
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Montage et suivi du développement de la communauté microbienne dans une plateforme de microwell Array
Dans cet article
Résumé
Le développement des communautés microbiennes dépend d'une combinaison de facteurs, y compris l'architecture environnementale, l'abondance des membres, les traits et les interactions. Ce protocole décrit un environnement synthétique et microfabricant pour le suivi simultané de milliers de communautés contenues dans les puits de femtolittes, où des facteurs clés tels que la taille de niche et le confinement peuvent être approchés.
Résumé
Le développement des communautés microbiennes dépend d'une combinaison de facteurs déterministes et stochastiques complexes qui peuvent modifier radicalement la répartition spatiale et les activités des membres de la communauté. Nous avons développé une plate-forme de micropuits qui peut être utilisée pour assembler et suivre rapidement des milliers de communautés bactériennes en parallèle. Ce protocole souligne l'utilité de la plate-forme et décrit son utilisation pour le suivi optique du développement de communautés simples à deux membres dans un ensemble de tableaux au sein de la plate-forme. Cette démonstration utilise deux mutants de Pseudomonas aeruginosa , une partie d'une série de mutants développés pour étudier la pathogénicité de la sécrétion de type VI. Les inserts chromosomiques des gènes mCherry ou GFP facilitent l'expression constitutive de protéines fluorescentes avec des longueurs d'onde d'émission distinctes qui peuvent être utilisées pour surveiller l'abondance et l'emplacement des membres de la communauté dans chaque micropuits. Ce protocole décrit un procédé détailléD pour assembler des mélanges de bactéries dans les puits du réseau et utiliser l'imagerie par fluorescence temporelle et l'analyse d'image quantitative pour mesurer la croissance relative de chaque population membre au fil du temps. L'ensemencement et l'assemblage de la plate-forme des micropuits, les procédures d'imagerie nécessaires à l'analyse quantitative des communautés microbiennes au sein du tableau et les méthodes qui peuvent être utilisées pour révéler les interactions entre les espèces d'espèces microbiennes.
Introduction
Les communautés microbiennes sont façonnées à la fois par des facteurs déterministes tels que la structure de l'environnement et les processus stochastiques associés à la mort cellulaire, à la division, à la concentration de protéines, au nombre d'organites et à la mutation 1 . Dans l'environnement naturel, il est presque impossible d'analyser l'impact individuel de ces influences sur la composition et l'activité de la communauté. Obscurcis par des structures naturelles et enterrés dans un milieu chimique et biologique, l'identification des membres de la communauté et la résolution de leur répartition spatiotemporelle dans l'environnement naturel sont extrêmement difficiles. Néanmoins, les efforts récents ont souligné l'importance de l'organisation spatiale sur la fonction communautaire et soulignent la nécessité de tenir compte de l'abondance et de l'organisation des membres dans les études en cours 2 , 3 , 4 .
IlEst clair que l'environnement chimique local ( c'est-à-dire la disponibilité des nutriments et des métabolites secondaires), la structure physique ( p. Ex., L' architecture du sol, les racines des plantes, les particules de l'océan ou les microvilles intestinales), la présence ou l'absence d'oxygène et l'introduction de Les espèces pathogènes affectent la composition, l'architecture et la fonction des communautés microbiennes 5 , 6 , 7 , 8 , 9 , 10 , 11 . Néanmoins, les techniques traditionnelles pour les cultures qui négligent de capturer ces facteurs continuent de prévaloir. La composition de la communauté ( par exemple, la présence d'espèces co-dépendantes), l'attachement physique, la concentration de la molécule de signalisation et le contact direct des cellules-cellules sont autant de facteurs importants pour façonner une communauté microbienne et peuvent être perdus dans cConditions culturelles conventionnelles. Ces propriétés sont difficiles à reproduire dans une culture liquide en vrac ou sur une plaque de gélose. La disponibilité de techniques microfluidiques, de micropatternation et de nanofabrication qui permettent la réplication des principales caractéristiques physiques et chimiques des environnements naturels a permis à de nombreux chercheurs de créer des communautés bactériennes pour étudier leurs interactions 12 , 13 , 14 et de développer des environnements synthétiques qui Imiter les conditions naturelles 4 , 15 , 16 , 17 , 18 , 19 , 20 .
