
Method Article
Spatiotemporal Analysis of Microglial Ca2+ Activity at Single-Cell Resolution
In This Article
Summary
In this paper, we describe a protocol for in vivo imaging of microglial Ca2+ activity and subsequent analysis of its spatiotemporal dynamics. This method enables thorough characterization of how microglia respond to changes in the brain environment, appropriately capturing the fine spatiotemporal scales at which such events occur.
Abstract
Microglia are the sole resident immune cells in the central nervous system. Their morphology is highly plastic, changing depending on their activity. Under homeostatic conditions, microglia possess a highly ramified morphology. This facilitates their monitoring of the surrounding environment through the continuous extending and retracting of their processes. During brain injury and inflammation, however, microglia become activated and undergo dramatic morphological changes, retracting their ramified processes and swelling their cell body. This facilitates activities such as migration and phagocytosis, which microglia undertake to navigate the brain environment to a less pathological state.
This close relationship between microglial morphology and changes in their activity have enabled considerable insights into various microglial functions. However, such morphological and activity changes are themselves phenomena that can result from any number of intracellular signaling pathways. Moreover, the time-lag between stimulus and response, as well as the highly compartmentalized morphology of microglia, make it difficult to isolate the causative mechanisms that underpin function. To solve this problem, we developed a genetically modified mouse line in which a highly sensitive fluorescent Ca2+-indicator protein is specifically expressed in microglia.
After describing methods for in vivo microglial Ca2+ imaging, this paper presents a structured analysis approach that classifies this Ca2+ activity to rationally defined subcellular regions, thus ensuring that the spatial and temporal dimensions of the encoded information are meaningfully extracted. We believe that this approach will provide a detailed understanding of the intracellular signaling rules that govern the diverse array of microglial activities associated with both higher brain functions and pathological conditions.
Introduction
Microglia are the resident immune cells in the central nervous system (CNS) and play important roles in maintaining a homeostatic brain environment and in regulating neural circuit formation during brain development1,2. A unique feature of microglia in the CNS is that their morphology is highly plastic; however, distinct morphological phenotypes can be associated with particular functions. Furthermore, the transformation between morphological phenotypes is highly dynamic, occurring on rapid time scales in response to changes in the surrounding environment3,4.
Under homeostatic physiological conditions, microglia assume a highly ramified morphology, with multiple processes radiating outward in all directions. These ramified processes themselves demonstrate high motility, continuously extending and retracting3,4. Such activity is primarily directed toward periodic contact with neuronal synapses, axons, and somas to monitor neuronal activity5,6,7,8,9. However, when the brain is injured, microglia quickly detect this abnormality, and as a first step in their adaptive response, direct the extension of their processes toward the corresponding locale3,4. Where microglia are required to undertake phagocytosis of dead cells and metabolites, they assume an amoeboid-like morphology, shortening their processes and enlarging their cell bodies, as part of their transition into the immunologically activated phenotype10,11.
However, whilst the dramatic morphological changes of microglial processes are easily detected, finer scale changes of the cell soma are significantly more difficult to capture, especially at a temporal resolution that is physiologically relevant. Furthermore, morphological changes themselves only represent the integrated result of any number of intracellular signaling pathways. This is problematic for a goal of tracking functional activity and mechanistically linking a stimulus with the end response it provokes.
Given its widespread role as a second messenger, examining intracellular Ca2+ dynamics better captures the associated spatiotemporal information when studying dynamic cell processes. Such an approach is applicable to microglia given that they express a variety of ionotropic and metabotropic receptors linked to downstream intracellular Ca2+ elevation. Indeed, in vivo Ca2+ imaging has been used to characterize spatiotemporal aspects of microglial activities in real time, successfully correlating changes in microglial Ca2+ activity with brain injury, inflammation, and both hyper- and hypoactivity in neurons12,13,14,15,16. For example, Ca2+ elevations associated with microglial process extension in response to hyper/hypoactive neuronal activity likely reflect the underlying Ca2+-dependent actin polymerization process16. Furthermore, in vivo Ca2+ imaging can also be readily combined with pharmacological approaches. For example, whilst microglia express both P2X (ionotropic) and P2Y (metabotropic) receptors, local application of P2Y agonists mimics and subsequently desensitizes the microglial Ca2+ response to damaged neighboring neurons13, thus implying the greater relevance of P2Y signaling to neuronal damage detection.
To date, previous reports examining microglial Ca2+ activity have employed region of interest (ROI)-based analysis methods. A drawback of these approaches is that they are still too coarse to be able to resolve the spatiotemporal dynamics of Ca2+ activity at the level of individual microglial processes. Thus, this protocol describes both conventional ROI-based methods for analyzing microglial Ca2+ activity and newer event-based approaches, which can extract individual Ca2+ events in microglial processes. Before this, we provide a general guide for in vivo two-photon imaging to appropriately capture microglial Ca2+ activity for detailed analysis.
Protocol
All animal experiments were approved by the National Institute for Physiological Sciences Animal Research Committees and were in accordance with the National Institutes of Health guidelines. For all experiments, 8-10-week-old male mice were raised under a 12/12 h light/dark cycle with ad libitum access to food and water. To visualize Ca2+ activity in microglia, ionized Ca2+ binding adapter molecule 1 (Iba1)-tetracycline transactivator (Iba1-tTA) mice were crossed with tetracycline operator-GCaMP6 (tetO-GCaMP6) mice17,18. Thus, in the absence of tetracycline-analog supplementation, the Iba1 promoter drives the expression of GCaMP6, exclusively in microglia. For all experiments, doxycycline dietary supplementation was stopped at 6 weeks after birth. At the end of all experiments, mice were euthanized by isoflurane overdose followed by cervical dislocation. See the Table of Materials for details related to all materials, animals, and reagents used in this protocol.
1. Surgical preparation of mice for in vivo two-photon imaging; day 1
- Perform all surgical procedures inside a laminar air flow cabinet to maintain sterile working conditions. Before starting surgery, sterilize the cabinet interior with UV light for 5 min.
- Sterilize all work surfaces, the surgery frame, and stereotaxic instruments by wiping them down with 70% ethanol.
- Sterilize all surgical instruments (scissors, forceps, razor blade, tweezers) and the custom-made head-plate to be attached to the mouse's skull by immersing them in 1% chlorhexidine gluconate solution.
- Anesthetize the mouse with ketamine (7.4 mg kg−1, intraperitoneally [i.p.]) and xylazine (10 mg kg−1, i.p.). Return it to its home cage until the anesthesia takes hold. Confirm the full induction of anesthesia by loss of the toe-pinch reflex.
- Sterilize the scalp with 1% chlorhexidine gluconate. Shave away the fur with a razor blade.
- Secure the mouse within the surgery frame via stereotaxic instruments.
- Apply vet ointment to the eyes to prevent dryness whilst under anesthesia.
- Apply 2% xylocaine jelly to the scalp for pain management. Wait for 5 min.
- Remove the scalp with scissors and expose the skull. Clean away the periosteum and dry the exposed skull surface by rubbing with cotton swabs.
NOTE: The exposed skull areas must be completely dry to ensure strong bonding with the custom-made head-plate. - Secure the custom-made head-plate to the skull with dental cement.
- Once the cement has set, fill in any gaps between the skull surface and the borders of the custom-made head-plate with additional dental cement.
- Waterproof the cement and skull surfaces by applying acrylic-based dental adhesive resin cement.
- Return the mouse to its home cage, placing it on a warming pad. Monitor the mouse until it regains sufficient consciousness to maintain sternal recumbency (within 2 h).
NOTE: Mice should be fully recovered by the next day and can then be housed with other animals.
2. Surgical preparation of mice for in vivo two-photon imaging; day 2
- Perform all surgical procedures inside a laminar air flow cabinet to maintain sterile working conditions. Before starting surgery, sterilize the cabinet interior with UV light for 5 min.
- Laminate together two glass coverslips of different dimensions (top glass: 3 mm × 3 mm; bottom glass: 2 mm × 2 mm) with UV-curable optical grade resin.
NOTE: The double coverslip provides long-term protection to the brain region exposed by the cranial window whilst enabling chronic optical access. Thus, its dimensions may be altered to suit the brain region to be imaged. - Sterilize all work surfaces and the surgery frame by wiping them down with 70% ethanol.
- Sterilize all surgical instruments (steel drill, tweezers, surgical needle hook) by immersing them in 1% chlorhexidine gluconate solution.
- Anesthetize the mouse with isoflurane (4% induction, 1.2%-1.5% maintenance). Secure the mouse within the surgery frame via its head-plate.
- To make a cranial window over the primary motor cortex, mark a square with dimensions of 2 mm × 2 mm centered 0.2 mm anterior to, and 1 mm lateral from, the Bregma skull landmark.
- Thin the skull along the border of the marked square using the steel drill.
NOTE: When close to the desired thickness, the thinned skulled areas will appear transparent when wetted with saline, and hairline cracks will start to appear. - After confirming that the entire border of the marked square has been appropriately thinned, carefully insert the surgical needle hook just below the skull surface, orienting its tip toward the center of the square. Gently lift the square skull piece with the hook and use tweezers to peel it away from the rest of the skull. If bleeding occurs, continuously wash the exposed brain surface with saline until it subsides completely.
- Place the double coverslip onto the exposed brain surface, orienting the side with the smaller coverslip toward the brain. Ensure the edges of the larger coverslip are in contact with the borders of the cranial window.
- Using a silicon-tipped glass rod mounted in a manipulator, gently press down on the double coverslip to ensure that it makes good contact with the brain surface.
- Fill the gap between the double coverslip, skull, and brain surface with UV-curable resin and irradiate with UV light until hardened (~20 s). Slowly raise the silicon-tipped glass rod away from the double coverslip.
- Allow the mouse to recover, placing it on a warming pad. Monitor the mouse until it regains sufficient consciousness to maintain sternal recumbency (30 min).
- Proceed to step 3 or return the mouse to its home cage.
NOTE: After proficient surgery, the dura and all underlying blood vessels will be completely intact with no bleeding. In this case, inflammation is minimal and it is possible to immediately proceed to step 3. If unsure, wait 1-3 weeks to ensure any inflammation has fully resolved.
3. Data collection using in vivo two-photon imaging
- Turn on the imaging setup in advance to ensure that the laser has sufficient time to warm up and stabilize. Tune the laser to emit at a wavelength of 920 nm, which is the two-photon spectrum that optimally excites the GCaMP6 fluorophore.
- Position the mouse under a 25x microscope objective lens and habituate it for 30 min. If necessary, anesthetize the mouse with isoflurane during imaging (4% induction, 1.2%-1.5% maintenance).
NOTE: Isoflurane may not be appropriate for some research applications, as it affects microglial process motility and Ca2+ activity19,20,21. - Set the zoom to 1x, then search for microglia expressing GCaMP6 in the cranial window area. Search at a depth between 100 µm and 300 µm below the brain surface.
- Once an appropriate GCaMP6-expressing microglia has been found, maximize the zoom so that Ca2+ activity can be captured with single cell resolution.
- Visually confirm that Ca2+ activity is clearly captured and adjust the laser power and imaging gain if necessary.
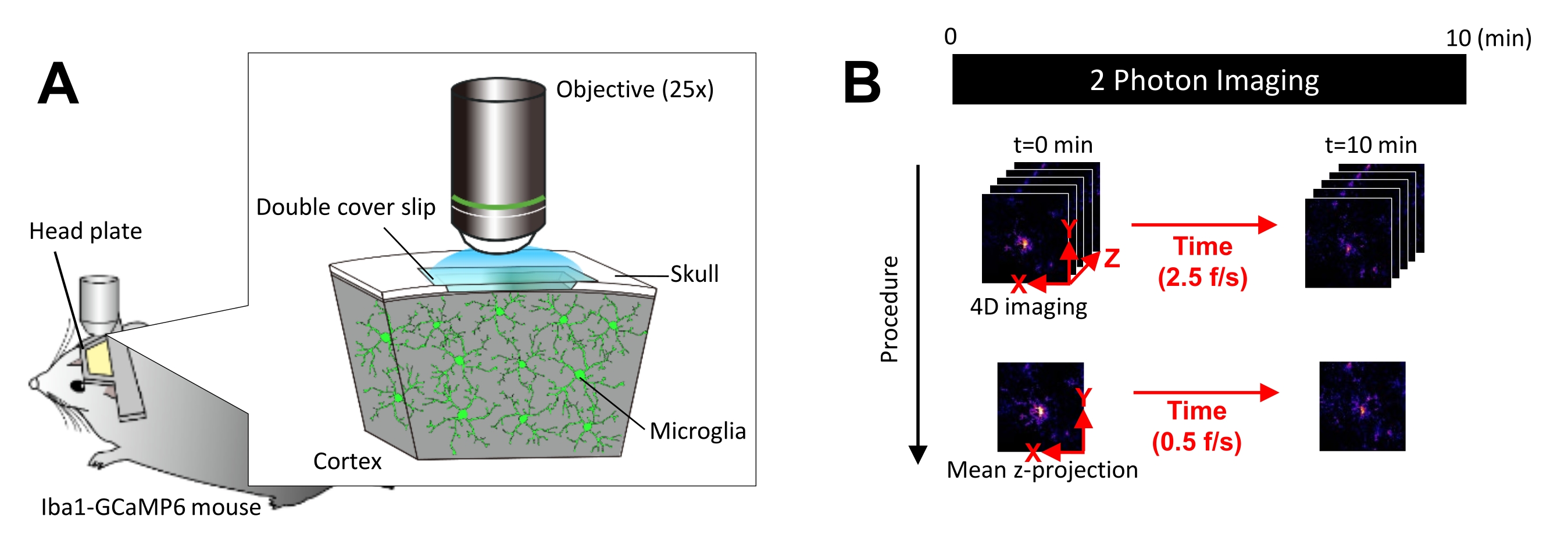
NOTE: Laser power should be minimized as much as possible to avoid photobleaching and injury to microglia. - Perform four-dimensional (4D) image acquisition as follows (steps 3.6.1-3.6.3; Figure 1B):
- Set the image dimensions to XY area = 512 × 512 pixels, 0.25 µm/pixel; Z area = five z-planes, 3 µm z-step (Figure 1B).
- Set the acquisition rate to 2.5 frames/s.
NOTE: Z-scanning at this speed is facilitated by a piezo nano-positioning system. - Acquire data until at least 10 individual Ca2+ events are observed (usually 10 min).
4. Preparation for analysis (motion correction, mean/max z-projection)
- Before proceeding to analyze microglial Ca2+ activity, correct the 4D images for motion-related artifacts (image registration) using the ECC image alignment algorithm22 within the MATLAB programming environment (R2020a). Download the code from: https://www.mathworks.com/matlabcentral/fileexchange/27253-ecc-image-alignment-algorithm-image-registration.
- Create a reference 3D image by registering all images within the first time frame using the imregister MATLAB operation (standard image processing toolbox).
- Register all subsequent images, matching them with the corresponding z-plane of the reference 3D image using the ecc function (ECC image alignment algorithm library).
- Generate mean intensity z-projections from the registered images using the mean MATLAB operation. If the imaging signal is too weak, generate maximum intensity z-projections instead using the max MATLAB operation.
NOTE: The signal to noise ratio is worse when using maximum intensity z-projections. - Proceed to either step 5 or 6. Use the z-projections generated in step 4 as the target of analysis for mapping the spatiotemporal dynamics of Ca2+ activity in steps 5 and 6.
5. ROI-based analysis
- Using the z-projections generated in step 4, identify microglial processes that maintain a stable area profile (stable area) throughout the imaging period for subsequent analysis of Ca2+ activity as follows (steps 5.3-5.6):
- Generate separate maximum intensity t-projections of the registered 4D images from 2 min samples taken at the start and end of the imaging period using the max MATLAB operation.
- Binarize the t-projections to generate polygons of the microglial morphology corresponding to the start and end of the imaging period using the imbinarize MATLAB operation. Use the automatically set default threshold.
NOTE: If the Ca2+ signal is weak during the start and end of the imaging period, binarization may generate polygons with missing borders. In this case, manually draw the missing borders using the pencil tool in ImageJ. - Overlay the t-projection polygons using the imadd MATLAB operation. The overlapping region(s) represent the stable area (Figure 2B).
- Within identified stable areas, use the drawpolygon MATLAB operation to manually define and trace ROIs for each of the primary microglial processes and any obvious second-order sub-branches.
- Track the absolute fluorescence intensities averaged across an individual ROI over all time frames (Figure 2C,D).
- From the time-series of absolute fluorescence intensities, calculate the relative change in fluorescence intensity (ΔF/F) time series according to equation (1). This time series represents the normalized microglial Ca2+ dynamics at the level of individual ROIs.
ΔF/F = (F(t) - F0) / F0 (1)
Where F(t) is the recorded fluorescence intensity at a given time and F0 is the 10th percentile of fluorescence intensity in all time frames (Figure 2E). - Identify candidate Ca2+ firing events occurring in a given individual ROI as follows (steps 5.5.1-5.5.3):
- Apply type I finite impulse response (FIR) low-pass filtering on the ΔF/F time-series using the fir1 MATLAB operation. Set the cut-off frequency to the Nyquist value (half the sampling rate).
- Visually inspect the filtered trace to confirm that the individual trace peaks correspond to bursts of Ca2+ activity in the registered 4D images (Figure 2F).
- Identify candidate Ca2+ firing events as concave down inflections in theΔF/F time-series trace. Define a baseline threshold as the median ΔF/F value within upper and lower ceilings that exclude the maximum and minimum 10% amplitudes over all time frames. Define a detection threshold of three SDs above the baseline threshold (Figure 2F,H).
- Identify true Ca2+ firing events from candidates as follows (steps 5.5.1-5.5.2):
- Calculate the slope of each candidate event by differentiating across the corresponding filtered ΔF/F time series frames using the numerical gradient MATLAB operation.
- Subsequently identify true Ca2+ events based on the rise-time of the profile. Define a baseline threshold as the average of slope values across all candidate events. Define a detection threshold of three SDs above the baseline threshold (Figure 2G,H).
- Characterize true Ca2+ events as follows (steps 5.6.1-5.6.3):
NOTE: The following is not an exhaustive list of parameters that can be used to characterize microglial Ca2+ events. Parameters of interest depend on the purpose of the study.- Derive the maximum amplitude of a true Ca2+ firing event as the ΔF/F value of the corresponding ΔF/F time-series frame.
- Derive the mean amplitude of a true Ca2+ firing event as the average ΔF/F value across the entire corresponding ΔF/F time-series subset of frames.
- Derive the frequency of true Ca2+ events as the number of events divided by imaging time.
6. Event-based analysis
- Perform event-based analysis on the z-projections from step 4 using the AQuA library within the MATLAB programming environment. Download the code from: https://github.com/yu-lab-vt/AQuA.
NOTE: A general use walkthrough for the AQuA library is available from: https://drive.google.com/file/d/1a3lhe0dUth-5J1-S2fZlPOCZlPbeuvUr/view. Detailed documentation for the AQuA library is available from: https://drive.google.com/file/d/1CckDLbrkw16b7MPlOQdYpZciIz80Snm_/view. - After launching MATLAB, switch from the default working directory folder to AQuA's designated working directory folder using the cd operation.
- Load the registered 4D images into the AQuA analysis pipeline as follows (steps 6.3.1-6.3.2):
- Launch the AQuA GUI. Type aqua_gui in the MATLAB command window in AQuA's designated working directory folder.
- Within the AQuA GUI, click new project and select the registered images to be analyzed. Specify the data type (GCaMPInVivo_cyto_Lck_) and imaging parameters (temporal resolution seconds per frame = 1.993; spatial resolution µm per pixel = 0.25; exclude pixels shorter than this distance to border = 5). Click open to load the data.
- Define landmarks for the subsequent analysis pipelines by tracing out areas of interest. Usually, landmarks are based on the cell boundary and area of the cell body.
- Subsequently, detect candidate Ca2+ events by running the automated analysis pipelines for the following parameters: active signal, super voxel, event detection, clean events, and merge events.
NOTE: A detailed explanation about each of these parameters and their candidate score outputs is provided in the AQuA walkthrough. Briefly, these parameters adjust the Ca2+ event detection threshold as follows: active signal = fluorescence amplitude, super voxel = clustering of fluoresce in 3D space, event detection = rise/decay kinetics, clean events = signal to noise ratio, and merge events = temporal separation of events. - Visually inspect the detected Ca2+ events (automatically overlayed onto the original registered images). If necessary, adjust the parameter settings of the analysis pipelines by considering the image quality in terms of the above parameters; if the image quality for a given parameter is good, a higher threshold may be set, and vice versa.
- Once all the Ca2+ events have been detected appropriately, characterize these events within the main MATLAB environment as follows (steps 6.8-6.14):
- Export the AQuA analysis output files.
NOTE: A detailed explanation about the output files, the extracted features that are used to define Ca2+ events, and the underlying parameters of these extracted features is provided in the AQuA documentation. - (optional) Categorize all the Ca2+ events into two groups: 1) events starting at the soma, and 2) events starting at the processes.
- Access the amplitude of individual Ca2+ events within the res.dffMat MATLAB structure of the AQuA analysis output file (.mat file).
- Derive the frequency of individual Ca2+ events using the res.fts.loc.x3D MATLAB operation (AQuA library).
- Derive the duration of individual Ca2+ events using the res.fts.curve.width11 MATLAB operation (AQuA library).
- Derive the area of individual Ca2+ events using the res.fts.basic.area MATLAB operation (AQuA library).
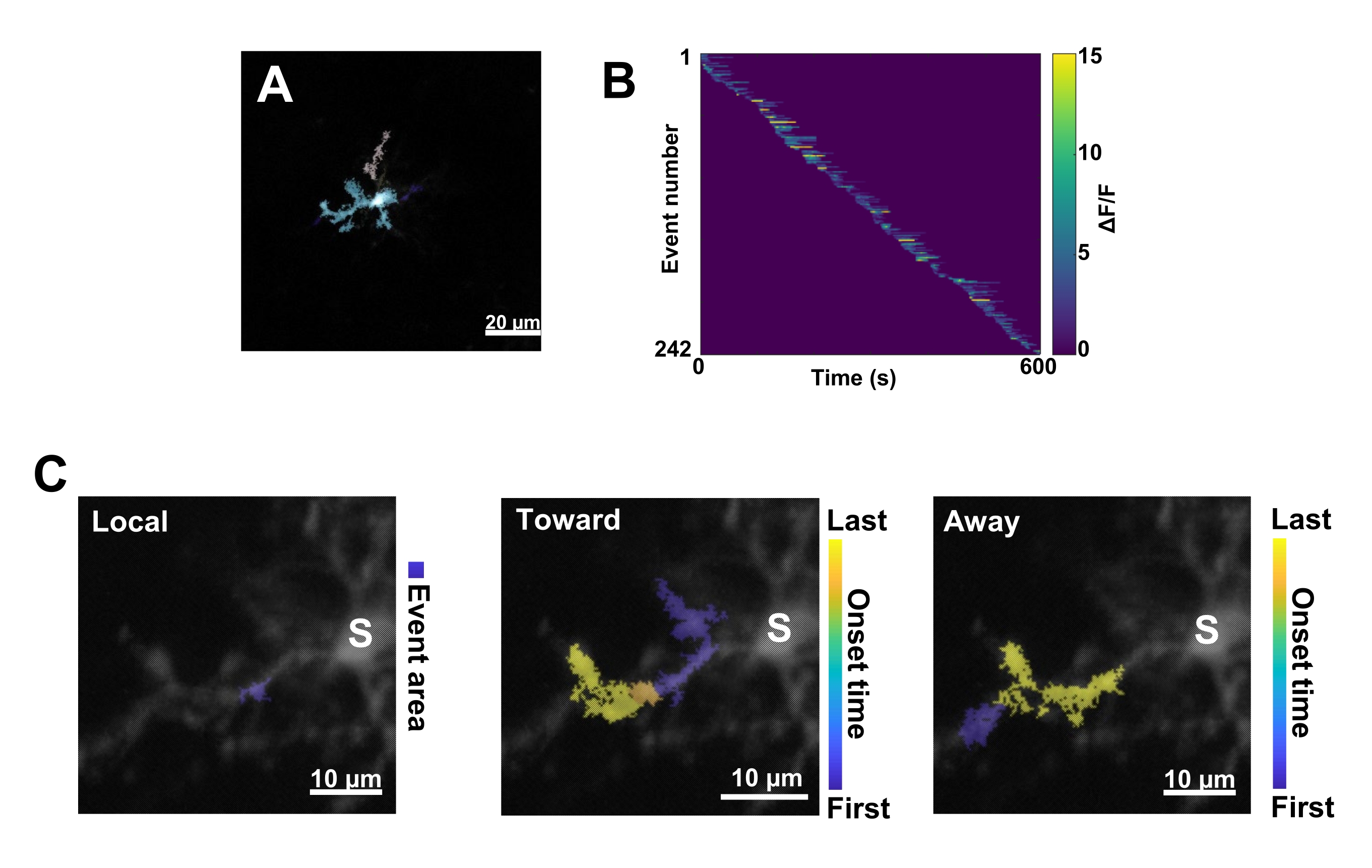
- Categorize all the Ca2+ events as: 1) local events, 2) propagative events traveling toward the soma, and 3) propagative events traveling away from the soma. To do this, use the res.fts.region.landmarkDir.chgToward and res.fts.region.landmarkDir.chgAway MATLAB operations (AQuA library) (Figure 3C).
Results
In transgenic mice exclusively expressing GCaMP6 (Ca2+-sensitive fluorescent protein) in microglia, we typically observe diverse patterns of microglial Ca2+ activity (Figure 2A). Importantly, even within a single microglia, the patterns of Ca2+ activity can differ dramatically between processes.
To quantify such process-to-process differences in the spatiotemporal dynamics of microglial Ca2+ activity, stable areas must first be identified and then divided into finely segmented ROIs (Figure 2B,C). For each ROI, parameters of Ca2+ activity must be derived and quantified, such as amplitude and frequency, by extracting features such as local amplitudes and trace slopes from the fluorescence intensity time series (Figure 2D-G).
Next, individual Ca2+ events must be examined by applying the accurate quantification algorithm AQuA (Figure 3A). From such event-based analyses, vast differences are typically observed in the origin, amplitude, duration, location, and flow direction characteristics of individual Ca2+ events (Figure 3B). If focusing on analyzing Ca2+ activity dynamics in microglial processes, a classification scheme of local events, events traveling toward the soma, and events traveling away from the soma is informative (Figure 3C).

Figure 1: Experimental setup for in vivo microglial Ca2+ imaging. (A) Experimental setup. An Iba1-tTA × tetO-GCaMP6 mouse with microglia-specific GCaMP6 expression. By inserting a cranial window in the mouse's skull, microglial Ca2+ activity can be observed in vivo using two-photon microscopy. (B) Experimental schedule and analysis procedure. 4D images are acquired for at least 10 min as five-frame z-stacks. The frame acquisition rate is 2.5 frames/s. Before analyzing microglial Ca2+ activity, the five-frame z-stacks are converted into 2D z-projections by taking the average (or maximum) intensity. The z-projection playback rate is 0.5 frames/s. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: ROI-based analysis for microglial Ca2+ activity. (A) Mean GCaMP6 intensity projection over 10 min for an individual microglia. (B) Stable areas (white) are defined by an overlay of binarized, maximum GCaMP6 intensity t-projections derived from 2 min samples taken at the start (magenta) and end (green) of an imaging period. (C) Stable areas are further segmented into regional ROIs. Individual colors indicate individual ROIs. (D) ΔF/F traces of all individual ROIs in C. Note the variation in activity patterns between ROIs. (E) Original trace of a ΔF/F time series derived from absolute intensity values for a single ROI. (F) The same ΔF/F time series after low-pass filtering. Candidate Ca2+ events are detected by an amplitude cut-off threshold (red line) defined as baseline + three SDs. The baseline (green line) is defined as the median value across the entire ΔF/F time-series within an upper and lower ceiling that excludes the maximum and minimum 10% of ΔF/F values. (G) The slope trace derived from the filtered ΔF/F time series in F. True Ca2+ events are sorted from candidate Ca2+ events based on a slope cut-off threshold (red line) defined as baseline + three SDs. The baseline (green line) is defined as the average value across the entire slope time-series within an upper and lower ceiling that excludes the maximum and minimum 10% of slope values. (H) Candidate Ca2+ events identified by amplitude criteria in F are indicated in orange. True Ca2+ events sorted from candidate Ca2+ events by slope criteria in G in red. Note that some candidate Ca2+ events have been merged based on the slope criteria. The corresponding filtered ΔF/F time series is overlayed below for reference. The black line indicates zero amplitude (ΔF/F). Mean and maximum amplitude of true Ca2+ events are derived as the mean and maximum of their corresponding peaks in the filtered ΔF/F time series. Frequency (events/min) is derived as the number of true Ca2+ firing events divided by the imaging period (10 min). Scale bars = 20 µm (A,B), 10 µm (C). Abbreviation: ROI = region of interest. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Event-based analysis for microglial Ca2+ activity. (A) Representative images of events detected using the AQuA algorithm. The colors indicate individual event areas detected at one particular time point. (B) Representative normalized Ca2+ activity (ΔF/F) in individual events sorted by the order of onset. The right bar indicates color-indicated ΔF/F. (C) Representative activity-footprints of events propagating toward and away from the soma or local events. For local events, the event shape is shown in blue. For propagative events, the event onset time is indicated by the blue-yellow scale. Since AQuA initially detects Ca2+ events individually, propagative events are identified subsequently based on the overlapping spatial locations and time series of multiple individual Ca2+ events. Note that this is the same dendritic branch used for the ROI analysis in Figure 2E-H. Scale bars = 20 µm (A), 10 µm (C). Abbreviation: ROI = region of interest; S = soma. Please click here to view a larger version of this figure.
{kind=link}
Discussion
This paper introduces an improved approach for imaging microglial Ca2+ activity with a high spatiotemporal resolution. This method is sensitive enough to detect different types of microglial Ca2+ activity at the level of single ramified processes, readily distinguishing between local and propagative events.
In the general method for in vivo two-photon imaging of microglial Ca2+ activity, careful attention must be paid to the following points to maximize imaging quality. First, as microglia are extremely sensitive to injury, it is important to minimize directly touching the surface of the brain with surgical tools during surgery. Key indications that surgery has been proficiently performed are intact blood vessels and dura and very minimal bleeding during surgery. Second, secure attachment of the head-plate to the mouse's skull and good contact between the double coverslip and brain surface greatly reduce motion-related artifacts whilst imaging. This is especially important when imaging with high spatiotemporal resolutions and in fully awake mice. Whilst the analysis pipeline reliably compensates for motion-related artifacts arising from the heartbeat, respiration, and general drift, it is less robust when handling significant geometric distortions that arise from sudden large movements.
The two analysis methods described here offer different advantages and are suited to different research questions. In ROI-based analysis, the user predefines the ROI (such as individual processes), allowing for the aggregate dynamics of Ca2+ activity of this ROI to be extracted. Thus, it is most suited to situations where phenomena are expected to be localized to a subcellular area that has both well-defined morphological boundaries and a relatively large area (i.e., a process branch). In event-based analysis, individual events are defined based on the spatiotemporal dynamics of the microglial Ca2+ activity itself and must then be placed in the context of user-defined landmarks within the microglia for their function to be interpreted. Thus, it is most suited to situations where assumptions about phenomena localization cannot be made or where the area of interest is relatively small (i.e., a process tip). As such, event-based analysis offers improved spatiotemporal resolution when characterizing microglial Ca2+ activity as compared to previous methods.
In these mice, the only fluorescent marker expressed by microglia is the Ca2+ indicator GCaMP6. Thus, in regions where Ca2+ activity is low, microglial morphology must be extracted by combining multiple time frames, which can degrade temporal resolution. However, this limitation can be overcome by expressing a separate red stably-fluorescent protein in microglia. Notably, novel adeno-associated viruses capable of transfecting microglia have recently been described23,24,25.
How microglial Ca2+ activity is altered by the surrounding environment is an emerging topic of interest. In particular, microglial Ca2+ activity appears to show significant correlations with neuronal activity, though the functional significance of this has yet to be thoroughly characterized. Thus, combining neuronal activity manipulation with the imaging and analysis methods for microglial Ca2+ activity presented here should yield new insights into microglial physiology and further advance our understanding of the roles that microglia play in physiological and pathological states.
Disclosures
The authors declare no conflicts of interest associated with this manuscript.
Acknowledgements
We are grateful to Prof. Kenji Tanaka (Keio University, Tokyo, Japan) for providing Iba1-tTA mice and tetO-GCaMP6 mice. This work was supported by Grants-in-Aid for Young Scientists (B) [16K19001 (to H.H.)], Grants-in-Aid for Early-Career Scientists [18K14825 (to H.H.)], Grant-in-Aid for Scientific Research (B) [21H03027 (to H.H.)], Grant-in-Aid for Transformative Research Areas (A) [21H05639 (to H.H.)], Grant-in-Aid for Scientific Research (A) [17H01530, 20H00500 (to J.N.)], and JST CREST Grant [JPMJCR1755 (to J.N.)], Japan.
Materials
| Name | Company | Catalog Number | Comments |
| 2% xylocaine jelly | AstraZeneca, UK | ||
| B6(129S6)-Tg(Aif1-tTA)54Kftnk | RIKEN RBC, Japan | RBRC05769 | Iba1-tTA mice |
| B6;129-Actb(tm3.1(tetO-GCaMP6)Kftnk) | RIKEN RBC, Japan | RBRC09552 | tetO-GCaMP6 mice |
| Forceps | Fine science tools, US | 13008-12 | |
| G-CEM ONE | GC corporation, Japan | ||
| Glass capillary | Narishige, Japan | GDC-1 | |
| ImageJ | NIH, US | ||
| Isofulrane | Pfizer, US | ||
| Ketamin | Daiichi-Sankyo, Japan | ||
| Kwik-sil | World Precision Instruments, US | KWIK-SIL | |
| MATLAB, 2020a | MathWorks, US | ||
| Micro cover glass (2 x 2 mm, No.3) | Matsunami, Japan | custum-made | Bottom glass for cranial window |
| Micro cover glass (3 x 3 mm, No.0) | Matsunami, Japan | custum-made | Upper glass for cranial window |
| N25X-APO-MP | Nikon, Japan | N25X-APO-MP | Objective lens (25x) |
| Norland optical adhesive | Edmund optics, US | 6101 | |
| Piezo nano-positioning system, Nano-Drive | Mao City Labs, US | ||
| Razor blade | Feather, Japan | FA-10 | |
| Scissors | Fine science tools, US | 14060-11 | |
| Steel drill | Minitor, Japan | BS1201 | |
| Stereotaxic instruments | Narishige, Japan | SR-5M-HT | |
| Super-bond (C&B kit) | Sun Medical, Japan | 4560227797382 | |
| Surgical needle hook | Fine science tools, US | 10065-15 | |
| Ti:Sappire laser, MaiTai DeepSee | Spectra Physics, US | Mai Tai eHP DS | |
| Tweezers | Fine science tools, US | 11051-10 | |
| Tweezers | Fine science tools, US | 11255-20 | |
| Two-photon microscope | Nikon, Japan | A1R-MP | |
| UV craft resin | Kiyohara, Japan | UVR | |
| Xylazine | Bayer, Germany |
References
- Miyamoto, A., et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nature Communications. 7, 12540(2016).
- Schafer, D. P., et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent. Neuron. 74 (4), 691-705 (2012).
- Davalos, D., et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 8 (6), 752-758 (2005).
- Nimmerjahn, A., Kirchhoff, F., Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308 (5726), 1314-1318 (2005).
- Cserep, C., et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 367 (6477), 528-537 (2020).
- Kato, G., et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro. 3 (3), (2016).
- Li, Y., Du, X. -F., Liu, C. -S., Wen, Z. -L., Du, J. -L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 23 (6), 1189-1202 (2012).
- Tremblay, M. -E., Lowery, R. L., Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. Plos Biology. 8 (11), 1000527(2010).
- Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience. 29 (13), 3974-3980 (2009).
- Oppenheim, R. W. Cell death during development of the nervous system. Annual Review of Neuroscience. 14 (1), 453-501 (1991).
- Sierra, A., Abiega, O., Shahraz, A., Neumann, H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Frontiers in Cellular Neuroscience. 7, 6(2013).
- Brawek, B., et al. A new approach for ratiometric in vivo calcium imaging of microglia. Scientific Reports. 7 (1), 6030(2017).
- Eichhoff, G., Brawek, B., Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochimica et Biophysica Acta. 1813 (5), 1014-1024 (2011).
- Liu, L., et al. Microglial calcium waves during the hyperacute phase of ischemic stroke. Stroke. 52 (1), 274-283 (2021).
- Pozner, A., et al. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Frontiers in Molecular Neuroscience. 8, 12(2015).
- Umpierre, A. D., et al. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife. 9, e56502(2020).
- Ohkura, M., et al. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. Plos One. 7 (12), e51286(2012).
- Tanaka, K. F., et al. Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Reports. 2 (2), 397-406 (2012).
- Liu, Y. U., et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nature Neuroscience. 22 (11), 1771-1781 (2019).
- Stowell, R. D., et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nature Neuroscience. 22 (11), 1782-1792 (2019).
- Sun, W. L., et al. In vivo two-photon imaging of anesthesia-specific alterations in microglial surveillance and photodamage-directed motility in mouse cortex. Frontiers in Neuroscience. 13, 421(2019).
- Evangelidis, G. D., Psarakis, E. Z. Parametric image alignment using enhanced correlation coefficient maximization. IEEE Transactions on Pattern Analysis and Machine Intelligence. 30 (10), 1858-1865 (2008).
- Rosario, A. M., et al. Microglia-specific targeting by novel capsid-modified AAV6 vectors. Molecular Therapy. Methods & Clinical Development. 3, 16026(2016).
- Maes, M. E., Wogenstein, G. M., Colombo, G., Casado-Polanco, R., Siegert, S. Optimizing AAV2/6 microglial targeting identified enhanced efficiency in the photoreceptor degenerative environment. Molecular Therapy. Methods & Clinical Development. 23, 210-224 (2021).
- Lin, R., et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nature Methods. 19 (8), 976-985 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved