
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Imagerie et analyse automatisées pour la quantification des macropinosomes marqués par fluorescence
Dans cet article
Résumé
Les essais automatisés utilisant des microplaques multi-puits sont des approches avantageuses pour identifier les régulateurs de voie en permettant l’évaluation d’une multitude de conditions dans une seule expérience. Ici, nous avons adapté le protocole bien établi d’imagerie et de quantification des macropoinosomes à un format de microplaques à 96 puits et fournissons un aperçu complet de l’automatisation à l’aide d’un lecteur de plaques multimode.
Résumé
La macropinocytose est une voie d’absorption de la phase liquidienne non spécifique qui permet aux cellules d’internaliser de grandes cargaisons extracellulaires, telles que des protéines, des agents pathogènes et des débris cellulaires, par endocytose en vrac. Cette voie joue un rôle essentiel dans une variété de processus cellulaires, y compris la régulation des réponses immunitaires et le métabolisme des cellules cancéreuses. Compte tenu de cette importance dans la fonction biologique, l’examen des conditions de culture cellulaire peut fournir des informations précieuses en identifiant les régulateurs de cette voie et en optimisant les conditions à utiliser dans la découverte de nouvelles approches thérapeutiques. L’étude décrit une technique d’imagerie et d’analyse automatisée utilisant un équipement de laboratoire standard et un lecteur de plaques multimode d’imagerie cellulaire pour la quantification rapide de l’indice macropinocytaire dans les cellules adhérentes. La méthode automatisée est basée sur l’absorption de dextran fluorescent de haut poids moléculaire et peut être appliquée à des microplaques de 96 puits pour faciliter l’évaluation de plusieurs conditions dans une expérience ou des échantillons fixes montés sur des couvercles en verre. Cette approche vise à maximiser la reproductibilité et à réduire la variation expérimentale tout en étant à la fois rapide et rentable.
Introduction
La voie endocytaire non spécifique de la macropinocytose permet aux cellules d’internaliser une variété de composants extracellulaires, y compris les nutriments, les protéines, les antigènes et les agents pathogènes, grâce à l’absorption en vrac du liquide extracellulaire et de ses constituants1. Bien qu’importante pour la biologie de nombreux types de cellules, la voie de la macropinocytose est décrite comme jouant un rôle essentiel dans la biologie tumorale, où, grâce à l’absorption macropinocytaire, les cellules tumorales sont capables de survivre et de proliférer en présence d’un microenvironnement appauvri en nutriments2,3. L’absorption des macromolécules extracellulaires, y compris l’albumine et la matrice extracellulaire, et des débris cellulaires nécrotiques, fournit une source alternative de nutriments pour la production de biomasse en créant des acides aminés, des sucres, des lipides et des nucléotides par le biais du catabolisme de cargaison médié par la fusion de macropinosomes et de lysosomes4,5,6,7,8.
L’induction et la régulation de la macropinocytose sont complexes et peuvent varier en fonction du contexte cellulaire. Jusqu’à présent, plusieurs inducteurs de la macropinocytose ont été identifiés et comprennent des ligands, tels que le facteur de croissance épidermique (EGF), le facteur de croissance dérivé des plaquettes (PDGF), la galectine-3 et Wnt3A9,10,11,12,13. En outre, les conditions de culture qui imitent le microenvironnement tumoral peuvent déclencher l’activation de la voie. Les tumeurs de l’adénocarcinome canalaire pancréatique (PDAC) sont privées de nutriments, en particulier pour l’acide aminé glutamine, ce qui fait que les cellules cancéreuses et les fibroblastes associés au cancer (CAF) dépendent de la macropinocytose pour la survie7,13,14,15. De plus, les stress tumoraux, tels que l’hypoxie et le stress oxydatif, peuvent activer cette voie de piégeage16. En plus des nombreux influenceurs extrinsèques qui peuvent induire la macropinocytose, une variété de voies intracellulaires contrôlent la formation de macropinosomes. La transformation oncogène médiée par Ras est suffisante pour initier la machinerie macropinocytaire, et plusieurs types de cancer présentent une macropinocytose constitutive oncogène pilotée par Ras4,5,9,17. Alternativement, l’activation ras de type sauvage et les voies indépendantes de Ras ont été identifiées pour activer la macropinocytose dans les cellules cancéreuses et les CAF10,11,15,18. L’utilisation de divers modèles in vitro en combinaison avec des traitements inhibiteurs a abouti à l’identification de plusieurs modulateurs de macropinocytose, qui comprennent des échangeurs sodium-hydrogène, la petite GTPase Rac1, la phosphoinositide 3-kinase (PI3K), la kinase activée par p21 (Pak) et la protéine kinase activée par AMP (AMPK)4,13,15 . Cependant, étant donné la multitude de facteurs et de conditions décrits qui régulent la macropinocytose, il est concevable que beaucoup plus de modulateurs et de stimuli restent inconnus. L’identification de nouveaux modulateurs et stimuli peut être facilitée par l’évaluation automatisée d’une multitude de conditions dans une seule expérience. Cette méthodologie peut faire la lumière sur les facteurs impliqués dans la formation de macropoinosomes et peut permettre la découverte de nouvelles petites molécules ou de produits biologiques qui ciblent cette voie.
Ici, nous avons adapté notre protocole précédemment établi pour déterminer l’étendue de la macropinocytose dans les cellules cancéreuses in vitro à un format de microplaque à 96 puits et à une imagerie et une quantification automatisées19,20. Ce protocole est basé sur la microscopie fluorescente, qui est devenue une norme dans le domaine pour déterminer la macropinocytose in vitro et in vivo4,5,6,7,9,10,11,12,13,15,16,17,18, 19,20,21,22. Les macropoinosomes peuvent être distingués des autres voies endocytaires par leur capacité à internaliser de grandes macromolécules, telles que le dextran de haut poids moléculaire (c.-à-d. 70 kDa)2,3,4,20,21,22,23. Ainsi, les macropinosomes peuvent être définis par l’absorption de fluorophores fluorophores marqués extracellulairement 70 kDa dextran. En conséquence, les vésicules macropinocytaires se manifestent sous forme d’amas intracellulaires de puncta fluorescents de tailles allant de 0,2 à 5 μm. Ces puncta peuvent être imagés au microscope et ensuite quantifiés pour déterminer l’étendue de la macropinocytose dans la cellule - « l’indice macropinocytaire ».
Dans ce protocole, les étapes essentielles pour visualiser les macropinosomes dans les cellules adhérentes in vitro sur une microplaque de 96 puits et des couvercles à l’aide d’équipements de laboratoire standard sont décrites (Figure 1). En outre, les instructions pour automatiser l’acquisition d’images et la quantification de l’indice macropinocytaire à l’aide d’un lecteur de plaques multimode d’imagerie cellulaire sont fournies. Cette automatisation réduit le temps, les coûts et les efforts par rapport à nos protocoles décrits précédemment19,20. En outre, il évite l’acquisition et l’analyse d’images involontairement biaisées et améliore ainsi la reproductibilité et la fiabilité. Cette méthode peut facilement être adaptée à différents types de cellules ou de lecteurs de plaques ou être utilisée pour déterminer d’autres caractéristiques des macropoinosomes, telles que la taille, le nombre et l’emplacement. La méthode décrite ici est particulièrement adaptée au dépistage des conditions de culture cellulaire qui induisent la macropinocytose, à l’identification de nouveaux modulateurs ou à l’optimisation des concentrations médicamenteuses d’inhibiteurs connus.

Figure 1 : Schéma du test automatisé pour déterminer l’indice macropinocytaire dans les cellules adhérentes. Créé à l’aide de BioRender. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
1. Préparation des matériaux
- Dissoudre 70 kDa de dextran marqué avec du FITC ou de la tétraméthylrhodamine (TMR) dans du PBS pour obtenir une solution à 20 mg/mL. Conserver les aliquotes à -20 °C.
- Dissoudre le DAPI dans le ddH2O pour obtenir une solution à 1 mg/mL. Conserver les aliquotes à -20 °C.
ATTENTION : Le DAPI est un cancérogène potentiel et doit être manipulé avec précaution. - Le jour de la fixation, préparez du formaldéhyde frais de qualité ACS à 3,7% dans du PBS.
ATTENTION : Le formaldéhyde est un fixateur, cancérogène connu et toxique s’il est inhalé. Faites la solution dans une hotte chimique pour éviter l’inhalation et manipulez avec soin. - Préparez des couvercles lavés à l’acide.
- Utilisez un bécher de 500 mL et un bain-marie pour chauffer 28 g de couvercles en verre borosilicate d’un diamètre de 12 mm et d’une épaisseur #1,5 pendant 24 h à 56 °C dans 100 mL de 1 M HCl. Scellez le bécher avec une pellicule de plastique pour éviter une évaporation importante.
ATTENTION: HCl est un acide fort qui est très corrosif et doit donc être manipulé avec soin et utilisé dans une hotte chimique pour éviter l’inhalation. - Laver les couvercles avec de l’eau distillée. Répétez le lavage 4 fois. Ensuite, lavez les couvercles avec de l’éthanol à 95%. Répétez le lavage 4 fois.
- Conservez les couvercles lavés à l’acide dans un plat de culture cellulaire à température ambiante pour une utilisation future en maintenant la stérilité par immersion dans de l’éthanol à 95%. Scellez le plat avec un parafilm pour éviter une évaporation importante.
- Utilisez un bécher de 500 mL et un bain-marie pour chauffer 28 g de couvercles en verre borosilicate d’un diamètre de 12 mm et d’une épaisseur #1,5 pendant 24 h à 56 °C dans 100 mL de 1 M HCl. Scellez le bécher avec une pellicule de plastique pour éviter une évaporation importante.
2. Préparation des cellules
- À l’aide d’une plaque de culture tissulaire confluente de 10 cm avec les cellules adhérentes d’intérêt, aspirer le milieu et rincer les cellules avec 5 mL de DPBS, préavertissé à 37 °C.
- Détachez les cellules de la plaque en ajoutant 1,5 mL de trypsine préavertie à 0,25 % et en incubant à 37 °C.
REMARQUE: Le temps d’incubation de la trypsine nécessaire pour détacher les cellules d’intérêt doit être déterminé empiriquement et peut être confirmé en observant le détachement au microscope optique conventionnel. - Recueillir les cellules dans un tube centrifuge de 15 mL et ajouter 4,5 mL de milieu complet pour éteindre la trypsine.
- Abattez les cellules par centrifugation pendant 3 min à 200 x g et aspirez le surnageant.
- Remettre en suspension la pastille cellulaire dans un volume adéquat de milieu complet préavertissé pour obtenir une suspension à cellule unique pour l’ensemencement.
- Procéder à l’ensemencement des cellules sur une plaque de 24 puits avec des couvercles ou un format de microplaque de 96 puits (Figure 1).
REMARQUE: Le nombre de cellules à ensemencer doit être déterminé empiriquement pour chaque lignée cellulaire, car les taux de prolifération et la taille varient d’une lignée cellulaire à l’autre. Ce protocole a été optimisé pour les cellules cancéreuses adhérentes avec une confluence cellulaire de 80% le jour du marquage des macropinosomes. La confluence cellulaire peut affecter la capacité macropinocytaire, et cela devrait également être déterminé empiriquement.- Plaque à 24 puits avec format de couvercle
- Ajouter des couvercles à une plaque de culture tissulaire de 24 puits, utiliser des pinces pour saisir un seul couvercle du bain d’éthanol. Tapotez le couvercle sur la paroi intérieure de la plaque pour éliminer l’excès d’éthanol et placez le couvercle à plat sur le fond d’un puits.
- Laissez l’éthanol s’évaporer et lavez le couvercle 2 fois avec du DPBS.
- Ensemencez les cellules au-dessus du couvercle en ajoutant 500 μL de suspension cellulaire à chaque puits. Placez les cellules dans un incubateur cellulaire à 37 °C avec 5 % de CO2 jusqu’à ce que la confluence cellulaire atteigne 60 % à 80 % la veille du marquage des macropoinosomes.
- La veille du marquage des macropinosomes, aspirer les milieux des puits et ajouter 500 μL de milieux préavertis sans sérum à chaque puits et placer les cellules dans un incubateur cellulaire à 37 °C avec 5% de CO2 pendant 16-24 h.
REMARQUE: Selon les conditions à étudier, les milieux sans sérum sont recommandés pour réduire les effets des facteurs de croissance qui peuvent induire une macropinocytose et qui sont normalement présents dans le sérum. Cependant, il faut considérer que la famine sérique peut affecter d’autres processus cellulaires, tels que la prolifération et l’autophagie. Comme le sérum résiduel peut affecter la capacité macropinocytaire des cellules, ainsi que l’activité inhibitrice, l’élimination du sérum peut être améliorée en rinçant doucement les cellules 1 ou 2 fois avec 500 μL de DPBS préavertis.
- Format de microplaque à 96 puits
- Transférer la suspension cellulaire dans un réservoir de réactif de 25 mL. À l’aide d’une pipette multicanal (8 ou 12 canaux), ensemencez 100 μL de la suspension cellulaire dans chaque puits d’une microplaque de criblage noire à haute teneur de 96 puits avec une oléfine cyclique optiquement claire ou un fond de verre.
- Placez les cellules dans un incubateur cellulaire à 37 °C avec 5 % de CO2 jusqu’à ce que la confluence cellulaire atteigne 60 % à 80 % la veille du marquage des macropoinosomes.
- La veille de l’étiquetage des macroponosomes, retirez et jetez le support de chaque puits à l’aide d’une pipette multicanal (8 ou 12 canaux) ou d’un adaptateur d’aspiration multicanal pour les embouts standard fixés à une pompe à vide.
- À l’aide d’un réservoir de réactif et d’une pipette multicanal (8 ou 12 canaux), ajoutez doucement 100 μL de milieu préavertissé sans sérum à chaque puits. Placez les cellules dans un incubateur à 37 °C avec 5% de CO2 pendant 16-24 h.
REMARQUE: Selon les conditions à étudier, les milieux sans sérum sont recommandés pour réduire les effets des facteurs de croissance qui peuvent induire une macropinocytose et qui sont normalement présents dans le sérum. Cependant, il faut considérer que la famine sérique peut affecter d’autres processus cellulaires, tels que la prolifération et l’autophagie. Comme le sérum résiduel peut affecter la capacité macropinocytaire des cellules, ainsi que l’activité inhibitrice, l’élimination du sérum peut être améliorée en rinçant doucement les cellules 1 ou 2 fois avec 100 μL de DPBS préavertissé.
- Plaque à 24 puits avec format de couvercle
3. Étiquetage des macropinosomes
- Plaque à 24 puits avec format de couvercle
- Aspirer les puits et ajouter 200 μL de milieux sans sérum avec 1 mg/mL de dextran de haut poids moléculaire marqué au fluorophore (70 kDa). Placez les cellules dans un incubateur à 37 °C pendant 30 min.
REMARQUE: Selon les conditions à étudier, au lieu d’utiliser des milieux frais, il peut être préférable de réutiliser les milieux conditionnés pour la charge de dextrane, car ils contiendraient des facteurs sécrétés ou supplémentés, tels que l’EGF ou des composés inhibiteurs, respectivement, qui peuvent affecter la capacité macropinocytaire des cellules. - Aspirer le milieu et laver doucement mais rapidement les cellules 5 fois avec du PBS glacé à l’aide d’un flacon de lavage prérefroidi. Serrez fermement la plaque à la main pendant les lavages pour aider à déloger les agrégats de dextran qui se collent aux couvercles.
- Fixez les cellules en ajoutant 350 μL de formaldéhyde à 3,7% et en incubant pendant 20 min. Ensuite, aspirez la solution de fixation et lavez les cellules avec pbS deux fois.
- Colorez les noyaux avec 350 μL de 2 μg/mL DAPI dans PBS. Après 20 min, aspirer la solution de DAPI et laver les cellules avec du PBS trois fois.
- Collez les isolateurs en silicone côte à côte sur une lame de microscope pour obtenir un espacement uniforme et une localisation reproductible des couvercles, nécessaires à l’automatisation de l’imagerie (Figure 2A, B).
REMARQUE: La lame entière du microscope peut être remplie avec un total de 3 isolateurs. - Pour chaque couvercle, ajoutez une goutte de support de montage de fluorescence de durcissement sur la lame du microscope dans l’espace ouvert de l’isolateur (Figure 2C). Ramassez un couvercle à l’aide d’une pince et retirez l’excès de PBS en tapotant doucement le côté des couvercles sur une lingette non pelucheuse.
- Placez le couvercle à l’envers sur la goutte du support de montage (Figure 2D). Tapotez doucement le couvercle à l’aide de pinces fermées pour éliminer les bulles du support de montage (Figure 2E).
- Rangez les glissières dans un environnement sombre et laissez le support de montage sécher à température ambiante, ce qui prend généralement de 16 à 24 h. Les diapositives peuvent désormais être imagées ou stockées à -20 °C pendant 2 semaines maximum.
- Avant l’imagerie, retirez les isolateurs de la lame du microscope. Laissez les lames s’équilibrer à la température ambiante et nettoyez les couvercles à l’aide d’un applicateur à pointe de coton mouillé avec un nettoyant pour vitres sans ammoniaque. Par la suite, utilisez un applicateur propre à pointe de coton mouillé avec 70% d’éthanol pour nettoyer et laissez le couvercle sec.
- Aspirer les puits et ajouter 200 μL de milieux sans sérum avec 1 mg/mL de dextran de haut poids moléculaire marqué au fluorophore (70 kDa). Placez les cellules dans un incubateur à 37 °C pendant 30 min.

Figure 2: Placer des couvercles sur une lame de microscope avec des isolateurs en silicone. (A) Les isolateurs en silicone sont pressés et collés à une lame de microscope. (B) L’ensemble de la lame du microscope peut être rempli d’un total de 3 isolateurs, ce qui permet un espacement uniforme et une localisation reproductible des couvercles. (C) Pour chaque couvercle, ajouter une goutte de support de montage en fluorescence sur la lame du microscope dans l’espace ouvert de l’isolateur. (D) À l’aide d’une pince, prélevez un couvercle dans la plaque de 24 puits et placez-le à l’envers sur la goutte du support de montage. (E) Lorsque des bulles sont présentes entre le couvercle et la lame du microscope, tapotez doucement le couvercle à l’aide d’une pince fermée pour enlever les bulles. Créé à l’aide de BioRender. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Format de microplaque à 96 puits
- Aspirer les puits à l’aide d’un adaptateur d’aspiration multicanal attaché à un vide et ajouter 40 μL de milieu sans sérum avec 1 mg/mL de dextran de haut poids moléculaire marqué au fluorophore (70 kDa) aux puits. Incuber les cellules dans un incubateur cellulaire à 37 °C pendant 30 min.
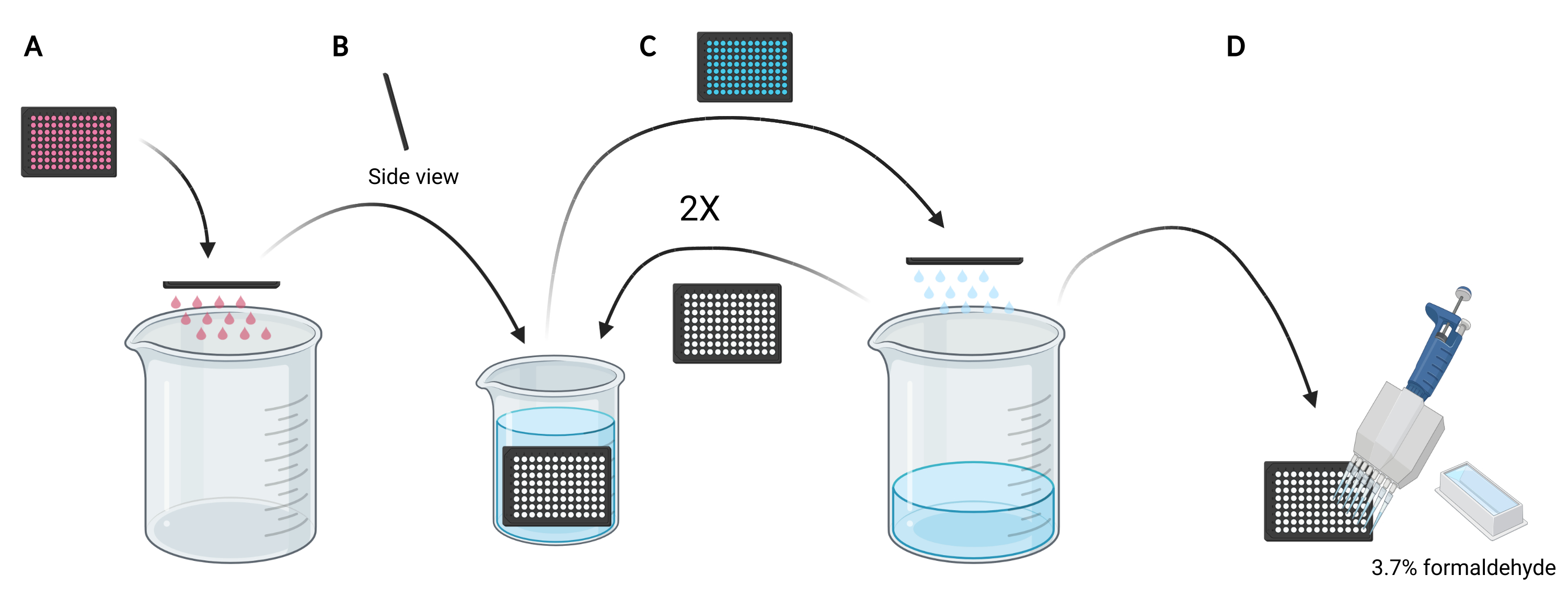
REMARQUE: Selon les conditions à étudier, au lieu d’utiliser des milieux frais, il peut être préférable de réutiliser les milieux conditionnés pour la charge de dextran, car ils contiendraient des facteurs sécrétés ou supplémentés, tels que l’EGF ou des composés inhibiteurs, respectivement, qui peuvent affecter la capacité macropinocytaire des cellules. - Jetez le support dans la microplaque en faisant glisser manuellement la plaque à l’envers dans un bécher vide de 5 L (Figure 3A).
- Rincez les cellules de la microplaque en immergeant lentement la plaque verticalement, à un léger angle, dans un bécher de 2 L rempli de PBS glacé (Figure 3B) et ensuite jetez le PBS dans la microplaque en faisant glisser la plaque à l’envers dans le bécher de 5 L (Figure 3C). Répétez 2 fois.
REMARQUE: Les cellules qui se fixent faiblement à la microplaque d’imagerie peuvent se détacher au cours de ce processus. Si nécessaire, les puits peuvent également être aspirés avec un adaptateur d’aspiration multicanal ou lavés plus doucement avec PBS à l’aide d’une pipette multicanal. Le traitement d’une microplaque de 96 puits nécessitera environ 2 L de PBS glacé. Si d’autres plaques doivent être analysées, utilisez un bécher plus grand et ajoutez 1 L de PBS glacé pour chaque plaque supplémentaire ou rafraîchissez le PBS glacé au besoin. - Après l’élimination du PBS du dernier rinçage, fixer les cellules pendant 20 min à température ambiante en ajoutant 100 μL de formaldéhyde à 3,7 % dans le PBS à chaque puits à l’aide d’un réservoir de réactif de 25 mL et d’une pipette multicanal (Figure 3D).
- Retirez la solution de fixation et lavez les cellules avec du PBS deux fois à l’aide de la technique d’immersion et de flicking. Colorez les noyaux avec 100 μL de 2 μg/mL de DAPI dans du PBS par puits.
- Après 20 min, rincer les cellules trois fois avec du PBS glacé en utilisant la technique d’immersion et de flicking décrite ci-dessus (étape 3.2.3). Retirez tout PBS résiduel en tapotant la microplaque à l’envers sur une lingette non pelucheuse et ajoutez 100 μL de PBS frais à chaque puits à l’aide d’un réservoir de réactif de 25 mL et d’une pipette multicanal. Imagez les cellules maintenant ou stockez-les recouvertes de lumière à 4 °C pendant une semaine.
REMARQUE: Alternativement, une solution de glycérol dans PBS (au lieu de PBS) peut être utilisée pour l’imagerie et le stockage afin de mieux stabiliser la fluorescence (Figure 4A). - Avant l’imagerie, laissez la plaque s’équilibrer à la température ambiante. Essuyez la microplaque avec une lingette non pelucheuse.
- Aspirer les puits à l’aide d’un adaptateur d’aspiration multicanal attaché à un vide et ajouter 40 μL de milieu sans sérum avec 1 mg/mL de dextran de haut poids moléculaire marqué au fluorophore (70 kDa) aux puits. Incuber les cellules dans un incubateur cellulaire à 37 °C pendant 30 min.

Figure 3 : Rinçage de la microplaque de 96 puits pour préparer la fixation. (A) Videz la microplaque du support dans un bécher de 5 L en les agitant manuellement. (B) Verticalement et à un léger angle, immergez lentement la microplaque dans un bécher de 2 L rempli de PBS glacé. (C) Videz la microplaque de PBS dans le bécher de 5 L en la faisant glisser manuellement. Répétez deux fois les étapes de lavage décrites au point B. (D) Après avoir vidé le PBS dans la microplaque pour la dernière fois, ajouter 100 μL 3,7% de formaldéhyde aux puits, à l’aide d’une pipette multicanal. Créé à l’aide de BioRender. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
4. Imagerie automatisée des macropoinosomes
Les images de macropinosomes peuvent être capturées à l’aide d’un microscope fluorescent standard, comme décrit précédemment19,20. Cependant, une telle procédure peut être améliorée en termes d’efficacité grâce à l’automatisation, en particulier lors de l’évaluation de nombreuses conditions de culture cellulaire différentes. L’automatisation de l’acquisition d’images peut être réalisée via un lecteur de plaques multimode d’imagerie cellulaire, ce qui réduit l’effort en réduisant les procédures de manipulation et, surtout, augmente la reproductibilité et la fiabilité des données en acquérant des images de manière impartiale. Plusieurs systèmes d’imagerie sont disponibles dans le commerce et les directions diffèrent d’un instrument à l’autre. Ici, l’acquisition d’images à l’aide d’un Cytation 5 est décrite. Cependant, le protocole ci-dessous peut être adapté à chaque instrument individuel en adhérant aux directives suivantes:
- Créez un protocole d’automatisation pour acquérir les images avec un objectif d’air 40x dans le canal de longueur d’onde du fluorophore dextran (FITC/TMR) et DAPI.
REMARQUE: L’inhibiteur de macropinocytose couramment utilisé EIPA présente une autofluorescence dans le canal FITC, en particulier lorsqu’il est précédemment excité dans le canal DAPI. D’autres composés testés pourraient également présenter une autofluorescence. Pour contourner ce problème, le fait de définir l’acquisition d’image pour qu’elle se produise d’abord dans le canal avec la longueur d’onde d’excitation la plus élevée (FITC/TMR) et en second dans le canal DAPI permet d’éviter cette occurrence. - Optimisez les paramètres d’exposition à l’aide d’un échantillon dont le niveau de macropinocytose est le plus élevé pour éviter la surexposition, ce qui peut entraîner une saturation du signal et une perte de données d’intensité. Utilisez des paramètres de mise au point qui localisent facilement et systématiquement l’échantillon pour produire des images de haute qualité.
- Acquérir plusieurs images dans chaque puits ou couvercle pour tenir compte de la variabilité de l’échantillon et obtenir une représentation précise de l’échantillon.
- Une fois les paramètres d’imagerie déterminés, utilisez les mêmes paramètres pour chaque échantillon de l’expérience.
- Suivez ces instructions pour l’acquisition d’images de macropoinosomes lors de l’utilisation d’un logiciel Cytation 5 et Gen5 :
- Plaque à 24 puits avec format de couvercle
- Démarrez le lecteur de plaques et insérez les lames de microscope à l’envers à l’aide du support de diapositive.
- Ouvrez le lecteur de microplaques et le logiciel d’imagerie, créez un nouveau protocole en cliquant sur Protocoles et Créez nouveau. Double-cliquez sur Procédure et sélectionnez le type de plaque.
REMARQUE: Si le type de plaque n’est pas disponible, ajoutez le type de plaque au logiciel en cliquant sur Système > Types de plaques > Ajouter une plaque et en utilisant les dimensions de plaque fournies par le fabricant. Pour faciliter l’utilisation, le gabarit pour deux lames de microscope avec trois couvercles espacés à l’aide des isolateurs en silicone est fourni dans le fichier supplémentaire 1. - Pour accéder aux paramètres d’imagerie, sélectionnez Actions > Image >'imageur inversé et cliquez sur OK. Utilisez l’objectif 40x, PL FL Phase avec large champ de vision et autofocus Binning.
- Pour le premier canal, sélectionnez le cube LED correspondant à l’étiquette fluorophore dextran (GFP ou RFP). Décochez sur Exposition automatique et cliquez sur le bouton de l’icône du microscope pour optimiser les paramètres d’exposition. Lorsque les paramètres d’exposition appropriés ont été déterminés, cliquez sur Enregistrer les paramètres.
REMARQUE: Ajustez les paramètres d’exposition à l’aide d’un échantillon dont le niveau de macropinocytose est le plus élevé pour éviter les images surexposées, ce qui peut entraîner une saturation du signal et une perte de données d’intensité. - Répétez l’étape précédente pour le deuxième canal à l’aide du cube LED DAPI.
- Définissez les paramètres de mise au point automatique pour chaque canal de fluorescence, sélectionnez Options de mise au point. Désélectionnez la méthode de mise au point par défaut et utilisez l’autofocus avec numérisation facultative et l’autofocus sans numérisation facultative pour le canal dextran-fluorophore et DAPI, respectivement. Cliquez sur OK pour enregistrer les paramètres.
REMARQUE: La distance de balayage peut être réduite à 200 μm et l’incrément à 20 μm pour augmenter l’efficacité de la mise au point automatique. Le scan optionnel est requis pour un autofocus adéquat sur les échantillons à faible fluorescence. Cela se produit normalement lors de l’analyse de conditions à faible macropinocytose, par exemple lorsque la macropinocytose est inhibée ou non présente de manière innée. - Utilisez l’option Définir les balises pour automatiser l’acquisition d’images dans différentes régions du couvercle. Cliquez sur l’icône du microscope et ajoutez des balises en cliquant dans la fenêtre d’image et en déplaçant la scène vers la région suivante. Lorsque le nombre approprié de régions a été sélectionné, passez à la barre de couverture suivante et répétez le processus. Pour finaliser, cliquez sur Enregistrer les paramètres.
REMARQUE : Pour obtenir une bonne représentation de la macropinocytose dans l’échantillon, sélectionnez environ 20 balises réparties uniformément sur la lèvre de couverture (Figure 4B). Moins de balises peuvent être utilisées, mais certaines images peuvent devoir être exclues de l’analyse d’image après l’acquisition en raison de différences de qualité, par exemple lorsque l’image est floue ou contient des bulles ou des taches et des taches fluorescentes. - Pour terminer le réglage des paramètres d’imagerie, cliquez sur OK. Pour créer une image des barres de couverture, sélectionnez Créer une nouvelle expérience et Lire maintenant dans les Outils de protocole. Enregistrez le protocole et l’expérience lorsque vous y êtes invité.
- Plaque à 24 puits avec format de couvercle

Figure 4 : Optimisation des conditions d’acquisition d’images. (A) L’augmentation de la concentration de glycérol augmente la fluorescence TMR-dextran, telle que déterminée dans les cellules AsPC-1 traitées avec EGF. (B) Exemple de coordonnées de balises d’imagerie pour l’acquisition automatique d’images lors de l’utilisation de la plaque à 24 puits avec format de couvercle. Le graphique à barres montre la fluorescence relative moyenne avec SEM de 5 expériences. La signification statistique a été déterminée par l’ANOVA bidirectionnelle, par rapport au PBS. ** p < 0,01; p < 0,001. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Format de microplaque à 96 puits
- Démarrez le lecteur de plaque et insérez la microplaque.
- Ouvrez le lecteur de microplaques et le logiciel d’imagerie, créez un nouveau protocole en cliquant sur Protocoles et Créez nouveau. Double-cliquez sur Procédure et sélectionnez le type de plaque.
REMARQUE: Si le type de plaque n’est pas disponible, ajoutez le type de plaque au logiciel en cliquant sur Système > Types de plaques > Ajouter une plaque et en utilisant les dimensions de plaque fournies par le fabricant. Pour faciliter l’utilisation, le modèle pour les microplaques CellCarrier-96 Ultra de PerkinElmer est fourni dans le fichier supplémentaire 2. - Pour accéder aux paramètres d’imagerie, sélectionnez Actions > Image > Image inversée et cliquez sur OK. Utilisez l’objectif 40x PL FL Phase avec large champ de vision et autofocus Binning.
- Pour le premier canal, sélectionnez le cube LED correspondant à l’étiquette fluorophore dextran (GFP ou RFP). Décochez sur Exposition automatique et cliquez sur le bouton de l’icône du microscope pour optimiser les paramètres d’exposition. Lorsque les paramètres d’exposition appropriés ont été déterminés, cliquez sur Enregistrer les paramètres.
REMARQUE: Lors de l’optimisation des paramètres d’exposition, un blanchiment par fluorescence important peut se produire. Cela peut entraîner des paramètres qui provoquent une surexposition lorsqu’un nouveau champ est imagé. Par conséquent, validez les paramètres d’exposition en vérifiant un champ encore non exposé et en vous assurant qu’aucune saturation du signal ne se produit aux paramètres sélectionnés. N’incluez pas les puits utilisés pour l’optimisation du paramètre d’exposition dans la quantification des macropinosomes, car la fluorescence a diminué à la suite du blanchiment pendant l’optimisation. Ajustez les paramètres d’exposition à l’aide d’un échantillon dont le niveau de macropinocytose est le plus élevé pour éviter les images surexposées, ce qui peut entraîner une saturation du signal et une perte de données d’intensité. - Répétez l’étape précédente pour le deuxième canal à l’aide du cube LED DAPI.
- Définissez les paramètres de mise au point automatique pour chaque canal de fluorescence, sélectionnez Options de mise au point. Désélectionnez la méthode de mise au point par défaut et utilisez l’autofocus laser. Capturez un scan de référence après avoir déterminé le plan focal pour une visualisation optimale des macropoinosomes et des noyaux. Cliquez sur OK pour enregistrer les paramètres.
REMARQUE: La distance de balayage peut être réduite à 400 μm et l’incrément à 3 μm pour augmenter l’efficacité de la mise au point automatique. Pour que l’option de mise au point automatique laser fonctionne correctement, nettoyez le fond de la plaque, séchez et essuyez la plaque avec une lingette non pelucheuse avant l’imagerie. L’autofocus laser est une méthode supérieure pour la mise au point car il nécessite un minimum de temps pour trouver le plan focal. D’autres méthodes de focalisation peuvent être utilisées, mais, comme aucun anti-décoloration n’a été ajouté aux puits, ces méthodes peuvent entraîner un blanchiment important des échantillons, ce qui aura un impact négatif sur la collecte des données. - Réglez le décalage horizontal et vertical sur zéro et sous Image unique, sélectionnez Montage sans chevauchement et utilisez 3 x 3 images, en fonction du nombre de cellules à inclure dans l’analyse.
REMARQUE: Selon la taille et la densité des cellules, plus ou moins d’images peuvent être prises pour obtenir une évaluation représentative de la macropinocytose dans tout l’échantillon. En évaluant la macropinocytose dans les cellules AsPC-1 ou MIA PaCa-2 dans des conditions variables, aucune différence dans l’interprétation des données entre un cadre photo 2 x 2 ou 4 x 4 n’est observée, bien que la variation entre les échantillons répliqués puisse augmenter en prenant moins de photos (Figure 5A, B). L’augmentation ou la diminution de la taille du cadre affectera le temps nécessaire pour scanner la plaque. Selon le temps d’exposition, une microplaque complète de 96 puits prendra environ 1 à 1,5 h pour numériser complètement à l’aide d’une image de 3 x 3. Une image de 2 x 2 et 4 x 4 réduira de moitié ou doublera ce temps, respectivement. - Pour terminer le réglage des paramètres d’imagerie, cliquez sur OK.
- Pour créer une image de la plaque, sélectionnez Créer une nouvelle expérience et Lire maintenant dans les Outils de protocole. Enregistrez le protocole et l’expérience lorsque vous y êtes invité.

Figure 5 : Conditions de contrôle pour l’évaluation de la macropinocytose dans les cellules PDAC. (A) Les cellules AsPC-1 présentent une macropinocytose en réponse à une stimulation EGF de 100 ng/mL pendant 5 min ou à une privation de glutamine pendant 24 h. Pour l’acquisition d’images, des cadres photo de 4 x 4, 3 x 3, 2 x 3 ou 2 x 2 ont été pris pour déterminer l’influence du nombre de photos sur la qualité des données. (B) Les cellules MIA PaCa-2 présentent une macropinocytose constitutive inhibée par un traitement de 30 minutes avec de l’EIPA de 75 μM ou un traitement de 2 heures avec de l’EHop-016 de 10 μM. Les cadres photo ont été pris comme dans A. Barre d’échelle = 25 μm. Les graphiques à barres montrent l’indice macropinocytaire relatif moyen avec SD de 1 expérience avec 4 répétitions. La signification statistique a été déterminée par l’ANOVA bidirectionnelle par rapport au +Q ou à l’état du véhicule. p < 0,001 Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
5. Détermination de l’indice macropinocytaire
L’indice macropinocytaire est l’étendue de la macropinocytose cellulaire qui est déterminée en quantifiant l’absorption de dextran fluorescent par cellule à l’aide d’une imagerie microscopique19. À cette fin, les images acquises sont utilisées pour déterminer la quantité de dextran internalisé en mesurant l’intensité totale de fluorescence ou la zone positive de fluorescence et le nombre total de cellules déterminé par la coloration DAPI. Cette analyse peut être effectuée avec des logiciels de traitement et d’analyse d’images open source, tels que Cell Profiler ou FIJI/ImageJ, comme décrit précédemment19,20. Cependant, lorsque vous travaillez avec un lecteur de plaques multimode, le logiciel fourni avec l’instrument peut inclure des applications d’analyse intégrées qui peuvent être utilisées pour calculer l’indice macropinocytaire. Dans certains cas, le pipeline d’analyse logicielle intégré peut ne pas être complètement apparent pour l’utilisateur. Il est donc recommandé de valider le logiciel à un stade précoce par rapport à une procédure non automatisée, telle que Cell Profiler ou FIJI/ImageJ. Ce protocole peut être adapté à d’autres outils logiciels de traitement et d’analyse d’images en respectant les instructions générales suivantes :
- Pour le DAPI et l’image dextran correspondante, soustrayez l’arrière-plan en appliquant la fonction appropriée, souvent appelée fonction de boule roulante. Ajustez les paramètres de sorte que le bruit de fond soit minimisé et qu’il y ait un effet de soustraction minimal ou nul sur le signal DAPI et dextran.
- À l’aide d’un champ avec un signal de dextran élevé, déterminez les paramètres du signal d’intensité, souvent appelé fonction de seuil, pour sélectionner les noyaux et déterminez le réglage du signal d’intensité minimale requis pour sélectionner uniquement les macropoinosomes.
- Pour l’image de dextran, calculez la fluorescence totale dans la sélection de macropoinosomes créée ou utilisez la sélection pour déterminer la surface totale positive pour le dextran.
- Pour l’image DAPI, utilisez la sélection pour déterminer le nombre de noyaux dans l’image afin de refléter le nombre de cellules présentes.
- Pour déterminer l’indice macropinocytaire, divisez la fluorescence ou la surface totale du dextran par le nombre de cellules déterminées par DAPI.
- Répétez ces étapes d’analyse pour toutes les images acquises en appliquant les mêmes paramètres numériques tout au long.
- Suivez ces instructions pour déterminer l’indice macropinocytaire lors de l’utilisation du logiciel Gen5 :
REMARQUE: Le pipeline d’analyse intégré a été validé et n’a détecté aucune différence de calcul par rapport à Fiji/ImageJ (Graphique 6A).- Une fois l’imagerie terminée, sélectionnez une image avec un niveau élevé de macropinocytose. Supprimez le signal d’arrière-plan, cliquez sur Processus (Figure supplémentaire 1A) et sélectionnez l’option Prétraitement de l’image .
- Pour le canal dextran, désélectionnez Auto et utilisez un diamètre de bille roulante de 5 μm, hiérarchisez les résultats fins et lissez l’image avec 1 cycle.
- Pour le canal DAPI, utilisez le prétraitement automatique et 1 cycle lisse. Cliquez sur OK et ajoutez l’étape de prétraitement de l’image au protocole; cliquez sur AJOUTER UNE ÉTAPE. Ensuite, sélectionnez l’image traitée sous le déploiement de l’image (Figure supplémentaire 1B) et cliquez sur le bouton Analyser (Figure supplémentaire 1C).
- Sous PARAMÈTRES D’ANALYSE, définissez le Type sur Analyse cellulaire. Sélectionnez le canal DAPI et cliquez sur Options (Figure supplémentaire 2A).
- Pour le masque principal, à l’aide de l’image DAPI traitée, créez un masque pour sélectionner des noyaux uniques. Utilisez l’arrière-plan sombre et l’option Auto . De plus, déterminez quels paramètres permettent la sélection du masque de noyaux simples et, une fois terminé, cliquez sur le bouton Appliquer pour déterminer si le masque est appliqué de manière appropriée.
REMARQUE : L’activation des options Fractionner les objets en contact et Remplir les trous dans les masques peut mieux fonctionner pour sélectionner des noyaux simples. Les tailles minimale et maximale des objets peuvent devoir être ajustées en fonction de la lignée cellulaire et sont le plus souvent définies dans la plage de 5 à 40 μm. Les objets Edge principaux peuvent être inclus et l’image entière doit être analysée. Le curseur peut être appliqué pour ajuster la sélection du masque à l’intensité du signal. - Ensuite, appliquez un masque secondaire pour optimiser les paramètres de sélection du puncta fluorescent macropinosome. Utilisez la fonction Mesurer dans un masque secondaire et développez le masque primaire de 40 μm en fonction de la taille des cellules.
- Utilisez la fonction Seuil et la méthode Seuil dans le masque pour sélectionner les zones dextran positives. Cliquez sur Appliquer pour déterminer si les paramètres sont appliqués correctement.
REMARQUE : Pour déterminer la valeur seuil, utilisez l’outil Afficher le profil de ligne (figure supplémentaire 2B) et tracez une ligne sur une zone dextran positif (figure supplémentaire 2C). Utilisez l’intensité mesurée pour déterminer le meilleur réglage pour créer un masque qui sélectionne les macropoinosomes et exclut le signal d’arrière-plan (Figure supplémentaire 2D). - Après avoir créé les masques appropriés pour sélectionner les noyaux et les macropoinosomes, cliquez sur l’onglet Mesures calculées et sélectionnez Sélectionner ou Créer des mesures d’intérêt au niveau de l’objet.
- Supprimez toutes les mesures présentes et ajoutez les mesures Intégrale et Zone pour l’analyse du masque secondaire. Cliquez sur OK et sélectionnez Calculer et afficher pour les nouvelles mesures. Lorsque vous avez terminé, cliquez sur OK et sélectionnez ADD STEP pour ajouter l’analyse et les calculs au protocole.
- Enregistrez le protocole finalisé pour une utilisation ultérieure, cliquez sur Fichier et Enregistrer le protocole sous.
- Une fois l’analyse des données terminée, sélectionnez les mesures d’intérêt et exportez les données pour déterminer l’indice macropinocytaire. Déterminez l’indice macropinocytaire comme suit :
Fluorescence de dextran par cellule = Objet Int_2[Dextran fluorophore]
Zone de dextran par cellule = Objet Area_2[Dextran fluorophore]
REMARQUE: Pour la plaque de 24 puits avec format de couvercles, les mesures reflètent la moyenne de l’indice macropinocytaire moyen par image. Alternativement, l’indice macropinocytaire peut être calculé manuellement pour l’ensemble de l’échantillon en divisant la somme de la « zone » ou de l'«intégrale » pour toutes les images par le « nombre total de cellules ». La différence entre ces approches dans le calcul de l’indice macropinocytaire est minime dans la plupart des contextes. Pour le format de microplaque à 96 puits, l’indice macropinocytaire est calculé comme la moyenne de l’ensemble de l’échantillon. - Enregistrez le protocole pour l’imagerie et l’analyse automatisée ultérieure. Réutiliser le protocole pour de futures expériences avec les mêmes fluorophores.
REMARQUE: Lors de l’utilisation de la fonction de mise au point automatique laser, un nouveau balayage de référence doit être effectué lorsqu’une lignée cellulaire différente doit être analysée, car les noyaux et les macropinosomes sont éventuellement localisés sur un plan différent. Chaque fois qu’une nouvelle expérience est réalisée à l’aide d’un protocole préalablement déterminé, les paramètres d’exposition de cette expérience doivent être optimisés.
6. Ajout de traitements
Les traitements cellulaires (petites molécules, produits biologiques, facteurs de croissance, métabolites, etc.) peuvent être incorporés à n’importe quelle étape du protocole, et le moment précis dépendra des objectifs et des objectifs de l’étude.
- Préparez les cellules comme dans la section 2.
- Juste avant d’ajouter les traitements d’intérêt, préparez les traitements et les contrôles appropriés à deux fois leurs concentrations finales dans des milieux sans sérum. Préparer les traitements dans un volume égal au volume du nombre de puits répliqués évalués.
NOTE: Compte tenu du rôle que les facteurs sécrétés peuvent jouer dans le contrôle des fonctions cellulaires, il peut être préférable de diluer les traitements d’intérêt dans les milieux conditionnés. À ces fins, il peut être utile d’ensemencer des plaques supplémentaires, telles que des boîtes de culture cellulaire de 6 cm ou 10 cm, lors de la préparation des cellules décrites à la rubrique 2 afin de générer le milieu conditionné pour la préparation des solutions de traitement. - Sans retirer le milieu du puits, ajoutez un volume de solution de traitement à chaque puits. Agiter la plaque pour assurer un bon mélange. Incuber les cellules pendant la durée souhaitée.
- Passez à la section 3.
REMARQUE: Lors de l’ajout du dextran, l’utilisation de milieux frais provoque l’élimination des traitements ajoutés, ce qui peut affecter le niveau de macropinocytose. Par conséquent, il peut être préférable d’ajouter du dextran directement aux puits sans aspirer ou bien rajouter les traitements ou réutiliser les milieux conditionnés pour préparer la solution de dextran.

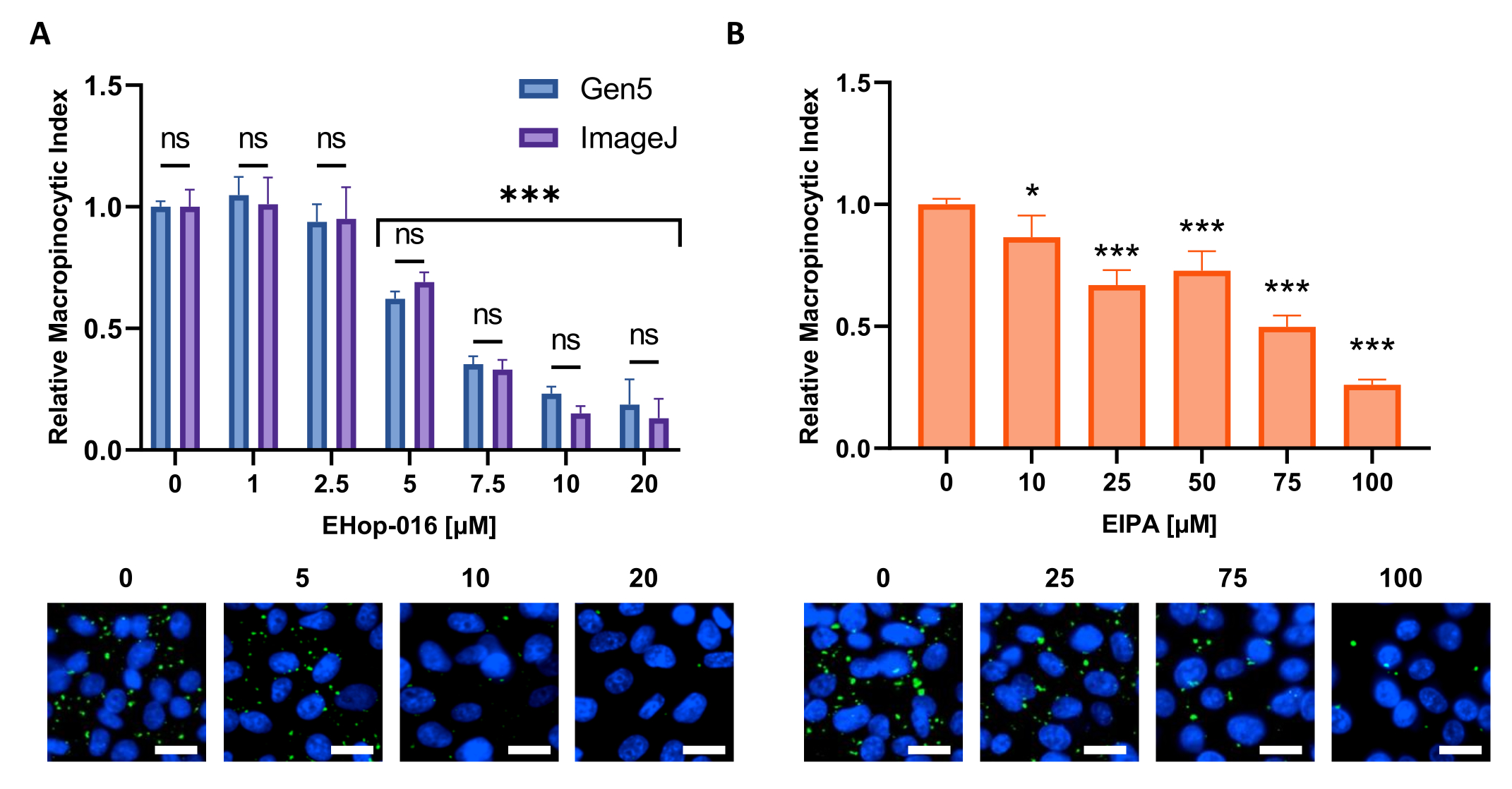
Figure 6 : Réalisation d’une courbe dose-réponse pour les inhibiteurs de la macropinocytose. Exemples de données obtenues lors de tests d’inhibiteurs connus de la macropinocytose dans une nouvelle lignée cellulaire. Les cellules PATU8998T ont été utilisées pour le format de microplaques à 96 puits et traitées pendant 2 h et 30 min avec les concentrations indiquées de (A) EHop-16 et (B) EIPA, respectivement. La comparaison des résultats obtenus par l’analyse d’images par le logiciel Gen5 ou ImageJ ne montre aucune différence significative entre les deux approches comme indiqué par ns dans (A). Barre d’échelle = 25 μm. Les graphiques à barres montrent la moyenne et le SD d’une seule expérience avec 4 réplications. La signification statistique a été déterminée par une ANOVA unidirectionnelle ou bidirectionnelle, par rapport aux conditions non traitées. * p < 0,05; p < 0,001. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Lorsque les étapes et l’ajustement du protocole décrit ci-dessus sont suivis en conséquence, les résultats expérimentaux finaux devraient fournir des informations sur la question de savoir si les conditions de culture cellulaire étudiées ou les inhibiteurs induisent ou réduisent la macropinocytose dans la lignée cellulaire d’intérêt. Pour renforcer la validité de ces résultats, l’inclusion de conditions de contrôle permettra d’examiner les résultats pour déterminer si l’expérience a été mené...
Discussion
La qualité des expériences et de l’acquisition des données dépend fortement de la qualité des réactifs, de l’optimisation des réglages et de la propreté des couvercles et des microplaques. Les résultats finaux devraient donner une variation minimale entre les répétitions; cependant, des variations biologiques se produisent naturellement ou peuvent être causées par un certain nombre de facteurs. La densité cellulaire peut amener les cellules à répondre plus ou moins aux inducteurs ou inhibiteurs de la ...
Déclarations de divulgation
C.C. est l’inventeur d’un brevet délivré intitulé « Cancer diagnostics, therapeutics, and drug discovery associated with macropinocytosis », brevet no 9 983 194.
Remerciements
Ce travail a été soutenu par des subventions NIH / NCI (R01CA207189, R21CA243701) à C.C. KMO.G. est récipiendaire d’une bourse postdoctorale TRDRP (T30FT0952). Le BioTek Cytation 5 fait partie du Sanford Burnham Prebys Cell Imaging Core, qui reçoit un soutien financier du NCI Cancer Center Support Grant (P30 CA030199). Les figures 1 à 3 ont été créées à l’aide de BioRender.
matériels
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin | Corning | 25053CI | 0.1% EDTA in HBSS w/o Calcium, Magnesium and Sodium Bicarbonate |
| 1.5 mL Microcentrifuge tube | Fisherbrand | 05-408-129 | |
| 10-cm Tissue culture dish | Greiner Bio-One | 664160 | CELLSTAR |
| 15 mL Centrifuge tube | Fisherbrand | 07-200-886 | |
| 2 L Beaker | Fisherbrand | 02-591-33 | |
| 24-well Tissue culture plate | Greiner Bio-One | 662160 | CELLSTAR |
| 25 mL Reagent reservoir | Genesee Scientific Corporation | 28-121 | |
| 500 mL Beaker | Fisherbrand | 02-591-30 | |
| 6-cm Tissue culture dish | Greiner Bio-One | 628160 | CELLSTAR |
| 8-Channel aspiration adapter | Integra Biosciences | 155503 | |
| 8-Channel aspiration adapter for standard tips | Integra Biosciences | 159024 | |
| 95% Ethanol | Decon Laboratories Inc | 4355226 | |
| Ammonia-free glass cleaner | Sparkle | FUN20500CT | |
| Black 96-well high-content screening microplate | PerkinElmer | 6055300 | CellCarrier-96 Ultra |
| Cotton-tipped applicator | Fisherbrand | 23-400-101 | |
| Coverslips | Fisherbrand | 12-545-80 | 12 mm diameter |
| Cytation 5 Cell Imaging Multi-Mode Reader | Biotek | CYT5FW | |
| DAPI | Millipore Sigma | 5.08741 | |
| Dextran 70 kDa - FITC | Life Technologies | D1822 | Lysine-fixable |
| Dextran 70 kDa - TMR | Life Technologies | D1819 | |
| DMSO | Millipore Sigma | D1435 | |
| DPBS | Corning | 21031CV | Without Calcium and Magnesium |
| Forceps | Fine Science Tools | 11251-20 | Dumont #5 |
| Formaldehyde, 37% | Ricca Chemical | RSOF0010-250A | ACS Reagent Grade |
| Glycerol | Fisher BioReagents | BP229-1 | |
| Hardening fluorescence mounting media | Agilent Tech | S302380-2 | DAKO |
| Hoechst 33342 | Millipore Sigma | B2261 | |
| Hydrochloric acid (HCl) | Fisher Chemical | A144-212 | Certified ACS Plus, 36.5%–38.0% |
| Lint-free wipes | Kimberly-Clark | 34155 | Kimwipes |
| Miscroscope slides | Fisherbrand | 12-544-1 | Premium plain glass |
| Multichannel pipette | Gilson | FA10013 | 8 channels, 0.5–10 µL |
| Multichannel pipette | Gilson | FA10012 | 12 channels, 20–200 µL |
| Multichannel pipette | Gilson | FA10011 | 8 channels, 20–200 µL |
| Parafilm M | Pechiney | PM996 | |
| Plastic wrap | Kirkland Signature | 208733 | Stretch-Tite |
| Silicone isolators | Grace Bio Labs Inc | 664107 | 13 mm Diameter X 0.8 mm Depth ID, 25 mm X 25 mm |
| Slide adapter | Biotek | 1220548 | |
| Wash bottle | Fisherbrand | FB0340922C |
Références
- Lin, X. P., Mintern, J. D., Gleeson, P. A. Macropinocytosis in different cell types: similarities and differences. Membranes. 10 (8), 21 (2020).
- Recouvreux, M. V., Commisso, C. Macropinocytosis: a metabolic adaptation to nutrient stress in cancer. Frontiers in Endocrinology. 8, (2017).
- Zhang, Y. J., Commisso, C. Macropinocytosis in cancer: a complex signaling network. Trends in Cancer. 5 (6), 332-334 (2019).
- Commisso, C., et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 497 (7451), 633-637 (2013).
- Kim, S. M., et al. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discovery. 8 (7), 866-883 (2018).
- Jayashankar, V., Edinger, A. L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nature Communications. 11 (1), (2020).
- Kamphorst, J. J., et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Research. 75 (3), 544-553 (2015).
- Olivares, O., et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nature Communications. 8, (2017).
- Seguin, L., et al. Galectin-3, a druggable vulnerability for KRAS-addicted cancers. Cancer Discovery. 7 (12), 1464-1479 (2017).
- Redelman-Sidi, G., et al. The canonical Wnt pathway drives macropinocytosis in cancer. Cancer Research. 78 (16), 4658-4670 (2018).
- Tejeda-Munoz, N., Albrecht, L. V., Bui, M. H., De Robertis, E. M. Wnt canonical pathway activates macropinocytosis and lysosomal degradation of extracellular proteins. Proceedings of the National Academy of Sciences of the United States of America. 116 (21), 10402-10411 (2019).
- Schmees, C., et al. Macropinocytosis of the PDGF beta-receptor promotes fibroblast transformation by H-RasG12V. Molecular Biology of the Cell. 23 (13), 2571-2582 (2012).
- Lee, S. -. W., et al. EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Developmental Cell. 50 (3), 381-392 (2019).
- Recouvreux, M. V., et al. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. Journal of Experimental Medicine. 217 (9), (2020).
- Zhang, Y., et al. Macropinocytosis in cancer-associated fibroblasts is dependent on CaMKK2/ARHGEF2 signaling and functions to support tumor and stromal cell fitness. Cancer Discovery. 11 (7), 1808-1825 (2021).
- Su, H., et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell. 39 (5), 678-693 (2021).
- Bar-Sagi, D., Feramisco, J. R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 233 (4768), 1061-1068 (1986).
- Mishra, R., et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. Journal of Clinical Investigation. 128 (10), 4472-4484 (2018).
- Commisso, C., Flinn, R. J., Bar-Sagi, D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nature Protocols. 9 (1), 182-192 (2014).
- Galenkamp, K. M. O., Alas, B., Commisso, C. Quantitation of macropinocytosis in cancer cells. Methods in Molecular Biology. 1928, 113-123 (2019).
- Wang, J. T. H., Teasdale, R. D., Liebl, D. Macropinosome quantitation assay. MethodsX. 1, 36-41 (2014).
- Lee, S. -. W., Alas, B., Commisso, C. Detection and quantification of macropinosomes in pancreatic tumors. Methods in Molecular Biology. 1882, 171-181 (2019).
- Williams, T., Kay, R. R. High-throughput measurement of dictyostelium discoideum macropinocytosis by flow cytometry. Journal of Visualized Experiments: JoVE. (139), e58434 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.