Ce protocole décrit une méthode pour fabriquer un dispositif de matrice de micropuits et fournit des procédures expérimentales détaillées qui peuvent être utilisées pour fonctionnerLes puits dans la matrice et la croissance des bactéries, à la fois en tant que colonies d'une seule espèce et dans les communautés multi-membres. Ce travail démontre également comment les bactéries modifiées pour produire des protéines rapporteurs fluorescentes peuvent être utilisées pour surveiller la croissance bactérienne dans les puits au fil du temps. Un tableau similaire a été présenté précédemment et a montré qu'il est possible de suivre la croissance des colonies à une seule espèce de Pseudomonas aeruginosa ( P. aeruginosa) dans les micropuits. En modulant la taille du puits et la densité d'ensemencement, les conditions de départ de milliers d'expériences de croissance peuvent varier en parallèle pour déterminer comment les conditions d'inoculation initiales affectent la capacité des bactéries à se développer 21 . Le travail actuel utilise une version légèrement modifiée du réseau de micro-puits qui se construit sur les travaux précédents en permettant la comparaison simultanée de tableaux multiples et en utilisant un protocole expérimental plus robuste. Le tableau utilisé dans ce travail contient plusieurs sous-arborescences, ou tableau ensemBles, contenant des puits de différentes tailles, allant de 15 à 100 μm de diamètre, qui sont disposés à trois pas différents ( c.-à-d. 2x, 3x et 4x le diamètre du puits). Les matrices sont gravées en silicium et la croissance des bactéries ensemencées dans les réseaux de silicium est activée en scellant les tableaux avec une lamelle qui a été revêtue d'un gel d'agarose à infusion moyenne. Les mutants P. aeruginosa conçus pour étudier le système de sécrétion de type VI sont utilisés dans cette démonstration.
Les résultats présentés ici se basent sur l'objectif ultime d'analyser les communautés de plusieurs membres dans les réseaux de micropuits, permettant aux chercheurs de surveiller l'abondance et l'organisation des bactéries in situ tout en contrôlant et sondant l'environnement chimique. Cela devrait finalement donner un aperçu des «règles» qui régissent le développement communautaire et la succession.
Protocole
1. Fabrication de matrices de silicium
- Revêtement de parylène
- Déposer entre 1-1,5 μm de parylène N sur des plaquettes de silicium en utilisant un système de revêtement de parylène vendu dans le commerce conformément aux spécifications et aux instructions du fabricant (réglages: point de consigne du vaporisateur = 160 ° C, point de consigne du four = 650 ° C).
REMARQUE: Environ 6 g de parylène N chargé dans une chambre donne des revêtements de 1 à 1,5 μm d'épaisseur.
- Déposer entre 1-1,5 μm de parylène N sur des plaquettes de silicium en utilisant un système de revêtement de parylène vendu dans le commerce conformément aux spécifications et aux instructions du fabricant (réglages: point de consigne du vaporisateur = 160 ° C, point de consigne du four = 650 ° C).
- Photolithographie
- Revêtir les plaquettes de parylène N-enduites avec un promoteur d'adhérence, 20% d'hexaméthyldisilazane (HMDS) et 80% d'acétate d'éther monométhylique de propylène glycol (PGMEA) (voir la Table des matériaux ) à 3 000 tr / min pendant 45 s. Remplissez une pipette de transfert de 2 mL avec un promoteur d'adhérence et faites saupoudrer sur toute la plaquette. Laissez la plaquette s'asseoir environ 10 s avant de la faire sécher.
- Remplissez une pipette de transfert de 2 mL avecNe photoresist (voir la Table des matériaux ) et distribuer la photoréserve au centre de la plaquette. Faire tourner à 3 000 tr / min pendant 45 s pour obtenir un revêtement résistant d'environ 1,5 μm d'épaisseur.
- Faire cuire doucement les échantillons sur une plaque chauffante à 115 ° C pendant 1 min.
- Utilisez un alignement de contact et un photomasque avec le motif de puits désiré pour exposer l'échantillon à la lumière ultraviolette. Exposez la plaquette enroulée par centrifugation à travers le photomasque à motifs pendant 6 s, en donnant une dose approximative de 60-80 mJ / cm 2 mesurée à 365 nm.
- Développez le motif en immergeant l'échantillon dans le promoteur (<3% d'hydroxyde de tétraméthylammonium dans l'eau, voir la Table des matériaux ) pendant 2 min. Rincer avec de l'eau DI et sécher avec de l'azote propre et sec.
NOTE: Les zones de photoresist exposées aux rayons UV doivent être effacées pendant le développement.
- Gravure ionique réactive
- Utiliser une gravure au plasma d'oxygène pour éliminer le parylène exposéTout le long du substrat en silicium.
REMARQUE: La recette peut être modulée pour modifier la vitesse de gravure du parylène. Pour les épaisseurs de parylène comprises entre 1 et 5 μm, utilisez une recette avec 60 mTorr, 20 ° C, 100 sccm O 2 , 10 W RF et 2 000 W ICP sur un outil RACI. Après la gravure et l'élimination de la couche de parylène exposée, la zone à motifs ( c'est-à-dire le silicium exposé) devrait être brillante et argentée. - Utiliser un procédé de gravure profonde RIE (DRIE, par exemple, Bosch DRIE) pour graver dans le silicium.
REMARQUE: Le taux de gravure et la durée détermineront la profondeur du puits. Un cycle complet du processus de Bosch (une étape de dépôt de 3 s: 20 mTorr, 15 ° C, 140 sccm C 4 F 8 , 10 W RF et 1 750 W ICP suivi d'un processus de gravure de 10 s: 20 mTorr, 15 ° C , 120 sccm SF 6 , 8 W RF et 1,750 W ICP) correspond à environ 1 μm de profondeur de gravure. Les puits utilisés dans cette démonstration vont de 3 à 3,5 μm de profondeur. - VÉgentez la profondeur de gravure en utilisant la profilométrie physique.
- Chargez l'échantillon dans un profilomètre physique (voir la table des matières ).
- Allumez le vide échantillon et appuyez sur le bouton de chargement manuel.
- Concentrez le système sur l'échantillon en appuyant sur le bouton "Focus". Placez une caractéristique appropriée pour la mesure sur l'écran de visualisation.
- Scannez l'échantillon. Nivelez le profil et mesurez la profondeur de la fonction.
- Enregistrez le taux de gravure et modulez les temps de gravure ultérieurs pour obtenir la profondeur souhaitée.
NOTE: Les mesures comprennent la profondeur du puits de silicium, l'épaisseur du parylène déposé et l'épaisseur du photorésist. La vérification de l'épaisseur de chaque couche tout au long de la procédure est nécessaire pour obtenir une profondeur de puits précise.
- Utiliser une gravure au plasma d'oxygène pour éliminer le parylène exposéTout le long du substrat en silicium.
2. Culture bactérienne et ensemencement ( figure 1a )
- Commencez les colonies sur Luria Broth (LB) des plaques de gélose à partir de stocks de glycérol et utilisent dans les deux semaines. Choisissez les colonies des souches désirées à partir des plaques d'agar LB et commencez des cultures de P. aeruginosa pendant la nuit. Incuber les cultures de nuit pendant environ 18 h à 37 ° C tout en agitant à 220 tr / min dans un milieu R2A.
NOTE: Les colonies doivent être prélevées dans les deux semaines suivant le placage pour s'assurer que les mutations et les gènes rapporteurs fluorescents sont retenus. Tous les travaux de P. aeruginosa devraient être effectués dans les conditions de BSL-2. - Utilisez un scribe de diamant pour sectionner la plaquette de silicium dans des puces individuels contenant les ensembles de différentes tailles et tableaux de pitch-well. Assurez-vous que chaque puce contient l'ensemble complet de tailles de puits et d'emplacements pour l'étude.

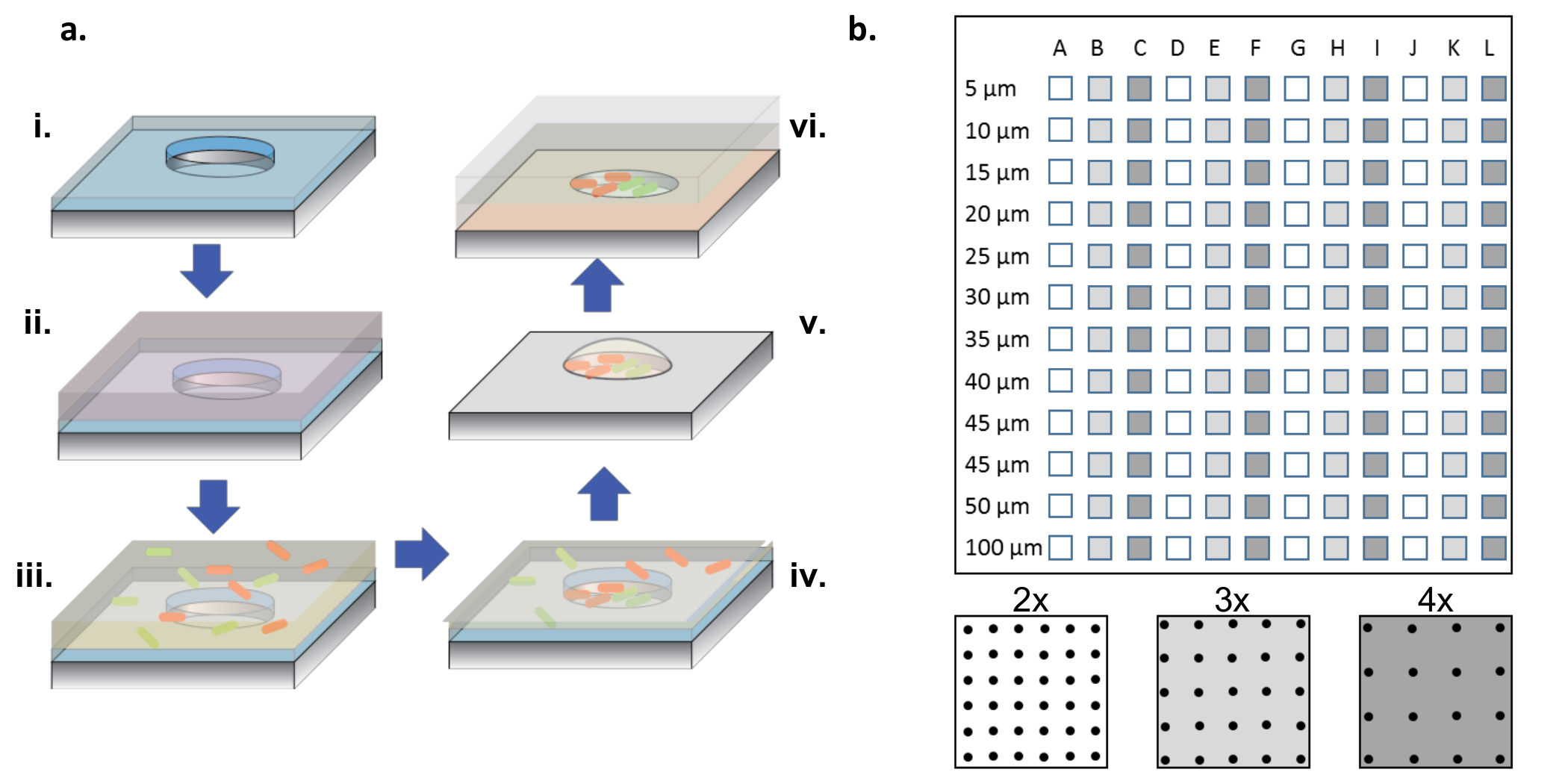
Figure 1: Procédure de fabrication et d'ensemencement cellulaire. (A) Arrays à micropuits aSont gravés dans des plaquettes de silicium revêtues d'une mince couche de parylène (i). Pour mouiller les puits et / ou fonctionnaliser la surface, une solution de protéine est ajoutée dans une gouttelette au dessus des arrays (ii). La solution de protéine est enlevée, les gaufrettes sont séchées et une nouvelle solution contenant les bactéries désirées est ajoutée (iii). La solution bactérienne est éliminée après une période d'incubation et les gaufrettes sont autorisées à sécher, laissant derrière elles des bactéries dans les puits et à la surface (iv). Les bactéries associées à la surface sont enlevées avec le décollage du parylène, laissant derrière eux des bactéries semées proprement dans les micropuits et encore viables en raison du milieu de glycérine à 2%, ce qui aide à maintenir les puits hydratés (v). Les copeaux de silicium sont ensuite placés vers le bas sur une lamelle en verre enduit d'un gel d'agarose, ce qui alimente une croissance bactérienne dans les micropuits (vi). ( B ) Mise en page de sous-réseaux sur un seul dispositif de silicium. Chaque sous-ensemble contient un ensemble de puits identiques. Le diamètre des micropuits dans tous les sous-élémentsLes râmes sont de 5-100 μm et sont organisés à 2x, 3x ou 4x le pas du diamètre du puits, qui est désigné par les couleurs blanc à gris foncé sur le schéma du panneau inférieur. Lorsque les profondeurs du puits sont peu profondes (<10 μm), les diamètres des puits de 5 et 10 μm sont rarement utiles, généralement en raison d'un manque de cellules qui colonisent ces très petits puits. Dans ce travail, seules les données des puits à 15-100 μm ont été analysées. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
REMARQUE: Comme le montre la figure 1b , une puce complète contient des sous-réseaux de puits, avec des diamètres allant de 5 à 100 μm, avec trois pas différents ( c'est-à-dire 2x, 3x et 4x le diamètre) en répétant 4 fois.
- Placer une gouttelette de 150 ul de 500 ug / ml d'albumine de sérum bovin (BSA) dans une solution de PBSEn haut du réseau pour mouiller les micropuits. Incuber la solution de BSA pendant 1 heure sur la puce à la RT dans une chambre humide.
- Créez la chambre en remplissant le fond d'une boîte à pointe de pipette vide avec une solution salée tamponnée au phosphate (PBS).
REMARQUE: D'autres substances, telles que des lectines spécifiques, peuvent être utilisées à la place de BSA pour fonctionnaliser la surface des micropuits.
- Créez la chambre en remplissant le fond d'une boîte à pointe de pipette vide avec une solution salée tamponnée au phosphate (PBS).
- Pendant l'incubation de puces de silicium avec une solution de BSA, centrifuger les cultures à 2500 tr / min (correspondant à une moyenne de 950 xg) pendant 5 min, puis les remettre en suspension dans 500 μL de milieu R2A frais avec 2% de glycérol.
- Déterminer la DO de la culture à l'aide d'un spectromètre UV-vis à 600 nm. Ajustez-le à une DO de 0,02 en utilisant 2% de milieu glycérol R2A.
REMARQUE: Le glycérol aide à éviter que les puits ne se dessèchent pendant le décollage du parylène.
- Déterminer la DO de la culture à l'aide d'un spectromètre UV-vis à 600 nm. Ajustez-le à une DO de 0,02 en utilisant 2% de milieu glycérol R2A.
- Après incubation, retirer la solution de BSA et rincer 3x avec du PBS en enlevant et en remplaçant la gouttelette liquideT couvrant le réseau de micropuits de silicium. Sécher sous azote.
- Ajouter 150 μL de cultures de 0,02 OD à chacun des tableaux secs placés dans une chambre humide. Incuber pendant 1 h à 4 ° C pour permettre aux bactéries d'adhérer aux parois du puits.
REMARQUE: La réfrigération n'est pas nécessaire pour l'incubation. Le temps d'incubation de 4 ° C peut être utilisé pour empêcher la croissance des bactéries avant l'apparition de l'imagerie afin de pouvoir visualiser l'organisation spatiale des communautés avant la croissance. L'incubation à température ambiante peut également être utilisée. Les deux protocoles donnent lieu à des courbes de croissance similaires.
3. Installation du microscope
- Avant de commencer l'incubation bactérienne sur les puces de silicium, allumez la chambre de contrôle environnementale (voir la table des matériaux) et ajustez les réglages sur le boîtier de commande afin que l'humidité (~ 100%) et la température (30-32 ° C, voir étape 3.2) peuvent être équilibrés avant d'ajouter des échantillons.
- Nivelez le support de l'échantillon et alignez leSur l'échantillon avec des lingettes de laboratoire trempées au PBS (voir la Table des matériaux ) pour augmenter l'humidité dans la chambre jusqu'au point de rosée. Réglez la température de la chambre à 30 ° C et celle du couvercle de la chambre à 32 ° C pour réduire la condensation sur le plan d'imagerie.
REMARQUE: Le porte-coulisseau s'insère dans la chambre des cellules vivantes avec un joint d'environ 1 cm d'épaisseur. Le support d'échantillon est nivelé avec l'aide d'un niveau de bulle placé sur le support d'échantillon. Le porte-échantillon peut être légèrement incliné et rester scellé dans le joint à niveau. - Alors que les cultures sont incubées sur les puces de silicium, allumez manuellement l'interrupteur d'alimentation pour la lampe au mercure au moins 30 minutes avant l'imagerie. Allume manuellement la caméra et le stade du microscope automatisé. Ouvrez le logiciel utilisé pour contrôler le microscope et les périphériques et assurez-vous que l'équipement est reconnu par le logiciel.
NOTE: L'agrandissement est 10X avec NA = 0.3.
- Préparer au micro-ondes des solutions d'agarose préalablement préparées ( c'est-à-dire 2% d'agarose dans le milieu R2A) jusqu'à ce qu'un état liquide soit atteint, environ 60 s.
- Mouillez l'arrière d'une lamelle de verre de diamètre extérieur de 75 mm x 22 mm, avec de l'éthanol et placez-le longitudinalement, centré sur une glissière en verre de 2 x 3 "(50 x 75 mm). Placez deux entretoises PDMS (épaisseur de ~ 1 mm) Le long des bords longs de la lamelle coulissante et déplacez le lamelle de verre de telle sorte qu'environ 1 mm de la lamelle couvre le bord de la glissière.
- Verser 5 ml de solution liquide d'agarose sur le couvercle de verre, juste pour le couvrir complètement, et placer une deuxième glissière en verre de 2 x 3 "sur le dessus pour assembler l'agar liquide entre la lamelle et la glissière.
REMARQUE: Ceci contrôle la profondeur de l'agarose, ce qui fait que l'épaisseur totale de la lamelle et de l'agarose durcie est égale en épaisseur aux entretoises PDMS. - Laisser reposer le sandwich en verre glacé en gel d'agarose au coulisseau à glissière jusqu'à ce que la solution d'agarose commence à se solidifier; Puis transférez-le au réfrigérateur. Après 15 min, retirer l'excès d'agarose solide et couper autour de la lamelle de verre. Déplacez-le sur un plat propre et placez-le dans un réfrigérateur jusqu'à l'utilisation.
5. Scellant les puits avec un couvercle et une imagerie enrobés d'agarose
- Une fois la période d'incubation des bactéries terminée, retirer le lamelle couché en agarose du réfrigérateur et préparer les copeaux de silicium, comme suit.
- Trempez les puces de silicium dans de l'eau ultrapure, une à la fois, pendant 10 s chacune. Placez-les sur leurs bords sur un essuyage ou un tissu de laboratoire jusqu'à ce que la plupart des excès de liquide aient été vidés des bords des frites.
- Couper un morceau de ruban pour faire correspondre la longueur de bord de chaque puce de silicium. Placez la bande sur le parylène qui recouvre le silicium et utilisez-le pour éliminer rapidement le revêtement de parylène.
- ImmedInverser immédiatement chaque puce pelée et placer chaque puce de sorte que le côté du réseau de micropuits (et entre en contact) avec le côté revêtu d'agarose d'une lamelle revêtue d'agarose. Veillez à ne pas déplacer ou à déplacer la puce après qu'elle touche l'agarose pour éviter la croissance de bactéries à l'extérieur des puits.
- Placez la lamelle de table de micropuits / agarose assemblée dans le support de glissière de la chambre de contrôle environnementale du plateau sur le microscope.
- Utilisez la lumière ambiante ou la lumière dirigée ( par exemple, une lampe de poche) pour localiser les réseaux d'intérêt. Utilisez le logiciel commercial qui contrôle l'étape automatisée pour sauvegarder ces positions (voir la table des matières). Éteignez la lumière ambiante ou dirigée après le stockage des positions.
- Dans le logiciel commercial, ouvrez le panneau "Acquisition ND".
REMARQUE: Ce panneau comprend un menu pour l'enregistrement automatique dans un répertoire spécifique, ainsi que l'acquisition d'images programmables. Pour ces expériences, Les menus "Heure", "XY" et "λ" sont utilisés. - Pour enregistrer les emplacements dans le logiciel, cliquez sur le "menu XY", puis cochez une case vide sur le côté gauche pour chaque position qui doit être sauvegardée. De plus, cliquez sur le bouton "Inclure Z".
- Acquérir des images dans le temps aux longueurs d'onde souhaitées et au grossissement 10 à l'aide des cubes de filtre à fluorescence appropriés (voir la Table des matériaux).
- Utilisez le logiciel de contrôle et les positions de matrices enregistrées pour vous déplacer dans chaque emplacement enregistré et pour vous concentrer sur les puits. Cliquez sur chaque emplacement XY dans la liste enregistrée et ajustez la mise au point en utilisant le filtre Green Flourescence Protein (GFP). Enregistrez la nouvelle position z en cliquant sur la flèche indiquant l'emplacement z.
REMARQUE: ce processus peut prendre beaucoup de temps. Envisagez de prendre la précaution d'augmenter le gain et d'utiliser le filtre de densité neutre pour réduire l'intensité de la lumière pour empêcher le photoblanchage. - Déterminer la distance de l'axe z entre les plans focaux pour chaque longueur d'onde en notant la différence dans la position de l'axe z lorsqu'il est concentré sur la surface du réseau. Choisissez 2-3 emplacements à partir du tableau avec la population de bactéries rouge / verte mélangées et concentrez-vous avec le filtre Red Fluorescence Protein (RFP).
- Soustrayez la distance entre les plans focaux en utilisant les filtres de fluorescence GFP et RFP et ajoutez le réglage du plan focal dans le menu "λ".
REMARQUE: par exemple, si le réseau apparaît concentré dans le canal GFP à une localisation z de 50 μm, et le même réseau apparaît concentré dans le canal RFP à 55 μm, ajoutez +5 à côté de la configuration optique RFP dans "λ "Menu.
- Soustrayez la distance entre les plans focaux en utilisant les filtres de fluorescence GFP et RFP et ajoutez le réglage du plan focal dans le menu "λ".
- Commencez l'acquisition de l'image à l'échéance.
NOTE: Pour les expériences présentées ici, les images RFP et GFP ont été acquises pour chaque position du tableau à des intervalles de 30 minutes en utilisant l'acquisition d'images multidimensionnelles via un logiciel commercial qui contrôleLa caméra, l'obturateur, la roue du filtre et l'étage motorisé.- Réglez l'intervalle à 30 min et la durée de l'expérience à 24 h dans le menu "Heure". Cliquez sur "Exécuter maintenant".
REMARQUE: Lorsque les cases "Heure", "XY" et "λ" sont cochées, l'exécution du programme passe la scène à l'image de chaque emplacement ( c'est-à-dire les emplacements XYZ enregistrés), prenez une image dans une seule longueur d'onde, déplacez la position z Pour tenir compte des différences de rayons focaux ( c.-à-d., Lambda ou contrôle de la longueur d'onde), prenez la deuxième image, passez à l'emplacement du tableau suivant (multipoint) et bouclez-les à des intervalles de 30 minutes (time-lapse).
- Réglez l'intervalle à 30 min et la durée de l'expérience à 24 h dans le menu "Heure". Cliquez sur "Exécuter maintenant".
- Utilisez le logiciel de contrôle et les positions de matrices enregistrées pour vous déplacer dans chaque emplacement enregistré et pour vous concentrer sur les puits. Cliquez sur chaque emplacement XY dans la liste enregistrée et ajustez la mise au point en utilisant le filtre Green Flourescence Protein (GFP). Enregistrez la nouvelle position z en cliquant sur la flèche indiquant l'emplacement z.
- Acquérir des images de contrôle d'illumination.
REMARQUE: utilisez les menus "ND Acquisition", "Time" et "XY" pour prendre des images de 4 sites, 25x chacun.- Prenez une série de 100 images "darkfield" en éteignant toutes les sources lumineuses et en prenantUne «image» d'une diapositive standard. Ces images capteront le bruit de la caméra. Utilisez le temps d'exposition le plus long utilisé pendant le timelapse (étape 5.3.3).
- Prenez une série de 100 images "champ d'illumination" en imagant une diapositive standard ( c'est-à-dire une intensité RFP ou GFP uniforme) dans quelques endroits différents pour capturer l'illégalité inégale aux conditions expérimentales données. Choisissez un temps d'exposition qui maximise le signal sans atteindre la saturation.
6. Analyse
- Traiter les piles d'images à l'aide d'un logiciel d'analyse d'image ( p. Ex., ImageJ).
- Convertissez les images acquises en format de fichier tiff en utilisant le logiciel commercial. Téléchargez des images dans le logiciel d'analyse d'image en cliquant sur "Fichier"> "Importer"> "Séquence d'image".
- Créez une "image de correction" en faisant la moyenne de toutes les images "champ sombre" et "champ d'illumination". SubtracT l'image moyenne "darkfield" de l'image moyenne "champ d'illumination" en choisissant "Process"> "Image Calculator". Sélectionnez les deux images, "Image1" et "Image2", puis "Soustraire" dans le champ "Opération". Cliquez sur "OK".
- Pour la moyenne, charger les images de correction (ou de champ sombre), cliquez sur "Image"> "Stacks"> "Projet Z"> "Projection moyenne".
- Effectuez l'enregistrement d'image si nécessaire. Ensuite, effectuez une soustraction de fond en cliquant sur "Traiter"> "Soustraire le fond". Entrez un rayon ( p. Ex., 125) dans le champ "rayon" et sélectionnez "paraboloïde coulissant".
- Effectuer la correction d'illumination en utilisant "Process"> "Calculator Plus". Choisissez les paramètres suivants: opération, division; I1, bien image; I2, image de correction; K1, image de correction signifie; Et k2, 0. Cliquez sur "CreA mangé une nouvelle fenêtre. "
REMARQUE: Cet ensemble de données n'a pas exigé d'enregistrement, mais dans d'autres travaux, ImageJ Plugin StackReg a été utilisé avec la transformation "Traduction". Pour la soustraction d'arrière-plan, utilisez le même rayon de paraboloïde coulissant pour chaque ensemble d'image. Par exemple, si les puits les plus grands ont un rayon de pixel de 100, utilisez un rayon supérieur à 100 ( par exemple, 125) pour chaque ensemble d'images.
- Déterminer la croissance de chaque souche dans les micropuits.
- Sélectionnez les régions d'intérêt (ROI) autour de chaque micropuits dans les tableaux souhaités en utilisant le plugin ImageJ "MicroArray".
- Dans le menu "MAP", cliquez sur "Réinitialiser la grille". Spécifiez les lignes, les colonnes et le diamètre (en fonction de la taille et du nombre du puits sur le tableau, voir Figure 1b ). Sélectionnez "cercle" dans le menu "ROI shape".
- Maintenez la touche "Alt" en sélectionnant le ROI supérieur gauche avec la souris pour vous déplacer tROI array. Maintenez la touche "shift" en sélectionnant le ROI en bas à gauche pour modifier la taille du tableau. Maintenez la touche "shift" en sélectionnant un ROI du côté droit du tableau, mais pas aux coins, pour modifier l'espacement des ROI.
- Utilisez les commandes ci-dessus pour adapter le tableau ROI sur les puits d'une image. Cliquez sur "Mesurer RT".
REMARQUE: le plugin exportera les mesures souhaitées de chaque ROI. Utilisez trois tailles de ROI, créant des anneaux concentriques autour des puits pour collecter localement le signal d'arrière -plan ( c'est-à-dire le signal de l'anneau intermédiaire soustrait de l'anneau extérieur) et les mesures de fluorescence ( c'est-à-dire le signal de la bague intérieure).
- Collectez les données dans un logiciel de calcul et calculez le signal de fond. Importez-le à un logiciel de script personnalisé pour une analyse plus approfondie.
- Sélectionnez les régions d'intérêt (ROI) autour de chaque micropuits dans les tableaux souhaités en utilisant le plugin ImageJ "MicroArray".
- Organisation et analyse des données
- Importez les données et organisez les donnéesRecueillis dans ImageJ dans une matrice dans l'ordre suivant pour tous les temps: colonne 1, numéro de sous-tableau; Colonne 2, rangée de puits; Colonne 3, colonne de puits; Colonne 4, intensité moyenne; Colonne 5, intensité de fond; Et la colonne 6, intensité moyenne - intensité de fond.
- Séparez les résultats de l'acquisition mCherry et GFP en différentes matrices. Stockez les résultats de chaque sous-tableau et chaque couleur dans une cellule différente dans un réseau de cellules.
REMARQUE: cette organisation simplifie le déplacement entre données d'image et résultats de mesure, nettoie les données et veille à ce que les mesures représentent avec précision les données.
- Séparez les résultats de l'acquisition mCherry et GFP en différentes matrices. Stockez les résultats de chaque sous-tableau et chaque couleur dans une cellule différente dans un réseau de cellules.
- Ajuster pour l'autofluorescence de P. aeruginosa .
NOTE: Dans les expériences impliquant la co-culture des souches GFP et mCherry, une puce mCherry-Only doit être analysée pour déterminer la relation entre mCherry et l'autofluorescence verte.- Déposez le signal mCherry-contre-GFP de tous les mCherry ΔretS &# 916; tse / i1-6 puits à tout moment-points pour déterminer la relation entre le signal mCherry et l'autofluorescence dans le canal GFP. Soustraire le signal d'autofluorescence des co-cultures.
- Tracez les trajectoires et adaptez une équation logistique modifiée à chaque trajectoire pour extraire des paramètres en utilisant un montage de moindres carrés dans un logiciel de tableur ou un logiciel de script personnalisé.
- Recherchez les corrélations entre et entre les paramètres de la trajectoire GFP et mCherry.
- Importez les données et organisez les donnéesRecueillis dans ImageJ dans une matrice dans l'ordre suivant pour tous les temps: colonne 1, numéro de sous-tableau; Colonne 2, rangée de puits; Colonne 3, colonne de puits; Colonne 4, intensité moyenne; Colonne 5, intensité de fond; Et la colonne 6, intensité moyenne - intensité de fond.
Résultats
La plate-forme expérimentale présentée ici est conçue pour des études à haut débit et à fort contenu des communautés bactériennes. Le design permet d'analyser simultanément des milliers de communautés, qui se développent dans des puits de différentes tailles. Avec cette conception de matrice de micropuits, on peut déterminer la dépendance de la composition de la communauté finale sur les densités de semis initiaux, la taille du puits et l'environnement chimique....
Discussion
Cet article présentait un périphérique de réseau de micropuits et des protocoles expérimentaux conçus pour permettre l'analyse par image de cellule animale à haut débit et à haute teneur du développement communautaire bactérien. Bien que la démonstration ait été axée sur l'étude des effets de la sécrétion de type VI médiée par le contact sur le développement de la communauté, les tableaux ont été conçus pour être flexibles et permettre l'étude d'une large gamme de communautés ...
Déclarations de divulgation
Les auteurs n'ont rien à dévoiler.
Remerciements
Les matrices de micropuits ont été fabriqués et caractérisés au Centre pour la Division des installations pour les utilisateurs de Nanophase Materials Sciences, Office of Basic Energy Sciences, US Department of Energy. Le soutien financier de ce travail a été fourni par l'intermédiaire du Fonds de recherche et de développement du directeur de laboratoire national Oak Ridge. Les auteurs souhaiteraient également remercier le J. Mougous Laboratory (Université de Washington, Seattle, WA) pour la fourniture de souches de P. aeruginosa utilisées dans ces études.
matériels
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Références
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.