Experiments. (104), e53084 (2015).
- Brighenti, S., Joosten, S. A. Friends and foes of tuberculosis: modulation of protective immunity. Journal of Internal Medicine. , 12778 (2018).
- Chattopadhyay, P. K., Roederer, M. Good cell, bad cell: flow cytometry reveals T-cell subsets important in HIV disease. Cytometry Part A. 77 (7), 614-622 (2010).
- Becht, E., et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology. 37, 38-44 (2019).
Erratum
Formal Correction: Erratum: Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection
Posted by JoVE Editors on 10/14/2020. Citeable Link.
An erratum was issued for: Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection. Author and affiliation information was updated.
The author and affiliation information was updated from:
Akhirunnesa Mily1, Sadaf Kalsum1, Marco Giulio Loreti1, Rokeya Sultana Rekha2, Jagadeeswara Rao Muvva1, Magda Lourda1,3, Susanna Brighenti1
1Center for Infectious Medicine (CIM), Department of Medicine Huddinge, ANA Futura, Karolinska Institutet
2Clinical Microbiology, Department of Laboratory Medicine (Labmed), ANA Futura, Karolinska Institutet
3Childhood Cancer Research Unit, Department of Women's and Children's Health, Karolinska Institutet
to:
Akhirunnesa Mily1,2, Sadaf Kalsum1, Marco Giulio Loreti1, Rokeya Sultana Rekha3, Jagadeeswara Rao Muvva1, Magda Lourda1,4, Susanna Brighenti1
1Center for Infectious Medicine (CIM), Department of Medicine Huddinge, ANA Futura, Karolinska Institutet
2Infectious Diseases Division, International Centre for Diarrhoeal Disease Research, Bangladesh
3Clinical Microbiology, Department of Laboratory Medicine (Labmed), ANA Futura, Karolinska Institutet
4Childhood Cancer Research Unit, Department of Women's and Children's Health, Karolinska Institutet
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.