
Method Article
Application of Mouse Parthenogenetic Haploid Embryonic Stem Cells as a Substitute of Sperm
In This Article
Summary
This article aims to demonstrate the use of parthenogenetic haploid embryonic stem cells as a substitute for sperm for the construction of semi-cloned embryos.
Abstract
In organisms with sexual reproduction, germ cells are the source of totipotent cells that develop into new individuals. In mice, fertilization of an oocyte by a spermatozoon creates a totipotent zygote. Recently, several publications have reported that haploid embryonic stem cells (haESCs) can be a substitute for gametic genomes and contribute to embryos, which develop into mice. Here, we present a protocol to apply parthenogenetic haESCs as a substitute of sperm to construct embryos by intracytoplasmic injection into oocytes. This protocol consists of steps for preparing haESCs as sperm replacement, for injection of haESC chromosomes into oocytes, and for culture of semi-cloned embryos. The embryos can yield fertile semi-cloned mice after embryo transfer. Using haESCs as sperm replacement facilitates genome editing in the germline, studies of embryonic development, and investigation of genomic imprinting.
Introduction
In mammals, gametes are the only cells that transmit genetic information to the next generation. Fusion of an oocyte and a spermatozoon forms a diploid zygote that develops into an adult animal. Mutations in gametic genomes are thereby inherited by the offspring and drive genetic variation in species1. Introduction of mutations in the germline has been applied to produce genetically modified animals for diverse biological studies including the characterization of gene function and disease modelling. Both oocytes and spermatozoa are terminally differentiated and highly specialized cells that have ceased proliferation. Therefore, direct modification of gametes is technically difficult, and specialized approaches have been developed. Genetic modifications can be introduced into the mouse germline by the injection of genetically modified ESCs into blastocysts, where they integrate into the developing embryo and colonize the germline. Additionally, genetic modification of zygotes using genome editing approaches, including the CRISPR/Cas9 system, has become widely adopted2.
Recently, an outstanding approach has been reported, which applies haESCs as a substitute for a gametic genome3,4,5,6,7,8. HaESCs are stem cell lines derived from the inner cell mass of parthenogenetic or androgenetic haploid blastocysts and possess a single set of chromosomes4,7,9,10. It has been demonstrated that both parthenogenetic and androgenetic haESCs can contribute to the genome of semi-cloned mice after intracytoplasmic injection into oocytes. In contrast to other approaches, the genomes of haESCs can be directly modified in culture owing to their self-renewal capacity. Introducing genetic modifications into the germline by replacing sperm with haESCs is an important method for biological studies. It provides for a possibility to separately culture and manipulate the maternal or paternal genome, which are derived from parthenogenetic or androgenetic haESCs, respectively. HaESCs can then be used as gametic genome replacement, which is especially advantageous for studies of genomic imprinting, allele-specific expression, and parental specific processes.
In mice, both maternal and paternal genomic information is required for normal embryo development11. Therefore, full-term pups could not be obtained when wild-type parthenogenetic haESCs (phaESCs) were injected to replace the sperm genome5,8. To overcome the developmental block, genomic imprinting of the maternal genome of parthenogenetic haESCs needs to be corrected to a paternal configuration. This can be achieved by manipulation of the differentially methylated regions (DMRs). To date, targeted deletions of the H19-DMR, Gtl2-Dlk1 IG-DMR, and Rasgrf1-DMR have been studied to repress maternally expressed genes in phaESCs3,5,8,12. These studies demonstrated that deletions of both the H19-DMR and the IG-DMR are sufficient to convert a maternal into a paternal imprint configuration that can substitute for sperm chromosomes. Intracytoplasmic injection of phaESCs that carry the two DMR deletions into oocytes yielded semi-cloned pups with a frequency between 5.1% and 15.5% of transferred 2-cell embryos.
This protocol is based on the application of phaESCs with deletions of both H19-DMR and IG-DMR, which we term double-knockout phaESCs (DKO-phaESCs). We provide detailed instructions for the modification of genomic imprinting in phaESCs to establish DKO-phaESC lines, for the injection of DKO-phaESCs into oocytes as a substitute for a sperm genome, for culture of semi-cloned embryos to blastocysts, and for obtaining semi-cloned mice. This protocol is a reference for researchers who require precise and direct manipulation of the paternal genome and generation of semi-cloned embryos and mice.
Protocol
All animal experiments were performed under the license ZH152/17 in accordance with the standards and regulations of the Cantonal Ethics Commission Zurich and the EPIC animal facility at the Institute of Molecular Health Sciences, ETH Zurich.
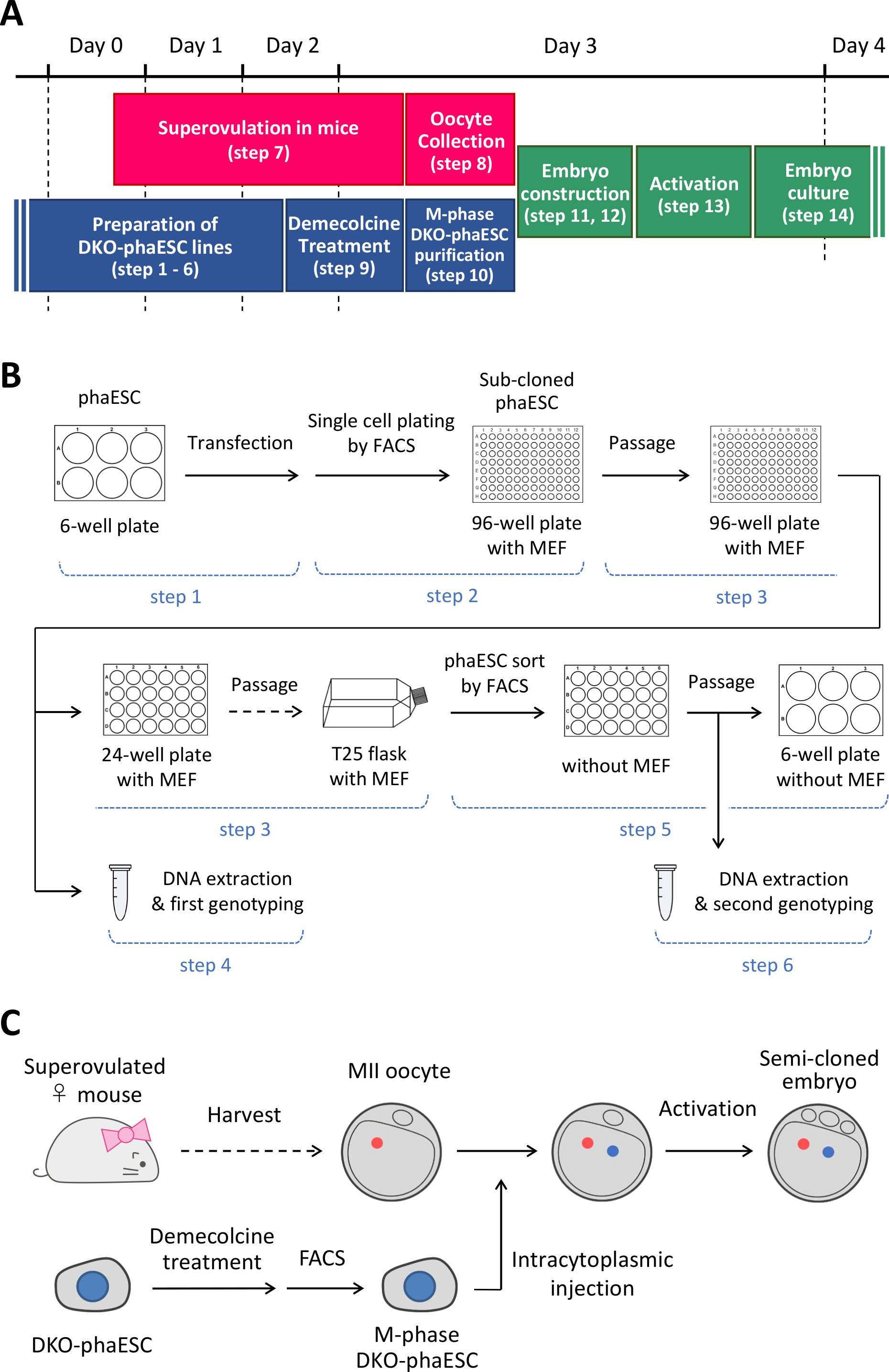
NOTE: This protocol starts with the deletion of the H19- and IG-DMRs in phaESCs. For details on how to establish phaESC lines, please refer to published reports10,13. An overview and timeframe of this protocol (steps 1–14) is provided in Figure 1A; media, solutions, and buffers are listed in Table 1. The procedure to establish DKO-phaESC lines (steps 1–6) is shown in Figure 1B, and the strategy for constructing semi-cloned embryos (steps 7–14) is depicted in Figure 1C.
1. Transfection of plasmids for deletion of H19-DMR and IG-DMR in phaESCs
- Prepare CRISPR/Cas9 plasmids for co-expression of Cas9 nucleases and guide RNAs to target deletions of H19-DMR and IG-DMR. Ligate four pairs of guide RNA oligos (H19-DMR-gRNA1-F, R; H19-DMR-gRNA2-F, R; IG-DMR-gRNA1-F, R; IG-DMR-gRNA2-F, R listed in Table 2) into pX330 plasmids.
NOTE: Refer to the published protocol on the detailed procedure for preparation of these 4 CRISPR/Cas9 plasmids14. Alternatively, plasmids available for general mouse strains are also accessible through a repository (Table of Materials). - Coat the surface of one well of a 6-well plate with 1 mL of 0.2% gelatin solution by incubation at 37 °C for 10 min.
- Plate 2 × 105 wildtype phaESCs on the gelatin-coated well in haESC medium without antibiotics, and incubate the plate at 37 °C in a 5% CO2 atmosphere for 1 day.
NOTE: Antibiotics are omitted from the medium to increase the efficiency of the subsequent lipofection. We used wildtype phaESCs at passage 10. We recommend using early passage phaESCs, but a variety of passage number have been successfully used8. The correlation between passage number and efficiency of obtaining semi-cloned embryos and mice is presently not known. - Transfect phaESCs in the well of a 6-well plate (from step 1.3) with 6 plasmids simultaneously using lipofection reagent: 50 ng piggyBac plasmid carrying a CAG-EGFP transgene, 50 ng piggyBac transposase plasmid, and 600 ng of each of the 4 CRISPR/Cas9 plasmids (from step 1.1). Refer to the manufacturer’s protocol on detailed procedure of the transfection.
NOTE: Two piggyBac plasmids are used to integrate a transposon for ubiquitous expression of enhanced green fluorescent protein (EGFP) into the genome of phaESCs. If GFP marking of the cells is not required, these two plasmids can be substituted by a CRISPR/Cas9 plasmid for transient expression of fluorescence proteins (e.g., pX458 plasmid) instead of one of the pX330 plasmids. Transient EGFP expression can then be used for sorting transfected cells. - Two days after the transfection, aspirate the medium, and add 800 µL of trypsin.
- Incubate the plate at 37 °C in a 5% CO2 atmosphere for 5 min. Then, add 2 mL of wash buffer to quench the trypsin, and pipette several times to obtain a single cell suspension.
- Transfer the cell suspension into a 15 mL tube.
- Centrifuge the tube at 160 x g for 5 min, and remove the supernatant.
- Resuspend the cell pellet in 400 µL of haESC maintenance buffer supplemented with 15 µg/mL Hoechst 33342.
NOTE: To reduce the potential toxicity of Hoechst 33342, 1 µg/mL Hoechst 33342 and 50 µM verapamil have been used instead of 15 µg/mL Hoechst 3334215. - Incubate the cell suspension at 37 °C in a 5% CO2 atmosphere for 15 min. After the incubation, transfer the cell suspension into a 5 mL tube through a cell strainer cap, and keep the tube at 4 °C until ready to use in the next step (section 2).
2. Single-cell plating of transfected phaESCs using a flow cytometer
- One day before sorting the transfected phaESCs, plate irradiated mouse embryonic fibroblasts (MEFs) on gelatin-coated 96-well plates at a density of 4 × 104 cells/cm2 in MEF medium. Typically, 6 plates are prepared to establish a phaESC line with targeted deletions. Incubate the plates at 37 °C in a 5% CO2 atmosphere.
NOTE: Irradiated MEFs are commercially available. We use MEFs derived from E12.5 embryos of DR4 mice. Although haESCs can grow on gelatin-coated plates without MEFs, we recommend MEFs for increasing the viability of sorted single haESCs. - On the day of sorting, aspirate the MEF medium from the 96-well plates, and add 120 µL of fresh haESC medium per well. Keep the plates at 37 °C in a 5% CO2 atmosphere.
- Set up a cell sorter with a 100 µm nozzle according to the manufacturer’s instructions. A 355 nm UV laser and a 488 nm blue laser are used for excitation of Hoechst 33342 and EGFP fluorescence, respectively.
NOTE: Alternatively, Hoechst 33342 can be detected by excitation with 405 nm. - Sort the transfected phaESCs in the 5 mL tube (from step 1.10) using a gate for collecting haploid cells in the G1/S phase that show EGFP expression. Deposit a single cell into each well of the 96-well plates from step 2.2.
NOTE: Detection of Hoechst 33342 staining generally distinguishes 3 peaks of cells with a 1n, 2n, and 4n DNA content, which correspond to haploid cells in G1 phase, a mixture of haploid cells in G2/M phase and diploid cells in G1 phase, and diploid cells in G2/M phase, respectively. Haploid cells at G1/S phase are identified as the peak with lower intensity of Hoechst 33342 fluorescence. A representative result and a sorting gate are shown in Figure 2A. - After plating, incubate the 96-well plates at 37 °C in a 5% CO2 atmosphere.
3. Sub-cloning of transfected phaESCs
- Three days after single-cell plating, colonies can be observed in several wells of the 96-well plates under a microscope. Mark the wells in which only single colonies grow.
NOTE: In our experience, single colonies were observed in 20%–40% of the wells of the 96-well plates. - On day 4 after single-cell plating, replace half of the medium with new haESC medium in the wells with single colonies.
- One day before passaging (at day 4 or 5 after single cell plating), plate irradiated MEFs on gelatin-coated 96-well plates at a density of 4 × 104 cells/cm2 in MEF medium. Keep the plates at 37 °C in a 5% CO2 atmosphere.
- After 5 or 6 days of single-cell plating, select wells of the 96-well plates containing single colonies with a diameter larger than 150 µm.
NOTE: Approximately 100 wells are preferably selected to establish a phaESC line with the targeted DMR deletions. - Aspirate the medium in the selected wells and add 30 µL of trypsin. Incubate the 96-well plates at 37 °C in a 5% CO2 atmosphere for 5 min. Then, add 30 µL of wash buffer to each well to quench the trypsin.
- Add 140 µL of haESC medium into each well, and pipette several times to obtain single cells.
- Aspirate the MEF medium from the wells of the 96-well plates prepared in step 3.3.
- Transfer the phaESCs from each well from step 3.6 into a fresh well of the new 96-well plate from step 3.7.
- Incubate the 96-well plates containing phaESC subclones at 37 °C in a 5% CO2 atmosphere.
- On the next day, aspirate all the medium from each well, and add 120 µL of new haESC medium. Incubate the plates at 37 °C in a 5% CO2 atmosphere.
- One day before the cells become confluent for passaging, plate the irradiated MEFs on gelatin-coated 24-well plates at a density of 4 × 104 cells/cm2 in MEF medium. Keep the plates at 37 °C in a 5% CO2 atmosphere.
- When phaESCs have become confluent for passaging, aspirate the medium and add 30 µL of trypsin. Incubate the 96-well plates at 37 °C for 5 min.
- Add 90 µL of wash buffer into each well to quench the trypsin. Pipette several times to obtain single cells.
- Aspirate the MEF medium from the wells of the 24-well plates from step 3.11, and add 600 µL of fresh haESC medium.
- Transfer 60 µL of the suspension of phaESC subclones from each well in step 3.13 into a new well of the 24-well plates from step 3.14. Keep the remaining suspension of each phaESC subclone for DNA extraction and genotyping in step 4.
- Incubate the 24-well plates with the phaESC subclones at 37 °C in a 5% CO2 atmosphere.
- One day before the cell cultures reach the density for passaging, plate the irradiated MEFs on gelatin-coated 6-well plates at a density of 4 × 104 cells/cm2 in MEF medium. Keep the plates at 37 °C in a 5% CO2 atmosphere.
- When phaESCs become confluent enough for passaging, aspirate the medium and add 250 µL of trypsin. Incubate the 24-well plates at 37 °C for 5 min.
NOTE: After genotyping in step 4.9, only phaESC lines with deletions of both the H19-DMR and the IG-DMR need to be passaged. - Add 750 µL of wash buffer into each well to quench the trypsin. Pipette several times to obtain a single cell suspension. Transfer the cell suspension into a 15 mL tube.
- Centrifuge the tube at 160 x g for 5 min, remove the supernatant, and resuspend the cell pellet in 2 mL of haESC medium.
- Aspirate the MEF medium from the wells of the 6-well plates from step 3.17. Transfer the phaESC suspension from each tube from step 3.20 into a new well. Keep the plates at 37 °C in a 5% CO2 atmosphere.
- Expand the cell clones by repeating steps 3.17 to 3.21 and increasing the plate size and the volumes of trypsin, wash buffer, and haESC medium. Prepare a T25 flask for each sub-cloned phaESC line of MEFs for step 5.
NOTE: We recommend freezing an aliquot of each sub-cloned phaESC line in 300 µL of freezing medium and keeping a cryostock in liquid nitrogen storage before proceeding to step 5.
4. First genotyping of sub-cloned phaESC lines with MEFs
- To extract genomic DNA from the remaining cell suspension from step 3.15, add 200 µL of lysis buffer to each well of the 96-well plates. Transfer the cell suspension to a 1.5 mL tube. Rinse each well with an additional 200 µL of lysis buffer to recover all remaining cells and collect in the same 1.5 mL tube.
- Incubate the 1.5 mL tube at 55 °C for 3 h with mixing.
- After incubation, add 460 µL of isopropanol to each 1.5 mL tube, and mix gently until a DNA precipitate becomes visible.
- Centrifuge the tubes at ≥ 10,000 x g for 5 min and remove the supernatant. Wash the DNA pellets with 200 µL of 70% ethanol.
- Centrifuge the tubes at ≥ 10,000 x g for 5 min and remove the supernatant.
- Dry the tubes in air for 10 min and then resuspend the DNA in 20 µL of water.
- Perform polymerase chain reaction (PCR) using thermostable DNA polymerase following the manufacturer’s protocol.
NOTE: The primer pairs for PCR are listed in Table 2 and are used as follows: H19-DMR-P1 and P3 (407 bp for deleted H19-DMR); H19-DMR-P2 and P3 (623 bp for wildtype H19-DMR); IG-DMR-P1 and P3 (319 bp for deleted IG-DMR); IG-DMR-P2 and P3 (492 bp for wildtype IG-DMR). The temperature profile of PCR for all primer pairs is as follows: 30 s 98 °C, 35 x (10 s 98 °C, 20 s 56 °C, 30 s 72 °C), 5 min 72 °C. The length of amplified DNA fragments for the H19-DMR and IG-DMR deletions shows some variation because of non-homologous end joining associated with CRISPR/Cas9-mediated editing. PhaESCs were cultured with MEFs, which contain wildtype H19-DMR and IG-DMR DNA. Therefore, primer pairs of H19-DMR-P2/P3 and IG-DMR-P2/P3, which amplify wildtype DNA fragments, are not informative. However, these primer pairs are included as controls and should give a band in all reactions. - Analyze the PCR fragments by agarose gel electrophoresis. Refer to the published protocol on the detailed procedure of the electrophoresis16.
- Identify cell lines with deletions of both H19-DMR and IG-DMR. A representative image of electrophoresis is shown in Figure 2B.
NOTE: In our case, eight cell lines with deletions of both H19-DMR and IG-DMR were identified among 135 sub-cloned phaESC lines.
5. Haploid cell purification of sub-cloned phaESC lines
- When the sub-cloned phaESC cultures in the T25 flasks from step 3.22 become dense enough for passaging, aspirate the medium and add 1.5 mL of trypsin. Incubate the flask at 37 °C for 5 min. Then, add 4.5 mL of wash buffer and pipette several times to obtain a single-cell suspension.
- Transfer each cell suspension into a 15 mL tube and centrifuge the tube at 160 x g for 5 min. Remove the supernatant and resuspend the cell pellets in 400 µL of haESC maintenance buffer supplemented with 15 µg/mL Hoechst 33342.
- Incubate the cell suspensions at 37 °C for 15 min. After the incubation, transfer the cell suspensions into a 5 mL tube through a cell strainer cap. Rinse the cell strainer cap with an additional 400 µL of haESC maintenance buffer, and collect the remaining cells in the same 5 mL tube. Keep the tube at 4 °C until ready to sort.
- Set up a flow cytometer with a 100 µm nozzle according to the manufacturer’s instructions.
NOTE: Hoechst 33342 can be detected by excitation at 405 nm. Here, a 355 nm UV laser is used for detection of Hoechst 33342. - Set up the cell suspension (from step 5.3) and a new 15 mL tube containing 2 mL of haESC maintenance buffer to collect sorted cells in the flow cytometer.
- Start the analysis and set up the sorting gate to collect haploid cells in the G1/S phase. Refer to the histogram in Figure 2A for identifying the G1/S phase phaESC population.
NOTE: Some sub-cloned phaESC lines may not contain any haploid cells because of complete diploidization or erroneous plating of diploid cells in step 2. If haploid cells are not observed in the G1/S phase, proceed to another sample without sorting. In our case, 5 cell lines contained haploid cells and 3 cell lines contained only diploid cells out of 8 sub-cloned phaESC lines. - After cell sorting, add 5 mL of wash buffer along the wall of the 15 mL collection tube. Centrifuge the tube at 160 x g for 5 min. Remove the supernatant.
- Select a plate of suitable size for culturing depending on the number of sorted cells. Use a single well of a 96-well plate, a 24-well plate, and a 12-well plate to culture 1,000–40,000 cells, 40,000–200,000 cells, and 200,000–400,000 cells, respectively. Resuspend the cell pellet in 120 µL, 600 µL, and 1.2 mL of haESC medium, respectively.
NOTE: Plate the cells at high density because low confluence can cause cell death of the cells after sorting. From this point onwards, phaESCs are cultured on gelatin-coated wells without MEFs to facilitate genotyping in step 6 and the subsequent application for intracytoplasmic injection from step 9. - After transferring the cell suspension into a gelatin-coated well of the appropriate size, incubate the plate at 37 °C in a 5% CO2 atmosphere.
- Continue expanding the phaESC cultures by repeating steps 3.18 to 3.21 with increasing plate sizes and increasing volumes of trypsin, wash buffer, and haESC medium. The cells are cultured on gelatin-coated wells without MEFs.
- For each sub-cloned phaESC line, prepare a culture in one well of a 24-well plate and one well of a 6-well plate for steps 6 and 9, respectively.
NOTE: Some cells of each sub-cloned phaESC line should be frozen in 300 µL of freezing medium as a cryostock in a liquid nitrogen storage tank before proceeding to step 9.
6. Second genotyping of sub-cloned phaESC lines without MEFs
NOTE: A second round of genotyping is performed to confirm that the sub-cloned phaESC lines possess deletions of both the H19- and IG-DMRs, and that wildtype alleles are absent after the removal of MEFs.
- Confirm under the microscope that the cultures in wells of the 24-well plates from step 5.11 are free from MEFs.
NOTE: If MEFs are observed, it is necessary to continue passaging the cultures until MEFs have disappeared to avoid contaminating the PCR with wildtype DNA from MEFs. - Aspirate the medium from confluent cultures and add 400 µL of lysis buffer per well of the 24-well plate. After pipetting several times, transfer the cell suspension to a 1.5 mL tube.
- Incubate the 1.5 mL tube at 55 °C for 3 h with mixing.
- After incubation, add 400 µL of isopropanol to the 1.5 mL tube, and mix gently until a DNA precipitate becomes visible.
- Centrifuge the tube at ≥ 10,000 x g for 5 min and remove the supernatant. Wash the DNA pellet with 200 µL of 70% ethanol.
- Centrifuge the tube at ≥ 10,000 x g for 5 min and remove the supernatant.
- Dry the tube in air for 10 min and then resuspend the DNA in 50 µL of water.
- Perform the genotyping PCR following step 4.7 and gel electrophoresis in step 4.8 to identify cell lines, which possess deletions of both the H19- and IG-DMRs and are free from wildtype alleles.
NOTE: An image of a typical second genotyping analysis is shown in Figure 2B for reference. In our case, all 5 cell lines selected after haploid cell purification (step 5) were free from wildtype alleles and possessed only the deletion alleles of the H19- and IG-DMRs. - Use the sub-cloned phaESC lines selected after this second genotyping as DKO-phaESC lines.
7. Superovulation of mice
- For production of MII oocytes, initiate superovulation by intraperitoneal injection of 5 IU of pregnant mare serum gonadotropin (PMSG) solution into each B6D2F1 female mouse (4–5 weeks old) 63–65 h before oocyte collection.
NOTE: The B6D2F1 mouse strain is recommended for this protocol because B6D2F1 oocytes tolerate intracytoplasmic injection well and show high developmental potential after the procedure17. - Forty-eight hours after PMSG injection, intraperitoneally inject 5 IU of human chorionic gonadotropin solution into each mouse.
8. Oocyte collection
- Prepare a 4-well plate containing 700 µL of hyaluronidase medium in one well and 700 µL of M2 medium in the remaining 3 wells. Additionally, prepare a 6-cm dish with 7 mL of M2 medium and a center-well dish with 900 µL of M16 medium. Pre-warm the plate and dishes at 37 °C in a 5% CO2 atmosphere.
- On the day of the intracytoplasmic injection, euthanize the superovulated females (from step 7.2) by either cervical dislocation or CO2 inhalation at around 8 AM in the morning. Dissect the oviducts using tweezers and scissors. Place the oviducts in the 6-cm dish with M2 medium.
- Release the cumulus-oocyte complexes (COCs) by tearing the ampulla of the oviducts with a 30 G needle. Transfer COCs into pre-warmed hyaluronidase medium and keep at 37 °C in a 5% CO2 atmosphere.
- After 2–3 min, collect cumulus-free oocytes with a mouth pipette and wash the oocytes 3 times by transferring them to fresh M2 medium in the other 3 wells of the 4-well plate.
- Collect metaphase II (MII) oocytes, which possess first polar bodies, in a center-well dish with M16 medium and keep the plate at 37 °C in a 5% CO2 atmosphere until use for intracytoplasmic injection in step 12.
9. Treatment and collection of DKO-phaESCs
- Prepare a DKO-phaESC culture in a well of a 6-well plate without MEFs at 60–80% confluency a day before the intracytoplasmic injection (from step 5.11).
- To induce cell cycle arrest in M-phase, aspirate the medium completely and add 2 mL of haESC medium containing 0.05 mg/mL demecolcine.
- After 8 h of demecolcine treatment, aspirate the medium and add 800 µL of trypsin.
- Incubate the plate at 37 °C in a 5% CO2 atmosphere for 5 min, then add 2 mL of wash buffer to quench the trypsin, and pipette several times to obtain a single-cell suspension.
- Transfer the cell suspension into a 15 mL tube. Centrifuge the tube at 160 x g for 5 min and remove the supernatant.
- Resuspend the cell pellet in 400 µL of haESC maintenance buffer containing 15 µg/mL Hoechst 33342.
- Incubate the cell suspension at 37 °C in a 5% CO2 atmosphere for 15 min. After the incubation, transfer the cell suspension into a 5 mL tube through a cell strainer cap, and keep the tube at 4 °C until cell sorting in step 10.
10. Purification of M-phase-arrested DKO-phaESCs
- Set up a flow cytometer with a 100 µm nozzle according to the manufacturer’s instructions.
NOTE: Hoechst 33342 can be detected by excitation at 405 nm. Here, a 355 nm UV laser is used for detection of Hoechst 33342. - Set up the M-phase-arrested DKO-phaESCs from step 9.7 and start the analysis. Select a suitable sorting gate for collecting the haploid M-phase cells (2n) from the sample treated with demecolcine.
NOTE: After demecolcine treatment, 2 cell populations are expected, corresponding to haploid and diploid M-phase-arrested cells as shown in Figure 3B. The cell cycle arrest after demecolcine treatment is complete, thus, no haploid 1n DNA peak is observed. This is important as the haploid M-phase cells and diploid G1 cells possess the same DNA content (2n) and would produce overlapping peaks. - Set up a 15 mL tube containing 2 mL of haESC maintenance buffer to collect the sorted cells in the flow cytometer. Start cell sorting.
- After cell sorting, add 5 mL of wash buffer along the wall of the collection tube. Centrifuge the tube at 160 x g for 5 min and remove the supernatant.
- Resuspend the cells in an appropriate volume of haESC maintenance buffer to obtain a final concentration of 5 x 105 cells/mL.
- Transfer the cell suspension into a 1.5 mL tube. Keep the tube on ice until ready for intracytoplasmic injection in step 12.
11. Preparation of holding and microinjection pipettes
NOTE: For performing the intracytoplasmic injection (step 12), several holding and microinjection pipettes are required (Figure 4A). These pipettes can be purchased on tailor-made demand from a commercial supplier or made from suitable glass capillaries using a micropipette puller and a microforge.
- Pull borosilicate glass capillaries on a micropipette puller. To pull borosilicate glass capillaries without filament (0.78 x 1.00 x 80 mm) the following parameters are given for a flaming horizontal puller (Table of Materials) as a reference, but will differ for other instruments and glass capillary types: Heat 510 (Ramp test 480), Pull 0, Velocity 150, Time 175 and Pressure 200 for holding pipettes; Heat 510 (Ramp test 480), Pull 90, Velocity 140, Time 125 and Pressure 200 for microinjection pipettes.
NOTE: The optimal parameters needs to be defined individually because several factors including humidity, the model of a micropipette puller and the lot of the glass capillaries can affect the shape of the injection pipettes. An elongated shape with a gradual taper should be aimed at. - Preparation of holding pipettes
- Set one pulled capillary prepared in step 11.1 to a microforge. Place the capillary over the glass bead on the filament and lower the capillary to make a contact with the glass bead while heating the filament.
- Break the capillary by turning off the heating and detaching from the glass bead such that its outer diameter is 60–100 µm.
- Position the capillary tip horizontally to face the glass bead on the filament.
- Heat the filament to allow the inner diameter of the capillary tip to melt to a diameter of 10–20 µm.
- Move the capillary so that the glass bead positions at a point ~1 mm from the capillary tip without contact. Heat the filament to allow the capillary to bend at a 20° angle. Dismount the capillary, termed a holding pipette, from the microforge.
NOTE: To measure the size of the capillary, an eyepiece reticle is preferably installed in the microforge.
- Preparation of microinjection pipettes
- Set one pulled capillary prepared in step 11.1 to a microforge. Place the capillary over the glass bead on the filament and lower the capillary to make a contact with the glass bead while heating the filament.
- Break the capillary by turning off the heating and detaching from the glass bead at a position that its outer diameter is 6 µm.
- Move the capillary so that the glass bead positions at a point ~1 mm from the capillary tip without contact. Heat the filament to allow the capillary to bend upwards at a 20° angle. Dismount the microinjection pipette from the microforge and store in a secure box for later use.
NOTE: The microinjection pipettes are prepared with the following specifications: outer diameter, 6 µm; inner diameter, 4.5–5 µm; bend angle, 20°. Defining the optimal design of the microinjection pipette is important for the success of the intracytoplasmic injection. Too large an inner diameter can prevent the rupturing of the plasma membrane of the donor DKO-phESCs (see discussion section). If the inner diameter is too narrow, it can impede smooth pipetting of the donor DKO-phESCs. A bend angle < 30° is preferable as a high bend angle impedes the effect of piezo pulses.
12. Intracytoplasmic injection of DKO-phaESCs
- Prior to the intracytoplasmic injection (from step 12.2), prepare a polyvinylpyrrolidone (PVP) solution by adding 5 mL of M2 medium into a 50 mL tube containing 0.6 g of PVP and agitating the tube on a rocker at 4 °C for 2 days. After the PVP has dissolved completely, the solution is sterile-filtered and stored at 4 °C.
- On the day of the intracytoplasmic injection, prepare a center-well dish with 900 µL of KSOM medium and pre-warm the dish at 37 °C in a 5% CO2 atmosphere.
- Prepare a micromanipulation dish by aligning drops of 5 µL of PVP solution and 20 µL of M2 medium on a lid of a 10-cm dish that is placed upside-down. Cover the drops with mineral oil, and place the dish on the stage of the injection microscope.
NOTE: The recommended arrangement of the micromanipulation dish is shown in Figure 4B. - Install a holding pipette onto the micromanipulator. Fill the microinjection pipette with the fluorocarbon oil by using a microloader tip, and mount it on the piezo actuator.
- Immerse the microinjection pipette in a drop with PVP solution and pipette up and down several times to coat the glass with PVP and make it less sticky. Load a small volume of PVP solution into the microinjection pipette, and move the pipette to a drop with M2 medium.
- Immerse the holding pipette in the M2 medium, and focus on the pipette in the bottom of the drop.
- Transfer approximately 2 µL of DKO-phaESC suspension from step 10.6 into the M2 medium drop.
- Transfer 10 MII oocytes from step 8.5 into the same M2 medium drop by using a mouth pipette.
- For injection, rotate an oocyte in the M2 medium drop so that the perivitelline space faces the microinjection pipette, and the MII plate is not located in the path of the microinjection pipette (Figure 4A). Hold the oocyte by applying negative pressure through the holding pipette.
NOTE: An MII plate is visually identified as a protrusion of ooplasm that is referred to as a “hump” and often located next to the first polar body. The MII plate contains the meiotic spindle with attached chromosomes. Touching of the microinjection pipette and the MII plate must be avoided as mechanical damage of the spindle and chromosomes can disrupt embryo development. - Load one DKO-phaESC into the tip of the microinjection pipette by applying gentle negative pressure. Rupture the plasma membrane of a DKO-phaESC by pipetting to avoid the injection of an intact DKO-phaESC (Figure 3C; see discussion).
NOTE: In case the plasma membrane of a DKO-phaESC is not ruptured by pipetting, discard the DKO-phaESC and load another DKO-phaESC. - Place the microinjection pipette in contact with the zona pellucida of the oocyte, and apply a small amount of negative pressure within the microinjection pipette.
- Apply piezo impulses (intensity, 20; frequency, 4) to break through the zona while pushing the tip of the microinjection pipette towards the perivitelline space. Confirm that the MII plate, containing a spindle and chromosomes, is not located in the path of the microinjection pipette.
NOTE: Empirically adjust the setting to the lowest piezo pulses for drilling through the zona to minimize the possibility of damage to the oolemma. - Discard the fragment of the zona pellucida from the microinjection pipette, and position the DKO-phaESC at the edge of the pipette.
- Penetrate the oocyte with the microinjection pipette so that the oolemma reaches the opposite side.
NOTE: Do not touch the MII plate to prevent damage to the spindle and chromosomes. - Apply a piezo pulse (intensity, 6; frequency, 1) to pierce the oolemma. Ensure that the oolemma relaxes along the shaft of the microinjection pipette.
NOTE: Empirically define the lowest setting of the piezo pulse for breaking the oolemma to minimize the damage to the oocyte. - Inject the DKO-phaESC with a minimal volume of medium into the ooplasm, and withdraw the microinjection pipette smoothly from the oocyte.
- Release the injected oocyte from the holding pipette, and place it on the side of the microdrop for later collection.
- Repeat the injection procedure from steps 12.9 to 12.17 for the other MII oocytes in the M2 medium drop.
NOTE: Avoid keeping the oocytes out of the incubator for more than 20 min. In our experience, a batch of 10 oocytes can be manipulated comfortably within 15 min after appropriate training. - Transfer the batch of injected oocytes from the M2 medium drop to a pre-warmed center-well dish with KSOM medium.
- Keep the dish for 1 h at 37 °C in a 5% CO2 atmosphere.
- Repeat the oocyte manipulation from steps 12.5 and 12.20 with additional groups of MII oocytes to obtain enough injected oocytes.
13. Activation of constructed semi-cloned embryos
- Prepare two center-well dishes with 900 µL each of KSOM medium and activation medium. Prepare a 4-well plate with 700 µL of KSOM medium in each well. Pre-warm the dishes and the plate at 37 °C in a 5% CO2 atmosphere.
- After 1 h in KSOM medium, transfer the injected oocytes from step 12.21 into the pre-warmed center-well dish with activation medium.
- Keep the dish for 6 h at 37 °C in a 5% CO2 atmosphere for activation.
- After activation, observe some semi-cloned embryos form three polar bodies, which are the first and the second polar bodies of the oocyte, and the pseudo polar body from the DKO-phaESC (Figure 3E).
- Wash the embryos 3 times by transferring them to new wells with KSOM medium in a 4-well plate.
- Transfer the embryos into the center-well dish with KSOM medium, and keep the dish at 37 °C in a 5% CO2 atmosphere for further development.
14. Development of constructed semi-cloned embryos
- After 1 day of culture in KSOM medium from step 13.6, several semi-cloned embryos reach the 2-cell stage (Figure 5A).
- For further development of preimplantation embryos in vitro, continue culturing the semi-cloned embryos in KSOM medium at 37 °C in a 5% CO2 atmosphere. Transfer the semi-cloned embryos to fresh KSOM medium at day 2. At day 4, several embryos will reach the blastocyst stage (Figure 5A).
- For derivation of semi-cloned mice, transfer 2-cell embryos from step 14.1 into the oviducts of pseudo-pregnant recipient females. Identify pseudo-pregnant females by mating to vasectomized males a day before the embryo transfer and select them on the basis of the presence of a clearly visible plug in the morning of the day for the embryo transfer (0.5 days post-coitum (dpc)). Around 19.5 dpc, full-term pups are naturally delivered from recipient females (Figure 5B).
Results
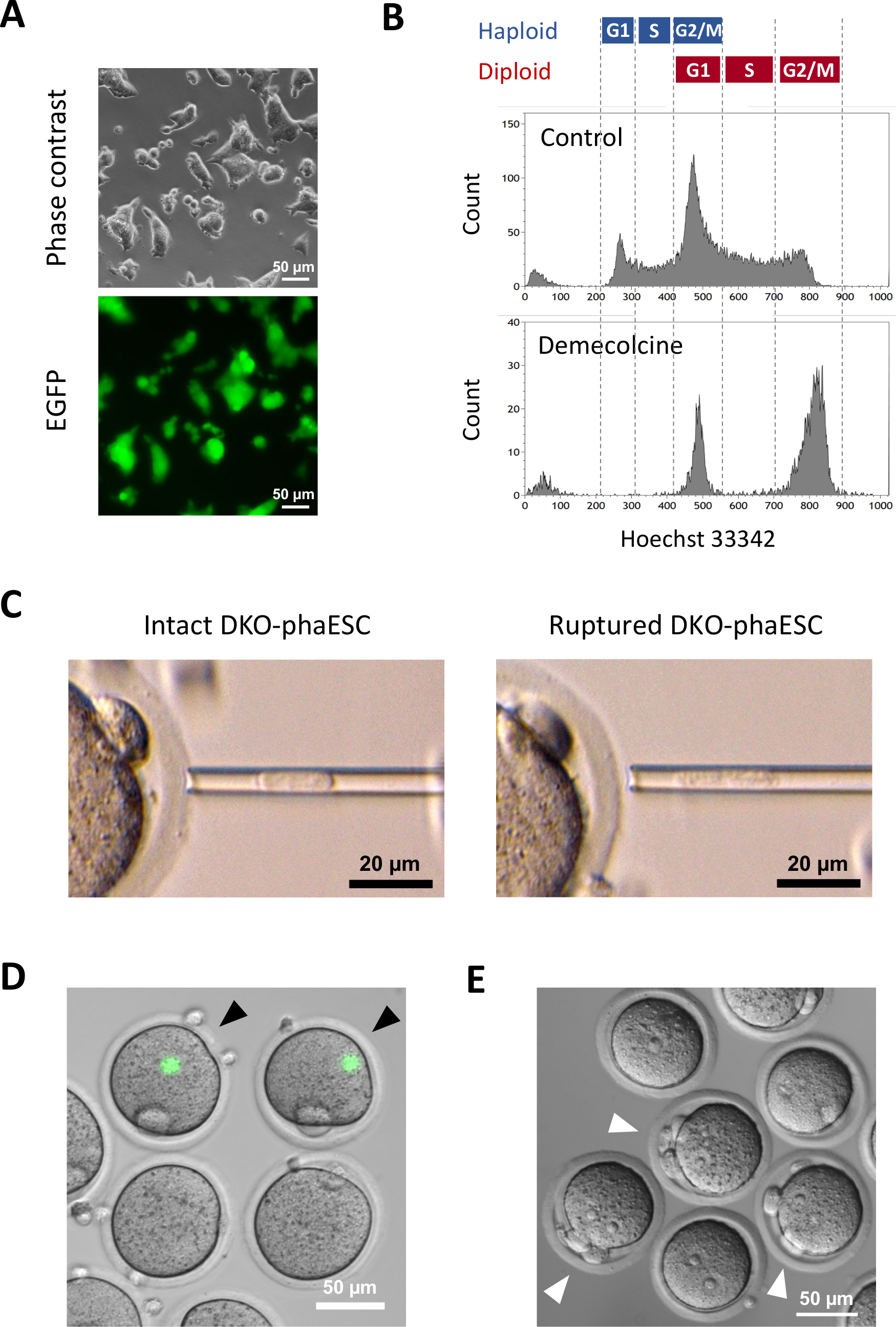
The purpose of this protocol is to apply haESCs as a substitute of sperm to obtain semi-cloned embryos and mice. For this purpose, a DKO-phaESC line carrying the CAG-EGFP transgene was generated and used for intracytoplasmic injection into MII oocytes. To obtain a suitable phaESC line with a paternal imprint configuration, we performed genetic engineering using Cas9 nucleases. A haploid ESC line contains haploid and diploid cells that arise due to an inherent tendency of haploid ESCs for diploidization10. A haploid chromosome set is a prerequisite for successful replacement of the sperm genome. DNA content analysis by flow cytometry shows the distribution of haploid and diploid cells at G0/G1-, S-, and G2/M-phases (Figure 2A).
To establish the DKO-phaESC lines, wildtype phaESC lines were transfected with piggyBac transposon constructs for stable transgenic EGFP expression and with CRISPR/Cas9 plasmids for obtaining deletions of the H19- and IG-DMRs. To exclude diploid ESCs and isolate only haploid ESCs expressing EGFP, a specific sort gate was defined (Figure 2A). Single haploid EGFP-positive cells were then plated into individual wells of 96-well plates to obtain sub-clones. MEF feeders were used to increase the plating efficiency and survival of the transfected phaESCs. After expansion of the cultures, an initial round of genotyping was carried out by PCR to identify sub-clones that carry deletions of both DMRs. After MEF feeders had been removed from the cultures, a second genotyping was performed to confirm the absence of wildtype alleles at the H19- and IG-DMRs (Figure 2B). From a total of 135 sub-clones, we obtained 5 haploid DKO-phESC lines that carried the deleted alleles and were free of wildtype alleles of both the H19-DMR and the IG-DMR.
A CAG-EGFP transgene was introduced into DKO-phaESCs to study their contribution to semi-cloned embryos by visualization of green fluorescence under a microscope (Figure 3A). For the intracytoplasmic injection, DKO-phaESCs were treated with demecolcine to arrest them in M-phase. Thus, the cell cycle of DKO-phaESCs was synchronized with that of MII oocytes. Flow cytometry analysis showed 2 populations corresponding to G2/M phase arrested haploid (2n) and diploid cells (4n) after the treatment with demecolcine (Figure 3B). Absence of the 1n haploid peak indicated that the cell cycle arrest was largely complete. M-phase haploid ESCs were then sorted and injected into oocytes. For this, a single DKO-phaESC was loaded into the microinjection pipette and injected into the cytoplasm of a MII oocyte (Figure 3C). The plasma membrane of the DKO-phaESC was ruptured by pipetting into the tip of the microinjection pipette.
After the injection, EGFP expression was rarely detected in the constructed semi-cloned embryos as the cytoplasm of DKO-phaESCs had dispersed in the large cytoplasm of the oocyte (Figure 3D). In rare cases, a round spot of intense EGFP expression could be observed within the ooplasm. This observation was likely caused by the inadvertent injection of intact DKO-phaESCs. Failure to rupture the DKO-phaESC cell membrane is likely not compatible with further embryo development and should be avoided. One hour after the injection, embryos were activated by treatment with strontium chloride18. Six hours after the initiation of activation, up to 3 polar bodies were observed under the microscope (Figure 3E). These polar bodies correspond to the first and second polar bodies of the oocyte and a pseudo polar body from the DKO-phaESC7. In addition, two pronuclei were observed under a differential interference contrast microscope, which resembled the pronuclear stage of zygotes after normal fertilization with sperm.
To demonstrate developmental competence, semi-cloned embryos were cultured to the blastocyst stage (Figure 5A). Furthermore, a full-term mouse was obtained after transferring semi-cloned 2-cell stage embryos into the oviducts of a recipient female (Figure 5B). As expected, the mouse derived from a semi-cloned embryo was a female as neither oocytes nor oocyte-derived phaESCs carry a Y chromosome. The semi-cloned mouse was overtly normal and produced healthy offspring when mated with a wildtype Swiss Webster male. Until now, the semi-cloned mouse has been alive for over 600 days without any apparent health problems.

Figure 1: Overview of the application of DKO-phaESCs as sperm replacement. (A) A time frame of the procedures of the protocol. (B) The scheme shows the steps to establish DKO-phaESC lines (steps 1–6). (C) The scheme shows the steps to construct semi-cloned embryos by intracytoplasmic injection of a DKO-phaESC into a MII oocyte (steps 7–14). Abbreviations: DKO = double-knockout; phaESC = parthogenetic haploid embryonic stem cell; FACS = fluorescence-activated cell sorting; MEF = mouse embryonic fibroblast. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Establishment of DKO-phaESC lines by CRISPR/Cas9-mediated deletions of H19- and IG-DMRs. (A) Flow cytometry analysis of phaESCs after transfection with piggyBac plasmids for stable EGFP expression and with 4 CRISPR/Cas9 plasmids. Non-transfected phaESCs are shown as control. The DNA profile (top) shows the cell cycle distribution of haploid and diploid cells. G1/S-phase haploid cells expressing EGFP are indicated by the green gate (bottom, right). (B) Genotyping of phaESC sub-clones that were grown on MEFs (first genotyping) and after removal of MEFs (second genotyping). Sub-cloned phaESC lines 1, 2, 3, and 4 represent wildtype cells, cells with a H19-DMR deletion, with an IG-DMR deletion, and with combined H19- and IG-DMR deletions, respectively. DKO-phaESC lines 5–8 possess both H19-DMR and IG-DMR deletions and are free from wildtype alleles. Abbreviations: DKO = double-knockout; phaESC = parthogenetic haploid embryonic stem cell; DMR = differentially methylated region; MEF = mouse embryonic fibroblast; EGFP = enhanced green fluorescent protein; WT = wildtype. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Injection of mitotically arrested DKO-phaESCs into MII oocytes. (A) Morphology of a DKO-phaESC culture that carries a CAG-EGFP transgene and deletions of the H19- and IG-DMRs; scale bar = 50 µm. (B) Representative flow cytometry analysis of DKO-phaESCs after arrest with demecolcine for 8 h. DKO-phaESCs without demecolcine treatment are shown as control. (C) Morphology of DKO-phaESCs in the microinjection pipette. A single intact DKO-phaESC before (left) and after (right) rupturing the plasma membrane by pipetting is shown; scale bar = 20 µm. (D) Constructed embryos at 1 h after injection of DKO-phaESCs into MII oocytes. A merged image of EGFP fluorescence and bright field is shown. Black arrowheads indicate round spots of intense EGFP expression after injection of intact DKO-phaESCs, which should be avoided; scale bar = 50 µm. (E) A differential interference contrast image of semi-cloned embryos 6 h after the initiation of activation with strontium chloride is shown. White arrowheads indicate embryos with 3 polar bodies including one pseudo polar body from the injected DKO-phaESC; scale bar = 50 µm. Abbreviations: DKO = double-knockout; phaESC = parthogenetic haploid embryonic stem cell; EGFP = enhanced green fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Scheme of the setup for intracytoplasmic injection of DKO-phaESCs into oocytes. (A) The arrangement of an injection pipette, a holding pipette, and an oocyte in the injection chamber is shown. α, bend angle of the microinjection pipette. (B) Layout of the drops in the micromanipulation dish for intracytoplasmic injection. Abbreviations: DKO = double-knockout; phaESC = parthogenetic haploid embryonic stem cell. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Development of semi-cloned embryos. (A) Preimplantation development of semi-cloned embryos in vitro. EGFP expression is initially observed in four-cell embryos at day 2 after intracytoplasmic injection. At day 4, intense EGFP expression can be observed in blastocysts; scale bar = 100 µm. (B) A semi-cloned mouse obtained after 2-cell embryo transfer to a recipient female. At 74 dpp, the semi-cloned mouse (arrowhead) delivered her first pups (asterisk) by natural birth after mating with a wildtype Swiss Webster male. Semi-cloned embryos and the semi-cloned mouse shown in this figure are identical with ones reported in Aizawa et al.3. Abbreviations: EGFP = enhanced green fluorescent protein; .dpp, days post-partum. Please click here to view a larger version of this figure.
{kind=link}
| Gelatin solution | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| Water | - | 500 mL | - |
| Gelatin | - | 1 g | 0.2% |
| Haploid embryonic stem cell (HaESC) medium | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| NDiff 227 | - | 10 mL | - |
| CHIR 99021 | 10 mM | 3 µL | 3 µM |
| PD 0325901 | 10 mM | 1 µL | 1 µM |
| LIF | 1 x 106 IU/mL | 10 µL | 1,000 IU/mL |
| Penicillin-Streptomycin | 100x | 100 µL | 1x |
| HaESC maintenance buffer | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| HaESC medium | - | 1 mL | - |
| HEPES solution | 1 M | 20 µL | 20 mM |
| Wash buffer | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| DMEM / F-12 | - | 100 mL | - |
| BSA fraction | 7.5% | 7.1 mL | 0.5% |
| MEF medium | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| DMEM | - | 500 mL | - |
| FBS | - | 56 mL | 10% |
| β-Mercaptoethanol | 14.3 mol/L | 3.9 µL | 100 µM |
| Penicillin-Streptomycin | 100x | 5.6 mL | 1x |
| Lysis buffer | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| Water | - | 8.25 mL | - |
| Tris-HCl (pH 8.5) | 1 M | 1 mL | 100 mM |
| EDTA | 0.5 M | 100 µL | 5 mM |
| SDS solution | 10% | 200 µL | 0.2% |
| NaCl | 5 M | 400 µL | 200 mM |
| Proteinase K | 20 mg/mL | 50 µL | 100 µg/mL |
| Activation medium | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| KSOM | - | 1 mL | - |
| Strontium chloride | 1 M | 5 µL | 5 mM |
| EGTA | 0.5 M, pH 8.0 | 4 µL | 2 mM |
| PVP solution | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| M2 medium | - | 5 mL | - |
| PVP | - | 0.6 g | 12% |
| PMSG solution | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| PBS | - | 450 µL | - |
| PMSG | 500 IU/mL | 50 µL | 50 IU/mL |
| hCG solution | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| PBS | - | 450 µL | - |
| hCG | 500 IU/mL | 50 µL | 50 IU/mL |
| Hyaluronidase medium | |||
| Component | Stock concentration | Volume / weight | Final concentration |
| M2 medium | - | 380 µL | - |
| Hyaluronidase | 10 mg/mL | 20 µL | 0.5 mg/mL |
| Abbreviations: LIF = leukemia inhibitory factor; MEF = mouse embryonic fibroblast; | |||
| FBS = fetal bovine serum; DMEM = Dulbecco's Modified Eagle Medium; BSA = bovine serum albumin; | |||
| EDTA = ethylenediaminetetraacetic acid; SDS = sodium dodecylsulfate; EGTA = ethylene-bis(oxyethylenenitrilo)tetraacetic acid; | |||
| PBS = phosphate-buffered saline; PVP = polyvinylpyrrolidone; hCG = human chorionic gonadotropin; | |||
| PMSG = pregnant mare serum gonadotropin. | |||
Table 1: Recipe of medium, buffer, and solution.
| Name | Sequence (5' to 3') | Application | |
| H19-DMR-P1 | GTG GTT AGT TCT ATA TGG GG | Genotyping | |
| H19-DMR-P2 | AGA TGG GGT CAT TCT TTT CC | Genotyping | |
| H19-DMR-P3 | TCT TAC AGT CTG GTC TTG GT | Genotyping | |
| IG-DMR-P1 | TGT GCA GCA GCA AAG CTA AG | Genotyping | |
| IG-DMR-P2 | CCA CAA AAA CCT CCC TTT CA | Genotyping | |
| IG-DMR-P3 | ATA CGA TAC GGC AAC CAA CG | Genotyping | |
| H19-DMR-gRNA1-F | CAC CCA TGA ACT CAG AAG AGA CTG | gRNA | |
| H19-DMR-gRNA1-R | AAA CCA GTC TCT TCT GAG TTC ATG | gRNA | |
| H19-DMR-gRNA2-F | CAC CAG GTG AGA ACC ACT GCT GAG | gRNA | |
| H19-DMR-gRNA2-R | AAA CCT CAG CAG TGG TTC TCA CCT | gRNA | |
| IG-DMR-gRNA1-F | CAC CCG TAC AGA GCT CCA TGG CAC | gRNA | |
| IG-DMR-gRNA1-R | AAA CGT GCC ATG GAG CTC TGT ACG | gRNA | |
| IG-DMR-gRNA2-F | CAC CCT GCT TAG AGG TAC TAC GCT | gRNA | |
| IG-DMR-gRNA2-R | AAA CAG CGT AGT ACC TCT AAG CAG | gRNA | |
Table 2: List of oligonucleotides.
Discussion
Cloning of mammals by somatic cell nuclear transfer (SCNT) has been pioneered in the 1990s19,20,21. These developments followed cloning studies conducted 30 years earlier in amphibians22. The considerable delay reflects the difficulty of embryology and genomic imprinting in mammals. The development of mammalian SCNT is the basis for the application of haESC for substituting sperm, which is detailed in this protocol.
Cell cycle synchronization is an important factor for the success of SCNT23. This is also the case for injection of haESC in this protocol. Introducing a donor genome into a recipient requires the cell cycle phases to be matched to avoid chromosomal breakage or aneuploidies that would abrogate embryo development. Semi-cloning has the additional complexity that two genomes and a cytoplast need to be compatible. A previous report has demonstrated that injection of M-phase-arrested androgenetic haESCs yielded better developmental rates of semi-cloned embryos than injection of G1-phase haESCs into oocytes7. This report suggests M-phase as a suitable synchronization point for semi-cloning. Accordingly, phaESCs were mitotically arrested in metaphase with demecolcine and injected into the ooplasm of MII oocytes, which were naturally arrested in metaphase II of meiosis. Importantly, M-phase arrest can be achieved in mouse ESCs with high efficiency, providing excellent synchronization between donor and recipient cell cycles.
During mitosis, the nuclear membrane breaks down and a spindle forms to which the replicated chromosomes attach. After injection of M-phase DKO-phaESCs, sister chromatids are segregated into a pseudo polar body and the zygote7 (Figure 3E). Consequently, a single set of chromosomes from a DKO-phaESC contributes to the semi-cloned embryo. It is critical that the sister chromatids of DKO-phaESC chromosomes can be correctly segregated after injection. The plasma membrane of an intact DKO-phaESC prevents segregation into a pseudo polar body. We did indeed observe rare cases where the plasma membrane of DKO-phaESCs was not ruptured, and embryos exhibited intact DKO-phaESCs in the ooplasm after injection (Figure 3D). Therefore, care must be taken to remove the plasma membrane of DKO-phaESCs by pipetting. During injection, it is equally important to avoid the disruption of the meiotic spindle of the oocyte, which could lead to chromosome segregation defects and induce aneuploidy as well.
In mammals, genomic imprinting limits the application of phaESCs as sperm replacement. Parthenogenetic haESCs possess a maternal configuration of genomic imprints, whereas sperm possess a paternal configuration. Therefore, generation of full-term pups has not occurred after the injection of wildtype phaESCs as sperm replacement. To overcome this limitation, deletions of the IG- and H19-DMRs are engineered in phaESCs. Modification of imprinted expression at the maternal Igf2-H19 and Gtl2-Dlk1 loci is sufficient to change the configuration of genomic imprints for allowing the generation of semi-cloned mice with a frequency of over 5.1%, based on transferred 2-cell embryos. These observations suggest that targeting two imprinted genes switches the genome of phaESCs into a functional paternal configuration that can replace sperm in mice. Nevertheless, a permanent genetic modification of the phaESCs is required for this strategy. As an alternative strategy, androgenetic haESC can be considered. Androgenetic haESCs are derived from the sperm genome and possess paternal imprints. There have been reports that wildtype androgenetic haESCs contributed as sperm replacement to generate full-term pups at a frequency between 1.3% and 1.9% of transferred embryos4,7,24. Full-term pups have also been obtained by injecting androgenetic haESCs with deletions of the IG- and H19-DMRs at a frequency of 20.2% of transferred 2-cell embryos24. The increased efficiency of semi-cloning using modified androgenetic haESCs is likely because imprints can become unstable in culture. Imprinting defects are corrected by the genetic deletions of critical DMRs.
Considering the difficulty in introducing genetic modifications directly into the oocyte or sperm genomes, haESCs are a valuable tool for manipulating the parental genomes separately. Using haESCs as a substitute of sperm provides a remarkable advantage for genome editing in the mouse germline. A recent study has combined CRISPR/Cas9-based genome editing with the application of haESCs for the characterization of imprinting regions that are critical for embryonic development12. This study analyzed the role of the Rasgrf1-DMR in combination with the H19-and IG-DMRs in the development of bimaternal mice, and the function of 7 different DMRs in the development of bipaternal mice. The method for substituting haESCs for sperm formed the basis for genetic screening approaches for identifying key amino acids within the DND1 protein in primordial germ cell development and for identifying genes in bone development24,25,26. Studies on genomic imprinting and genetic screening to identify key factors in embryonic development are considerable approaches for the application of haESCs as gametic genomes.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Dr. Giulio Di Minin for derivation of phaESC lines and Dr. Remo Freimann for flow cytometry operation. We also acknowledge Ms. Michèle Schaffner and Mr. Thomas M. Hennek for technical support with embryo transfer. This work was supported by the Swiss National Science Foundation (grant 31003A_152814/1).
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mLTube | Eppendorf | 0030 125.150 | haESC culture |
| 15 ml Tube | Greiner Bio-One | 188271 | haESC culture |
| 12-well plate | Thermo Scientific | 150628 | haESC culture |
| 24-well plate | Thermo Scientific | 142475 | haESC culture |
| 6-well plate | Thermo Scientific | 140675 | haESC culture |
| 96-well plate | Thermo Scientific | 167008 | haESC culture |
| β-Mercaptoethanol | Merck | M6250 | haESC culture |
| BSA fraction | Gibco | 15260-037 | haESC culture |
| CHIR 99021 | Axon | 1386 | haESC culture |
| Demecolcine solution | Merck | D1925 | haESC culture |
| DMEM | Gibco | 41965-039 | haESC culture |
| DMEM / F-12 | Gibco | 11320-033 | haESC culture |
| DR4 mouse | The Jackson Laboratory | 3208 | haESC culture |
| EDTA | Merck | E5134 | haESC culture |
| FBS | biowest | S1810-500 | haESC culture |
| Freezing medium | amsbio | 11897 | haESC culture |
| Gelatin | Merck | G1890 | haESC culture |
| Hepes solution | Gibco | 15630-056 | haESC culture |
| Irradiated MEF | Gibco | A34966 | haESC culture |
| LIF (Leukemia inhibitory factor) | (Homemade) | - | haESC culture |
| Lipofection reagent | Invitrogen | 11668019 | haESC culture |
| NDiff 227 | Takara | Y40002 | haESC culture |
| PD 0325901 | Axon | 1408 | haESC culture |
| Pen Strep | Gibco | 15140-122 | haESC culture |
| piggyBac plasmid carrying a CAG-EGFP transgene | Addgene | #40973 | haESC culture |
| piggyBac transposase plasmid | System Biosciences | PB210PA-1 | haESC culture |
| pX330 plasmid | Addgene | #42230 | haESC culture |
| pX458 plasmid | Addgene | #48138 | haESC culture |
| Trypsin | Gibco | 15090-046 | haESC culture |
| DNA polymerase | Thermo Scientific | F549S | Genotyping |
| dNTP Mix | Thermo Scientific | R0192 | Genotyping |
| Ethanol | Merck | 100983 | Genotyping |
| Hydrochloric acid | Merck | 100317 | Genotyping |
| Isopropanol | Merck | 109634 | Genotyping |
| Proteinase K | Axon | A3830 | Genotyping |
| SDS | Merck | 71729 | Genotyping |
| Sodium chloride | Merck | 106404 | Genotyping |
| ThermoMixer | Epeendorf | 5384000012 | Genotyping |
| Tris | Merck | T1503 | Genotyping |
| 5 mL Tube | Falcon | 352063 | Flow cytometry |
| 5 mL Tube with cell strainer cap | Falcon | 352235 | Flow cytometry |
| Hoechst 33342 | Invitrogen | H3570 | Flow cytometry |
| MoFlo Astrios EQ | Beckman Coulter | B25982 | Flow cytometry |
| 1 mL syringe | Codan | 62.1640 | Superovulation |
| 30-gauge 1/2 inch needle | Merck | Z192362-100EA | Superovulation |
| B6D2F1 mouse | The Jackson Laboratory | 100006 | Superovulation |
| hCG | MSD animal health | 49451 | Superovulation |
| PBS | Gibco | 10010015 | Superovulation |
| PMSG | Avivasysbio | OPPA1037 5000 IU | Superovulation |
| 10-cm dish | Thermo Scientific | 150350 | Embryo manipulation |
| 4-well plate | Thermo Scientific | 179830 | Embryo manipulation |
| 50 mL tube | Greiner Bio-One | 227261 | Embryo manipulation |
| 6 cm dish | Thermo Scientific | 150288 | Embryo manipulation |
| Center-well dish | Corning | 3260 | Embryo manipulation |
| Digital rocker | Thermo Scientific | 88880020 | Embryo manipulation |
| EGTA | AppliChem | A0878 | Embryo manipulation |
| Hyaluronidase | Merck | H4272 | Embryo manipulation |
| KSOM | Merck | MR-020P-5F | Embryo manipulation |
| M16 medium | Merck | M7292 | Embryo manipulation |
| M2 medium | Merck | M7167 | Embryo manipulation |
| Mineral oil | Merck | M8410 | Embryo manipulation |
| Glass capillary | Merck | P1049-1PAK | Embryo manipulation |
| Stereomicroscope | Nikon | SMZ745 | Embryo manipulation |
| Strontium chloride | Merck | 13909 | Embryo manipulation |
| 5ml syringe | Codan | 62.5607 | Microinjection |
| Borosilicate glass cappilary | Science product | GB100T-8P | Microinjection |
| CellTram 4r Oil | Eppendorf | 5196000030 | Microinjection |
| Fluorinert FC-770 | Merck | F3556 | Microinjection |
| Holding pipette | BioMedical Instruments | - | Microinjection |
| Inverted microscope | Nikon | Eclipse Ti-U | Microinjection |
| Microforge | Narishige | MF-900 | Microinjection |
| Microinjection pipette | BioMedical Instruments | - | Microinjection |
| Microlaoder tips | Eppendorf | 5252 956.003 | Microinjection |
| Micromanipulaor arms | Narishige | NT-88-V3 | Microinjection |
| Micropipette puller | Sutter Instrument | P-1000 | Microinjection |
| PiezoXpert | Eppendorf | 5194000016 | Microinjection |
| PVP (Polyvinylpyrrolidine) | Merck | PVP360 | Microinjection |
| Syringe filter | SARSTEDT | 83.1826.001 | Microinjection |
References
- Ellegren, H., Galtier, N. Determinants of genetic diversity. Nature Reviews: Genetics. 17 (7), 422-433 (2016).
- Huijbers, I. J. Generating genetically modified mice: A decision guide. Methods in Molecular Biology. 1642, 1-19 (2017).
- Aizawa, E., Dumeau, C. E., Freimann, R., Di Minin, G., Wutz, A. Polyploidy of semi-cloned embryos generated from parthenogenetic haploid embryonic stem cells. PloS One. 15 (9), 0233072 (2020).
- Li, W., et al. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature. 490 (7420), 407-411 (2012).
- Li, Z., et al. Birth of fertile bimaternal offspring following intracytoplasmic injection of parthenogenetic haploid embryonic stem cells. Cell Research. 26 (1), 135-138 (2016).
- Wan, H., et al. Parthenogenetic haploid embryonic stem cells produce fertile mice. Cell Research. 23 (11), 1330-1333 (2013).
- Yang, H., et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 149 (3), 605-617 (2012).
- Zhong, C., et al. Parthenogenetic haploid embryonic stem cells efficiently support mouse generation by oocyte injection. Cell Research. 26 (1), 131-134 (2016).
- Elling, U., et al. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell. 9 (6), 563-574 (2011).
- Leeb, M., Wutz, A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 479 (7371), 131-134 (2011).
- Surani, M. A., Barton, S. C., Norris, M. L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 308 (5959), 548-550 (1984).
- Li, Z. K., et al. Generation of bimaternal and bipaternal mice from hypomethylated haploid ESCs with imprinting region deletions. Cell Stem Cell. 23 (5), 665-676 (2018).
- Elling, U., et al. Derivation and maintenance of mouse haploid embryonic stem cells. Nature Protocols. 14 (7), 1991-2014 (2019).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Shuai, L., et al. Generation of mammalian offspring by haploid embryonic stem cells microinjection. Current Protocols in Stem Cell Biology. 31, 1-15 (2014).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. Journal of Visualized Experiments. (62), e3923 (2012).
- Behringer, R., Gertsenstein, M., Nagy, K. V., Nagy, A. . Manipulating the Mouse Embryo: A Laboratory Manual. , (2014).
- O'Neill, G. T., Rolfe, L. R., Kaufman, M. H. Developmental potential and chromosome constitution of strontium-induced mouse parthenogenones. Molecular Reproduction and Development. 30 (3), 214-219 (1991).
- Campbell, K. H., McWhir, J., Ritchie, W. A., Wilmut, I. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 380 (6569), 64-66 (1996).
- Wakayama, T., Perry, A. C., Zuccotti, M., Johnson, K. R., Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature. 394 (6691), 369-374 (1998).
- Wilmut, I., Schnieke, A. E., McWhir, J., Kind, A. J., Campbell, K. H. Viable offspring derived from fetal and adult mammalian cells. Nature. 385 (6619), 810-813 (1997).
- Gurdon, J. B. Adult frogs derived from the nuclei of single somatic cells. Developmental Biology. 4, 256-273 (1962).
- Campbell, K. H., Alberio, R. Reprogramming the genome: role of the cell cycle. Reproduction Supplement. 61, 477-494 (2003).
- Zhong, C., et al. CRISPR-Cas9-mediated genetic screening in mice with haploid embryonic stem cells carrying a guide RNA library. Cell Stem Cell. 17 (2), 221-232 (2015).
- Bai, M., et al. Targeted genetic screening in mice through haploid embryonic stem cells identifies critical genes in bone development. PLoS Biology. 17 (7), 3000350 (2019).
- Li, Q., et al. CRISPR-Cas9-mediated base-editing screening in mice identifies DND1 amino acids that are critical for primordial germ cell development. Nature Cell Biology. 20 (11), 1315-1325 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved