
Method Article
Isolation of Primary Cancer-Associated Fibroblasts from a Syngeneic Murine Model of Breast Cancer for the Study of Targeted Nanoparticles
* These authors contributed equally
In This Article
Summary
This paper aims to provide a protocol for the isolation and culture of primary cancer-associated fibroblasts from a syngeneic murine model of triple-negative breast cancer and their application for the preclinical study of novel nanoparticles designed to target the tumor microenvironment.
Abstract
Cancer-associated fibroblasts (CAFs) are key actors in the context of the tumor microenvironment. Despite being reduced in number as compared to tumor cells, CAFs regulate tumor progression and provide protection from antitumor immunity. Emerging anticancer strategies aim to remodel the tumor microenvironment through the ablation of pro-tumorigenic CAFs or reprogramming of CAFs functions and their activation status. A promising approach is the development of nanosized delivery agents able to target CAFs, thus allowing the specific delivery of drugs and active molecules. In this context, a cellular model of CAFs may provide a useful tool for in vitro screening and preliminary investigation of such nanoformulations.
This study describes the isolation and culture of primary CAFs from the syngeneic 4T1 murine model of triple-negative breast cancer. Magnetic beads were used in a 2-step separation process to extract CAFs from dissociated tumors. Immunophenotyping control was performed using flow cytometry after each passage to verify the process yield. Isolated CAFs can be employed to study the targeting capability of different nanoformulations designed to tackle the tumor microenvironment. Fluorescently labeled H-ferritin nanocages were used as candidate nanoparticles to set up the method. Nanoparticles, either bare or conjugated with a targeting ligand, were analyzed for their binding to CAFs. The results suggest that ex vivo extraction of breast CAFs may be a useful system to test and validate nanoparticles for the specific targeting of tumorigenic CAFs.
Introduction
During the past decades, it has become clear that killing tumor cells is usually not sufficient to eradicate malignancy, as the tumor microenvironment may prompt tumor relapse and induce therapeutic resistance1,2. A novel paradigm has then emerged: targeting tumor stroma to deprive the tumor of supporting factors and thus, boost the efficacy of chemotherapeutics3,4,5. In particular, cancer-associated fibroblasts (CAFs) are an interesting stromal target in many solid tumors6,7. CAFs are a very heterogeneous group of cells that interact with cancer cells and cells of the immune system through the secretion of growth factors, cytokines, and chemokines; build up and remodel the extracellular matrix; and enable metastasis formation8,9,10,11,12. Depending on the tumor type, CAFs show pro-tumorigenic functions, while other subtypes of CAFs seem to have tumor-suppressive functions13,14. To better clarify this dichotomy, a thorough characterization of CAFs from primary and metastatic tumors is important.
In this context, an emerging field of research has focused on the development of nanosized agents designed to target and/or destroy CAFs by delivering active molecules and drugs able to remodel the tumor microenvironment15,16,17,18. Several types of nanoparticles have been designed to achieve CAF ablation by cytotoxic drugs, to induce CAF-targeted photodynamic therapy, or to reprogram CAFs by reverting them to a quiescent state or inducing TNF-related apoptosis induced ligand expression, which induces apoptosis of neighboring cancer cells16,19. Moreover, the potential of many nanoparticles to actively target specific biological markers gives rise to the hope of selecting CAF subsets to target. Although its absolute specificity for CAF is still questioned, fibroblast activation protein (FAP) is one of the most promising targets of pro-tumorigenic stroma and is exploited to steer nanodrug delivery, thus paving the path for the development of CAF-targeting nanotherapeutics20,21,22.
This paper describes the isolation of primary CAFs from a syngeneic model of murine breast cancer and reports their use in the study of the targeting capability of nanoparticles engineered to recognize the CAF marker, FAP. Ferritin nanocages are used as candidate nanosystems to set up the method, as their specificity of delivery may be shaped by the surface exposure of targeting moieties23,24. Moreover, ferritins have been successfully proven to be excellent biocompatible shuttles for antitumor applications, triggering rapid accumulation of the payload in the tumor mass25,26,27. To date, preclinical studies of CAF-targeting nanosystems have involved in vitro testing on fibroblast cell lines stimulated in culture with transforming growth factor-beta to induce cell activation and the expression of some immunophenotypic features of CAFs28,29. This method is usually applied to immortalized cell lines (such as NIH3T3, LX-2) and is quite rapid and simple, yielding activated cells in a few hours or days. A limitation is that although in vitro stimulation induces the expression of some genes attributed to activated myofibroblasts, it cannot entirely recapitulate all the biological features of real CAFs, especially their heterogeneity in vivo.
Another strategy involves the extraction of primary CAFs from human or mouse tumor samples30,31. This ensures that CAF activation occurs in a physiological context, and that the heterogeneity of CAF subpopulations is maintained. According to the research objective, CAFs may be derived from different sources, thus offering the possibility to study the most reliable condition. The protocol reported here would be valuable for scientists who seek to perform a preliminary evaluation of the functionality of novel nanoparticles designed to target CAFs from a murine breast cancer model. Isolated CAFs would be useful for screening those nanoparticles that are promising enough to proceed for in vivo evaluation in animal models of cancer. This will be relevant during the first steps of nanoparticle production, driving nanotechnologists toward the refinement of nanoparticle design by mainly considering the strategy of ligand immobilization to achieve optimal targeting properties.
Protocol
1. Establishing a syngeneic 4T1 model of breast cancer
NOTE: The present protocol describes the isolation of primary CAFs from a mouse 4T1 breast tumor. The animal study described here has been approved by the Italian Ministry of Health (aut. number 110/2018-PR).
- Tumor cell culture and implantation
- Thaw 1 × 106 4T1-luc cells in a T75 flask with 10 mL of Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 1% L-glutamine. Add Mycoplasma removal agent (1:100) in the culture medium.
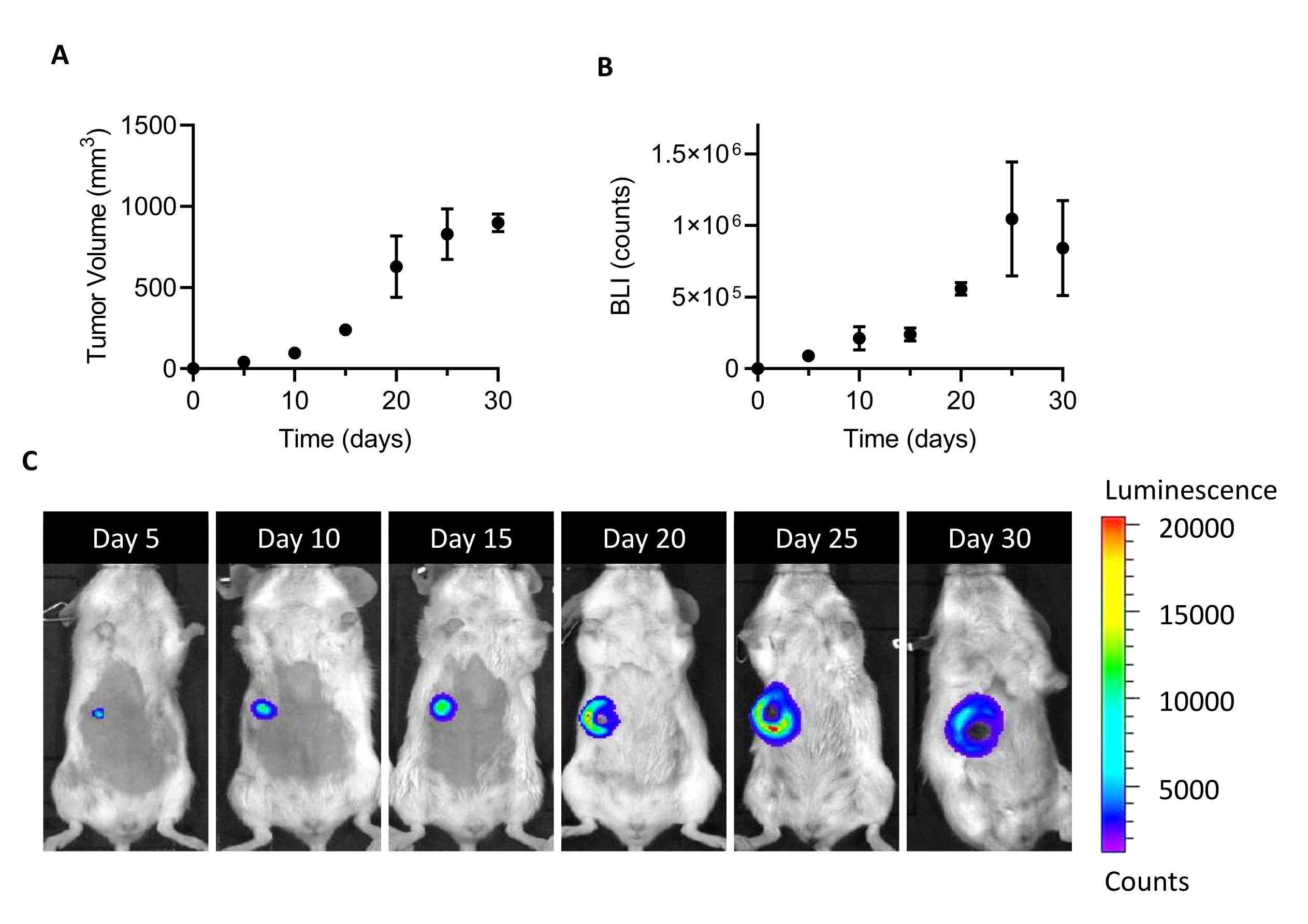
NOTE: 4T1-luc cells stably express luciferase and can be visualized by bioluminescence (BLI) upon proper stimulation with D-luciferin. This allows in vivo monitoring of the viability and proliferation of tumor cells upon implantation in mice (Figure 1). - Maintain the cells at 37 °C and 5% CO2 in a humidified atmosphere until ~80% confluence, by changing the medium every 2 days.
NOTE: Continue treatment with Mycoplasma removal agent for 1 week (~2/3 passages) before injection in mice to ensure that injected cells are mycoplasma-free. - On the day of the procedure, detach cells using 1 mL of trypsin-ethylenediaminetetraacetic acid (EDTA) solution (trypsin 1:250) for 5 min at 37 °C. Stop trypsin activity by adding culture medium, count the cells with trypan blue (1:1), and calculate the number of cells/mL.
- Pipette the volume corresponding to 1 × 106 cells and centrifuge for 5 min at 400 × g. Pour off the supernatant, and resuspend the pellet in 1 mL of RPMI 1640 base medium. Keep the cells on ice until ready for injection.
NOTE: For each mouse, 1 × 105 cells are needed; however, always calculate for an excess of 2 mice (corresponding to 2 × 105 cells) to ease syringe loading. - One day before the operation, shave the fur of 8 BALB/c mice (female, seven-week-old) to expose the area of the abdominal mammary glands on the right side. Use an electric shaver and apply a thin layer of depilatory cream for 4 min; then, wash away the cream with water and a piece of paper.
- Induce anesthesia by continuous inhalation of 2% isoflurane gas for ~5 min, and perform a subcutaneous injection in the region of the abdominal mammary glands with a 27 G tuberculin syringe. Turn the needle upwards to enter the skin subcutaneously; then, hold the skin upwards and slowly inject 100 μL of cell suspension (1 × 105 cells). Slightly turn the needle around before slowly retracting the syringe.
NOTE: Remember to bring the cell suspension to room temperature 15 min before injection.
- Thaw 1 × 106 4T1-luc cells in a T75 flask with 10 mL of Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 1% L-glutamine. Add Mycoplasma removal agent (1:100) in the culture medium.
- Tumor growth and dissection
- At 5 days post-cell-injection, inject 150 mg/kg D-luciferin intraperitoneally (100 μL) 5 min before imaging. Capture BLI images using an in vivo imaging system setting 10 s exposure, medium binning, and f/stop at 4. Define the luminescent area of the tumor as the region of interest (ROI), and quantify the total signal in the ROI (photon/sec/m2) using imaging software.
- Repeat the procedure of imaging (1.2.1) every 5 days from the injection to monitor tumor growth in terms of an increase in BLI.
- To establish the tumor volume, hold the mouse and expose its belly. Use calipers to measure the tumor length (L) and width (W) once a week. Calculate the tumor volume (V) using equation 1.
[V = (L x W2)/2] (1) - At 20 days post-cell-injection, sacrifice the animals by cervical dislocation, dissociate the tumors from the skin with scissors, and collect them in a tissue storage solution (see the Table of Materials).
NOTE: Tumor samples may be used immediately for dissociation into single cells (section 2) or stored at 4 °C for up to 48 h.
2. Tumor dissociation into single cells
NOTE: For the following steps, use sterile reagents and disposables in a laminar flow hood. Work with 4 tumors at a time.
- Place the tumor sample in a Petri dish, and carefully remove any fragment of skin, fat, and necrotic areas with the help of tweezers and scalpel. Then, reduce the tumor into small pieces of approximately 1–2 mm and transfer them to a tube (Figure 2A).
NOTE: 4T1 tumors become highly necrotic while growing, with a tendency to ulceration. It is important to carefully remove the necrotic areas to avoid any interference of debris with the next steps. - Prepare a digestion mix for tumor dissociation: mix 2.35 mL of RPMI 1640 medium, 100 μL of enzyme D, 50 μL of enzyme R, and 12.5 μL of enzyme A. Soak the tumor fragments in the solution and tightly close the tube.
NOTE: The indicated volumes of digestion mix are valid for tumors up to 1 g and may be adjusted for larger tumors according to their weight. 4T1 tumors grown for 20 days normally are in a range of 0.5–0.8 g. - Turn the tube upside down, checking that all the pieces of tumor stand in the bottom of the tube toward the cap. Attach the tube to a mechanical dissociator in the proper housing, and run a specific dissociation program designed for tough tumors (see the manufacturer’s instructions).
NOTE: Special tubes may be required to fit into the dissociator. C-tubes bear a rotor inside the cap to promote mechanical dissociation of the tissue. - Detach the tube, maintain it upside down, and incubate the sample at 37 °C for 40 min with gentle shaking. Then, attach the tube to the dissociator in the proper housing, and run the following specific dissociation program designed for tough tumors two times. Ensure there are no large pieces of tissue at the end of the procedure (Figure 2B).
NOTE: To dissociate the tumor, other enzyme cocktails could be used to degrade the extracellular matrix. However, in this protocol, a commercially available Tumor Dissociation kit containing an optimized enzyme mix (enzymes D, R, A) and a semi-automated dissociator that mechanically dissociates the tissue were used. This combination ensured a proper degradation of the extracellular matrix together with maintenance of cell integrity and cell surface epitopes. - Filter the sample through a 40 μm cell strainer in a 50 mL tube, wash the filter with 10 mL of RPMI 1640 medium, and centrifuge the tube for 7 min at 300 × g. If the cell pellet appears red, lyse erythrocytes by adding 1 mL of Ammonium-Chloride-Potassium (ACK) lysing buffer for 5 min at room temperature, wash with 10 mL of RPMI 1640, transfer the cell suspension in a 15 mL tube, and centrifuge again.
- Resuspend the pellet in 1 mL of PBE buffer composed of phosphate-buffered saline (PBS), 0.5% bovine serum albumin (BSA), and 2 mM EDTA, and count the cells with trypan blue (1:1). In case of cell clumps, filter the cell suspension through a 70 μm cell strainer, previously soaked with PBS.
NOTE: Especially when tumors are big, it is important to check the cell count and eventually to split each tumor sample in different tubes with no more than 107 cells in each tube before proceeding to step 2.7. - Prepare the dead cell removal binding buffer by diluting the 20x binding buffer stock solution (see the Table of Materials) with sterile double-distilled water. Wash cells with 5 mL of 1× binding buffer (Figure 2C).
NOTE: Keep the buffer at 4 °C. - Centrifuge for 7 min at 300 × g, and resuspend the cell pellet with 0.1 mL of dead cell removal microbeads (see the Table of Materials). Mix well and incubate at room temperature for 15 min.
- During the incubation with beads, prepare a magnetic stand under the hood and hang ferromagnetic separation columns (one column for up to 107 cells) on it with the tips pointing down. Equilibrate the columns with 0.5 mL of cold 1× binding buffer. Wait until the solution has flowed through under gravity.
NOTE: It is important to not exceed the number of cells per column to avoid any risk of clotting the column bed and reducing the recovery yield. When working with more than 107 cells, either use more columns according to the total number of cells or use bigger columns. In such cases, adjust the reagent volumes described in the following steps to the ones indicated by the manufacturer. - At the end of incubation, add 400 μL of cold 1× binding buffer to the bead/cell suspension, load the entire volume onto the column, and collect the effluent (corresponding to unlabeled cells) in a 15 mL tube.
- Wash the column four times with 0.5 mL of cold 1× binding buffer, and collect the total effluent (corresponding to the live cell fraction) in the same tube.
- Count the cells with trypan blue (1:1). Place 1 × 105 cells in two tubes for Fluorescence Activated Cell Sorting (FACS), and keep them at 4 °C until process validation (see step 4.1): use the first tube to set the analysis parameters and the regions of positivity (control tube) and the other one for biomarker analysis (sample tube). Use the remaining cells for CAFs extraction (see section 3).
NOTE: Removal of dead cells is important to reduce non-specific reactions with the microbeads in section 3. However, to improve the yield, avoid the removal of dead cells if the proportion of dead cells is <20%.
3. Extraction of primary CAFs from breast tumor
NOTE: For section 3, use the mouse Tumor-Associated Fibroblast Isolation kit containing Non-Tumor-Associated Fibroblast Depletion Cocktail and Tumor-Associated Fibroblast Microbeads suited for magnetic labeling of the cells (see the Table of Materials).
- Depletion of non-cancer-associated fibroblasts
- Moisten a 70 μm cell strainer with PBS, and filter the cell suspension to remove any clumps. Then, centrifuge the cells for 10 min at 300 × g and aspirate the supernatant.
NOTE: If a single tumor was divided into two samples for step 2.7, pool the cell suspension two by two before proceeding with step 3.1.2. - Resuspend the pellet in 80 μL of cold PBE buffer, and add 20 μL of Non-Tumor-Associated Fibroblast Depletion Cocktail. Mix well and incubate at 4 °C for 15 min in the dark.
- During the incubation with beads, prepare ferromagnetic depletion columns (1 per sample) on a magnetic stand, and equilibrate the columns with 2 mL of cold PBE buffer. Wait until the solution has flowed through.
- At the end of incubation, add 400 µL of cold PBE buffer to the bead/cell suspension, load the entire volume onto the column, and collect the effluent (corresponding to unlabeled cells) in a 15 mL tube.
- Wash the column twice with 2 mL of cold PBE buffer, and collect the total effluent in the same tube. Centrifuge for 10 min at 300 × g and resuspend in 0.1 mL of cold PBE buffer.
NOTE: Depletion of non-cancer-associated fibroblasts drastically reduces the total amount of cells recovered. It is generally necessary and recommended to pool cell suspensions from four different samples to obtain a visible pellet and enough cells for step 3.2. This will ensure an optimal yield. - Place 20 µL of the cell suspension in a FACS tube and keep it at 4 °C for process validation (see section 4). Use the remaining cells for CAF selection (see step 3.2).
- Moisten a 70 μm cell strainer with PBS, and filter the cell suspension to remove any clumps. Then, centrifuge the cells for 10 min at 300 × g and aspirate the supernatant.
- Positive selection of cancer-associated fibroblasts
- Add 20 μL of Tumor-Associated Fibroblast Microbeads to 80 μL of cell suspension. Mix well and incubate at 4 °C for 15 min in the dark.
- During incubation with beads, prepare ferromagnetic separation columns (1 per sample) on a magnetic stand, and equilibrate the columns with 0.5 mL of cold PBE buffer. Wait until the solution has flowed through.
- At the end of the incubation, add 400 μL of cold PBE buffer to the bead/cell suspension, load the entire volume onto the column, and let the unlabeled cells flow through into a 15 mL tube.
- Wash the column three times with 0.5 mL of cold PBE buffer, and collect the total effluent in the same tube.
NOTE: The flow-through contains unlabeled CD90.2-negative cells, which may be discarded as non-cancer-associated fibroblasts. - Remove the column from the magnet and place it in a 1.5 mL tube. Add 1 mL of cold PBE onto the column, and immediately push the plunger into the column to flush the cells out.
- Centrifuge for 10 min at 400 × g and resuspend in 0.1 mL of cold PBE buffer. Place 20 μL of cell suspension in a FACS tube and keep it at 4 °C for process validation (see section 4).
NOTE: After centrifuging, the pellet may be hardly visible, especially when the original tumors are small. Use conical bottom centrifuge tubes and be careful not to lose any cells when aspiring the supernatant. - Dilute the remaining cells in an appropriate volume of Dulbecco’s Modified Eagle’s Medium (DMEM)/Ham’s F-12 supplemented with 15% FBS, 2 mM L-glutamine, 1% P/S, and 1% non-essential amino acids, and seed the cells in a tissue-culture plate.
- Check the cell density under a microscope, and place the plate in an incubator at 37 °C and 5% CO2 to let the cells adhere and grow.
NOTE: If the cells are too dense, immediately split the cell suspension into two wells of the tissue culture plate before placing the plate in the incubator.
4. Process validation
- Flow cytometry
NOTE: Section 4.1 does not require sterile conditions and can be performed outside the laminar hood. Perform section 4.1 in parallel for samples collected in step 2.11 (dissociated tumor), step 3.1.6 (after depletion of non-cancer-associated fibroblasts), and step 3.2.6 (after enrichment of cancer-associated fibroblasts).- Centrifuge the FACS tubes prepared in steps 2.11, 3.1.6, and 3.2.6 for 10 min at 300 × g. Discard the supernatant.
NOTE: Prepare at least one additional tube, which contains unstained cells acting as a control to set the parameters for analysis. - In sample tubes, resuspend cells in 88 µL of PBE, and add 2 µL of anti-CD45 antibody conjugated with fluorescein isothiocyanate (FITC, 1:50 dilution, according to the manufacturer’s instructions) and 10 µL of anti-CD90.2 antibody conjugated with phycoerythrin (PE, 1:10 dilution, according to the manufacturer’s instructions). Mix thoroughly and incubate for 10 min at 4 °C in the dark. In the control tube (unstained), resuspend cells in 100 µL of PBE. Incubate for 10 min at 4 °C in the dark to replicate the procedure followed for antibody-stained cells.
NOTE: Temperature and time of incubation must be checked carefully, as a variation in such variables might result in poor or non-specific staining, thus altering the results. - After completing the incubation step, perform a wash by adding 1 mL of PBE. Centrifuge for 10 min at 300 x g. Discard the supernatant.
- Resuspend cells (all tubes) in 500 µL of PBS.
- Mix well and proceed with flow cytometry analysis. Set the channels to measure the fluorescence of the antibodies (i.e., FITC and PE).
- From the control tube, select all viable cells by drawing a first gate (P1) on the Forward versus Side scatter (FSC vs SSC) plot. Within P1, set a second gate (P2) comprising single cells only.
- Using the specific fluorescence channels of the antibodies (i.e., FITC and PE), set proper gates to discern positively stained cells.
- Start the analysis and record at least 10,000 events in P2. Read signals from both channels simultaneously.
NOTE: If the overall number of cells is less than expected, it could be preferable to read the whole tube. - (OPTIONAL) If further staining with additional antibodies/fluorophores is programmed, set an appropriate compensation matrix before initiating the analysis.
- Centrifuge the FACS tubes prepared in steps 2.11, 3.1.6, and 3.2.6 for 10 min at 300 × g. Discard the supernatant.
- Monitoring of cell morphology and characteristics
NOTE: Once seeded, the cells must be handled under sterile conditions.- On the day after seeding, visualize the cells under an optical microscope to check cell adhesion and morphology. Aspirate the supernatant and replace with fresh medium.
NOTE: CAFs are large spindle-shaped cells, which can be easily distinguished from the epithelial-like structure of 4T1 tumor cells. - Maintain the cells at 37 °C and 5% CO2 in a humidified atmosphere, changing the medium every 2 days.
- When cells reach ~80% confluence (approximately 4-6 days after seeding), detach the cells using TrypLE Select solution (see the Table of Materials) for 5 min at room temperature. Add fresh medium (4:1), centrifuge the cell suspension at 400 × g for 5 min, and resuspend the pellet in fresh medium.
- Split the cells 1:2, and expand the culture until the number of cells needed for the experiment is obtained.
- At each passage, check cell morphology and growth under an optical microscope. If cell morphology is not homogeneous (e.g., clones of small round-shape cells appearing in culture), analyze a sample of cells by flow cytometry as described in section 4.1. If homogeneous, proceed to step 4.2.7.
NOTE: Changes in cell morphology may indicate contamination by non-cancer-associated fibroblasts, which need to be removed to ensure culture purity. - Analyze the flow cytometry results and proceed according to one of the following scenarios: (i) if contamination of CD90.2-/CD45- cells occurs, collect all the cells, and repeat the positive selection by following step 3.2; (ii) if contamination of CD45+ cells also occurs, collect all the cells, and repeat the depletion in step 3.1; (iii) if no contamination occurs (only CD90.2+/CD45- cells are present), keep the cells in culture and proceed directly to 4.2.7.
NOTE: As repeating the depletion process with microbeads will reduce cell recovery, it is recommended only when cells do not need to be used immediately. - Count the cells with trypan blue (1:1), place 5 × 105 cells in two FACS tubes, and centrifuge for 10 min at 300 × g. Discard the supernatant, and resuspend the pellet in 0.5 mL of blocking buffer (PBS supplemented with 2% BSA, 2% goat serum) for 15 min at room temperature.
NOTE: One tube acts as a control consisting of unstained cells only, while cells in the second tube will be stained for flow cytometry analysis. - Centrifuge for 10 min at 300 × g, discard the supernatant and resuspend in 99 µL of blocking buffer. Add 1 µg of anti-FAP antibody to the sample tube and 1 mL of blocking buffer to the control tube. Mix thoroughly and incubate for 15 min at room temperature.
NOTE: FAP is a surface marker of reactive tumor stroma and may be used to identify and characterize CAFs. - Wash thrice with PBS, centrifuge, and incubate with the appropriate secondary antibody conjugated with a fluorescent dye (1 μg) in blocking buffer for 15 min at room temperature.
- Wash thrice with PBS, and analyze by flow cytometry (follow steps 4.1.5 to 4.1.7 by setting the appropriate channel to detect the fluorophore used). Then, use the cells for experiments (step 5.2).
- (OPTIONAL) Freeze a sample of cells in 1 mL of 90% FBS and 10% dimethyl sulfoxide (DMSO); store at -80 °C for further use.
- On the day after seeding, visualize the cells under an optical microscope to check cell adhesion and morphology. Aspirate the supernatant and replace with fresh medium.
5. CAFs targeting by engineered Ferritin nanoparticles
NOTE: A recombinant variant of human ferritin heavy chain (HFn) was used as bare nanoparticle or was conjugated with targeting moieties. Here, HFn nanoparticles functionalized with the variable portion of an anti-FAP antibody (Fab@FAP) were prepared by the NanoBioLab at University of Milano-Bicocca at two HFn:Fab@FAP molar ratios, 1:1 and 1:5, according to a previously described protocol32.
- Fluorescent labeling of HFn nanoparticles
NOTE: Both bare and functionalized HFn nanocages are fluorescently labelled with FITC.- Dissolve the FITC powder in 99% ethanol to obtain a concentration of 2 mg/mL.
NOTE: This solution should be prepared freshly. - Incubate 50 µL of the prepared solution (200 µg of FITC) for every 1 mg of protein (i.e., 100 µL of HFn at 10 mg/mL). Add 50 µL of 1 M sodium bicarbonate (NaHCO3), and adjust the volume to 500 µL with 0.1 M NaHCO3.
NOTE: Scale up the volumes according to the experimental needs. - Incubate the reaction mixture at room temperature, in the dark, and with continuous stirring for 1 h.
- Remove the excess of FITC from the conjugate by gel filtration using a spin desalting column (7 kDa MWCO).
- Assess the concentration of the recovered labelled protein by using a spectrophotometer. Estimate amounts of protein and dye by measuring the absorbance at 280 nm and at 488 nm, respectively.
- Dissolve the FITC powder in 99% ethanol to obtain a concentration of 2 mg/mL.
- Binding of HFn nanoparticles to CAFs
NOTE: Use CAFs from no later than passages 4–5 in culture.- Place 5 × 105 cells in each FACS tube, centrifuge for 10 min at 300 × g, and resuspend in 0.5 mL of PBS, 0.3% BSA supplemented with 0.1 mg/mL of the different preparations of nanoparticles (bare HFn, HFn-Fab@FAP 1:1, HFn-Fab@FAP 1:5) previously labeled with FITC.
NOTE: Run each condition in triplicate. Prepare an additional tube as a control unlabeled sample to which no nanoparticles are added. - Incubate for 2 h at 4 °C in the dark, centrifuge, and wash three times in PBS.
- Resuspend cells with 0.5 mL PBS, mix well, and analyze by flow cytometry. Set the channels to measure the fluorescence of FITC.
- After gating on live single cells, acquire 20,000 events. Use the control unlabeled tube to set the gate of FITC positivity and obtain the percentage of FITC+ events (corresponding to nanoparticle binding).
- Place 5 × 105 cells in each FACS tube, centrifuge for 10 min at 300 × g, and resuspend in 0.5 mL of PBS, 0.3% BSA supplemented with 0.1 mg/mL of the different preparations of nanoparticles (bare HFn, HFn-Fab@FAP 1:1, HFn-Fab@FAP 1:5) previously labeled with FITC.
6. Statistical analysis and experimental replicates
- Animals
- Perform three independent experiments of tumor growth and CAFs isolation, using 8 animals per single experiment, as described in 1.1.5. To optimize CAFs isolation yield, divide the excised tissues into subgroups (n=4) and process them, following steps described in section 2.
- HFn interaction with cells
- Evaluate the interaction of functionalized and non-functionalized HFn with target and non-target cells (CAFs and 4T1, respectively) in terms of the percentage of positively stained cells by fluorescently labelled nanocages. Report the results as average ± standard deviation of three independent experiments.
- Statistical analysis
- To calculate statistical significance in cell binding experiments with HFn and HFn-FAP, use the ordinary one-way ANOVA test.
Results
In vivo model set-up for optimal CAFs isolation
The injection of 105 4T1-luc cells into the mammary fat pad of female BALB/c mice leads to the growth of a detectable tumor mass at 5 days after implantation. By measuring the tumor volume by calipers and the tumor cell viability by BLI, tumor growth was monitored for one month after implantation. To find a sacrifice window that is adequate for CAFs isolation, an optimal compromise was sought between higher tumor size and BLI on the one hand and an emerging tumor ulceration and necrosis on the other hand (Figure 1). As a necrotic core appears 20 days after implantation, and it enlarges at 25 and 30 days (as documented by BLI images in Figure 1C), day 20 was set as the time point to optimize cell recovery after the isolation process. Even after carefully removing all visible necrotic areas during the first steps of tumor handling ex vivo, a high percentage of dead cells was found at the end of dissociation into single cells (Table 1). As this percentage may be relevant, especially with the increase in tumor size, dead cell removal is always necessary when working with the 4T1 model.
Optimization of CAFs isolation procedure, culture, and characterization
Two more passages are needed to isolate the population of CAFs (CD90.2+ CD45-) from the panel of collected viable cells: the depletion of non-tumor-associated fibroblasts and the enrichment of tumor-associated fibroblasts (Figure 2). The depletion bead cocktail efficiently removes CD45+ cells (accounting for 67.35% and 0.69% of total cells pre- and post-depletion, respectively, Table 2), and was always used to process one single tumor in every column. As shown in Table 1, the number of eluted cells dropped from an average of 3 × 106 to 1 × 105 after the depletion step. Due to this massive decrease in total cell number, it is convenient to pool the collected cells from at least 2 to a maximum of 4 tumors in a single tube before proceeding with the enrichment step. By doing so, an adequate number of cells was obtained for incubation with tumor-associated fibroblast beads and passed through a single separation column to obtain a final average of 93% of CD90.2+ CD45- cells (Table 2).
Once seeded onto tissue culture plates, these recovered cells attached to the plastic and revealed a large spindle-shaped morphology typical of fibroblasts (Figure 3A,B) and different from 4T1 tumor cells (Figure 3D). Not pooling tumors after depletion causes the cellular yield with tumor-associated fibroblast microbeads to be too low to establish a culture. In other cases, when the duration and temperature of the incubations with microbeads are not carefully maintained, some non-specific binding may occur. In such cases, the enrichment steps were less efficient and higher percentages of CD90.2- CD45+ and CD90.2- CD45- cells were recovered along with the CD90.2+ CAFs (5.97 ± 1.5 and 16.75 ± 1.1, respectively) (Figure 4 and Table 3). These contaminant cells were likely to be responsible for the presence in culture of small clones with different morphology (Figure 4B, black arrowheads) that grew faster than CAFs and prevailed over the primary CAF culture (Figure 4C). These suboptimal results confirmed the importance of always double-checking both CD90.2 and CD45 expression, as well as cell morphology at the end of the enrichment process and during cell growth in culture.
Use of isolated cells to evaluate CAF-targeting potential of engineered nanodrugs
The freshly isolated CAFs can be used for several applications ranging from basic research to pharmacological studies. This group’s aim is to develop HFn nanocages that can specifically target CAFs. HFn was functionalized with a specific anti-FAP antibody fragment (HFn-FAP) at two different protein:antibody ratios (a lower 1:1 and a higher 1:5), and their binding with CAFs was tested. As FAP was used as a surface biomarker of pro-tumorigenic CAFs, it was of fundamental importance to check FAP expression on isolated CAFs (Figure 3C). FAP expression was followed over 5 passages in culture to confirm that the primary CAF culture maintained its original characteristics.
FAP functionalization on HFn was found to contribute to a significant shift toward CAF targeting as compared to bare HFn, and the lower amount of antibody (1:1) was enough to observe this effect (Figure 5A). However, this was not observed with the tumor 4T1 cells used to set up the in vivo tumor model, wherein bare HFn showed higher binding than functionalized HFn (Figure 5B). This was most likely due to the absence of FAP overexpression in 4T1 cells and the preferential interaction of HFn with TfR1, which regulates HFn uptake in cells, as widely reported by this group27,33. These results confirm the usefulness of using primary cultures of breast CAFs to preliminarily screen the targeting capability of nanoparticles designed to tackle the tumor microenvironment.

Figure 1: Establishment of the 4T1 tumor model. Cells (105 cells/mouse) were injected into the mammary fat pad. Tumor growth was followed at days 5, 10, 15, 20, 25, and 30 after implantation (A) by measuring tumor volume with calipers and (B) by bioluminescence imaging. Tumor volumes and BLI are expressed as mm3 and counts, respectively. Results are reported as average ± SEM (n=6). (C) BLI representative images obtained 5, 10, 15, 20, 25, and 30 days after cell implantation confirm tumor growth until day 25, when it seems to reach a plateau. At the last time point of analysis (30 days), the BLI does not increase as compared to day 25. Starting at day 20, areas of necrosis and ulceration start to become visible in the central part of the tumor. Color Scale: Min = 1,194, Max = 20,462. Abbreviations: BLI = bioluminescence imaging; SEM = standard error of the mean. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Tumor sample preparation and flow cytometry characterization of isolated CAFs. Tumors were excised and reduced into (A) small pieces of approximately 1–2 mm with the help of a scalpel; (B) single cell suspension after tissue digestion and mechanical dissociation of an excised tumor; (C) cell pellet obtained after lysis of red blood cells; (D) flow cytometry analysis of CD45 and CD90.2 expression in cells obtained after removal of red blood cells and dead cells, (E) after depletion of non-cancer-associated fibroblasts, and (F) after enrichment of cancer-associated fibroblasts, where the majority of cells are CD90.2+ CD45- (blue rectangle). Abbreviations: CAFs= cancer-associated fibroblasts; CD = cluster of differentiation; PE-A = area of phycoerythrin; FITC-A = areas of fluorescein isothiocyanate. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Morphological analysis of CAFs and FAP expression. (A, B) CAFs morphology was checked throughout all passages of culture by optical microscopy (different levels of confluence at passage 2 and passage 5) and compared to (D) 4T1 tumor cells; scale bars = 10 µm. (C) Fibroblast activation protein (FAP) expression was evaluated by flow cytometry at the end of the isolation process on the collected CD90.2+ CD45- cells to confirm their molecular characteristics. A fluorescence threshold was set on unstained control cells (red graph) to quantify mean fluorescence intensity and the percentage of positive cells (FAP+) among antibody-stained cells (blue graph). Abbreviations: CAFs= cancer-associated fibroblasts; FAP = fibroblast activation protein; CD = cluster of differentiation; MFI = mean fluorescence intensity; Ab = antibody; FITC-A = area of fluorescein isothiocyanate; FAP+ = FAP-positive cells. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Example of a sub-optimal CAFs isolation process. (A) Flow cytometry evaluation after the final isolation step revealed the presence of contaminant CD90.2- CD45+ and CD90.2- CD45- cells. (B) These cells can be seen as clones with small round morphology upon seeding (black arrowheads and inset in the bottom left corner of the panel) that (C) prevailed over CAFs after the third passage in culture. Scale bars = 10 µm. Abbreviations: CAF = cancer-associated fibroblast; CD = cluster of differentiation; PE-A = area of phycoerythrin; FITC-A = areas of fluorescein isothiocyanate. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Binding of HFn nanocages on CAFs and 4T1. HFn nanocages were fluorescently labeled with FITC, functionalized with an anti-FAP antibody fragment (HFn-FAP) at two different protein-antibody ratios (1:1 and 1:5), and incubated with (A) target CAFs and (B) 4T1 cells at 4 °C for 2 h. Binding was evaluated by flow cytometry. (A) HFn-FAP binding with CAFs is significantly increased at both antibody fragment concentrations by three-fold as compared to bare HFn. (B) in contrast, a significantly higher binding of bare HFn was observed in 4T1 cells, where binding is not enhanced by FAP recognition. Results are reported as average ± standard deviation of three independent experiments. *** p = 0.0003; °° p = 0.0021 ; °°° p = 0.0008. Abbreviations: CAFs= cancer-associated fibroblasts; FAP = fibroblast activation protein; HFn = recombinant variant of human ferritin heavy chain used as bare nanoparticle or conjugated; HFN-FAP = HFn nanoparticles functionalized with the variable portion of an anti-FAP antibody prepared at two HFn:Fab@FAP molar ratios, 1:1 and 1:5. Please click here to view a larger version of this figure.
{kind=link}
| Cell Number (average ± SD) | Extraction Yield Total (per passage) | |
| Excised tumor (Post red blood cell removal) | 1.27 x 108 ± 9.81 x 107 | 100% |
| Excised tumor (Post dead cell removal) | 3.01 x 106 ± 9.61 x 105 | 2.38% |
| Post depletion cocktail | 1.15 x 105 ± 4.95 x 104 | 0.11% (3.82%) |
| Post enrichment | 3.00 x 104 ± 1.2 x 104 | 0.027% (26.09%) |
Table 1: Total cell count after each step of isolation of cancer-associated fibroblasts.
| Cell Distribution (%) | CD90.2+ CD45- | CD90.2+ CD45+ | CD90.2- CD45+ | CD90.2- CD45- |
| Excised tumor (Post dead cell removal) | 1.81 ± 0.98 | 10.78 ± 4.51 | 56.57 ± 14.05 | 28.20 ± 17.57 |
| Post depletion cocktail | 69.33 ± 16.75 | 0.14 ± 0.13 | 0.55 ± 0.63 | 39.89 ± 30.31 |
| Post enrichment | 93.14 ± 3.3 | 0.09 ± 0.1 | 0.09 ± 0.08 | 6.69 ± 3.4 |
Table 2: Cell distribution according to expression of CD90.2 and CD45 after each passage of isolation of cancer-associated fibroblasts.
| Cell Distribution (%) | CD90.2+ CD45- | CD90.2+ CD45+ | CD90.2- CD45+ | CD90.2- CD45- |
| Post Enrichment (no pooling) | 75.80 ± 2.9 | 1.49 ± 0.4 | 5.97 ± 1.5 | 16.75 ± 1.1 |
Table 3: CD90.2 and CD45 expression of cells collected after a sub-optimal cancer-associated fibroblast isolation experiment.
Discussion
CAFs are emerging as key players in remodeling extracellular matrix, promoting metastasis progression, and limiting drug access to the tumor site34. However, due to their heterogeneity, their roles are still controversial—some CAFs are tumorigenic, whereas some other subtypes seem to have a tumor-suppressive role. In this context, their isolation can be of extreme interest to shed more light on their much-debated role in cancer progression, which will have important clinical implications12,31. Moreover, successful CAF extraction from human and preclinical mouse tumor models would also facilitate the development of new CAF-targeting drugs. This paper reports a method to efficiently isolate and culture primary CAFs from a syngeneic preclinical model of breast cancer. Based on experience, three experimental steps mostly influence the success of the protocol.
The first one is working fast to avoid aggregation of cell suspensions associated with the risk of clotting in the column and slowing cell efflux: in fact, when cells that passed through the column were clustered together, the probability of a sub-optimal isolation process increased, and the final cultures contained “contaminant” cell clones. The second one is pooling cells from different tumors after the depletion passage to guarantee enough cells to be passed through in the final enrichment step. The final critical issue is plating the extracted cells at high cell densities, most likely starting from a single well of the 24-well plate to boost cell growth and colony expansion for up to 4–5 passages. If the cells were seeded at low densities, they expanded on the free surface and quickly stopped replicating.
Several studies have already described CAF extraction methods of both human and mouse origin, to study their role in promoting cancer development and invasiveness. This has been done in many types of tumors, including breast cancer, melanoma, cholangiocarcinoma, and pancreatic adenocarcinoma35,36,37,38. However, due to the lack of specific CAF markers and to their heterogeneity, the results of these processes are still sub-optimal.
Here, the isolated cells were used to validate the CAF-targeting ability of HFn nanocages functionalized with anti-FAP targeting moieties. The use of primary cultures of CAFs was a significant advantage, allowing the validation of the nanostrategy ex vivo with a much simpler and cost-effective binding experiment than doing it directly in vivo at this preliminary phase of nanodrug optimization.
In this sense, ex vivo testing on CAFs can precede in vivo animal experiments by allowing the screening and selection of the most promising nanoparticles for the optimal targeting of CAF. The CAF model presented here may also be used for preliminary efficacy studies, when loading nanoparticles with active drugs. Animals will be only used later to evaluate biodistribution, pharmacokinetics, and efficacy studies.
Several CAF-targeting nanodrugs are being developed, such as ferritin nanocages for photodynamic therapy in breast cancer and peptide-based particles to deliver doxorubicin in prostate cancer models39,40. However, not many studies have focused on the isolation of CAFs as cell platforms for nanodrug optimization.
This protocol has some limitations. First, it is a time-consuming protocol, requiring the preparation and purchase of several reagents and materials. Second, due to the scarcity of CAFs in the 4T1 breast tumor, their yield is low. Nanoparticle experiments should be planned and performed as soon as the required cell number is achieved, as primary CAFs undergo senescence and cannot be maintained in culture for a long time. In conclusion, this method of CAFs isolation, culturing, and characterization can be a powerful tool to accelerate the development of new targeted nanomedicines in the fight against cancer.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This study was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) under IG 2017-ID. 20172 project – P.I. Corsi Fabio. SM acknowledges Pediatric Clinical Research center “Romeo and Enrica Invernizzi” that supports her position. AB thanks AIRC (ID. 20172 project) and University of Milan for research fellowship. LS and MS postdoctoral and doctoral fellowships are supported by University of Milan.
Materials
| Name | Company | Catalog Number | Comments |
| ACK Lysing buffer | Lonza | 10-548E | Store at +15° to +30 °C. |
| Alexa Fluor 488 goat anti-human antibody | Immunological Science | IS-20022 | Protect from light. |
| Anti-FAP Fab fragment (3F2) | Creative Biolabs | TAB-024WM-F(E) | |
| BALB/c mice | Charles River | 028BALB/C | 7 weeks old female mice |
| Bovine Serum Albumin | Sigma-Aldrich | A7906 | Store at 2-8 °C. |
| CD45 antibody | Miltenyi Biotec | 130-110-796 | FITC-conjugated fluorescent antibody; clone REA 737; Store protected from light at 2–8 °C. |
| CD90.2 antibody | Miltenyi Biotec | 130-102-960 | PE-conjugated fluorescent antibody; clone 30-H12; Store protected from light at 2–8 °C. |
| Cell strainers 70 µm | VWR | 732-2758 | |
| CytoFLEX | Beckman Coulter | laser configuration B4-R2-V0 | |
| Dead cell Removal Kit | Miltenyi Biotec | 130-090-101 | contains 10 mL of Dead Cell Removal MicroBeads; 25 mL of 20× Binding Buffer Stock Solution. Store protected from light at 2−8 °C. |
| D-Luciferin | Caliper | 760504 | reagent for in vivo imaging |
| DMEM High Glucose w/o L-Glutamine w/ Sodium Pyruvate | Euroclone | ECB7501L | Warm at 37 °C in a water bath before use. |
| DMSO | Sigma-Aldrich | D2650 | |
| DPBS w/o Calcium, w/o Magnesium | Euroclone | ECB4004L | Keep at room temperature. |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | E9884 | |
| Fetal Bovine Serum | Euroclone | ECS0180L | Before use, heat at 56 °C for 30 min to kill all the complement proteins. |
| Fluorescein Istothiocyanate isomer I (FITC) | Sigma-Aldrich | F7250 | Store protected from light at 2–8 °C. |
| GentleMACS C Tubes | Miltenyi Biotec | 130-096-334 | They are used in combination with the gentleMACS Dissociator. |
| GentleMACS dissociator | Miltenyi Biotec | 130-093-235 | Two samples can be processed in parallel. Special protocols have been developed for various tissues. |
| Goat serum | Euroclone | ECS0200D | |
| Ham's F12 w/o L-Glutamine | Euroclone | ECB7502L | Warm at 37 °C in a water bath before use. |
| IVIS Lumina II imaging system | Caliper Life Sciences | This imaging system is easy to use for both fluorescent and bioluminescent imaging in vivo. Equipped with a Living Image Software for analysis | |
| LD columns | Miltenyi Biotec | 130-042-901 | Composed of ferromagnetic spheres, designed for stringent depletion of unwanted cells. Position the required number of columns on the magnetic Separation Unit and equilibrate with buffer before use |
| L-Glutamine | Euroclone | ECB3000D | 200 mM stock |
| Light/fluorescence microscope equipped with camera | Leica Microsystems | DM IL LED Fluo/ ICC50 W CameraModule | inverted microscope for live cells with camera |
| MACS MultiStand | Miltenyi Biotec | 130-042-303 | |
| MACS Separation Unit | Miltenyi Biotec | 130-090-976 | Position the magnet on appropriate stand before use. |
| MACS Tissue Storage Solution | Miltenyi Biotec | 130-100-008 | |
| MS columns | Miltenyi Biotec | 130-042-201 | Composed of ferromagnetic spheres, designed for positive selection of cells. Position the required number of columns on the magnetic Separation Unit and equilibrate with buffer before use. |
| Mycoplasma Removal Agent | Euroclone | ECMC210A | Dilute in culture medium 1:100. |
| NaHCO3 | Invitrogen | A10235 | |
| Nanodrop spectrophotometer | Thermo Fisher Scientific | ND-2000C | |
| Non essential aminoacids | Euroclone | ECB3054D | |
| Penicillin-Streptomycin | Euroclone | ECB3001D | |
| Recombinant Human apoferritin H-homopolymer (HFn) | Molirom | MLR-P018 | |
| RPMI 1640 w/o L-Glutamine | Euroclone | ECB9006L | Warm at 37 °C in a water bath before use. |
| Tumor-Associated Fibroblast Isolation kit | Miltenyi Biotec | 130-116-474 | Contains 1 mL of Non-Tumor-Associated Fibroblast Depletion Cocktail, mouse; 1 mL of CD90.2 (Tumor-Associated Fibroblast) MicroBeads,mouse. Store protected from light at 2-8 °C. |
| Tumor Dissociation Kit | Miltenyi Biotec | 130-096-730 | Contains lyophilized enzymes (D, R, A) and buffer A; reconstitute enzymes according to the manufacturer's instructions, aliquot and store at - 20 °C. |
| Trypan blue 0.4% | Lonza | 17-942E | dilute 1:1 with a sample of cell suspension before counting the cells |
| TrypLE select | Gibco | 12563-029 | use at room temperature |
| Trypsin-EDTA | Lonza | BE17-161E | Warm at 37 °C in a water bath before use |
| T75 Primo TC flask | Euroclone | ET7076 | |
| Zeba Spin Desalting Columns | Thermo Fisher Scientific | 89890 | 7K MWCO, 2 mL |
| 1.5 mL tubes | Biosigma | CL15.002.0500 | |
| 15 mL tubes | Euroclone | ET5015B | |
| 4T1-luc2 | Caliper | Mouse mammary gland cancer cell line stably transfected with firefly luciferase gene | |
| 50 mL tubes | Euroclone | ET5050B |
References
- Quail, D. F., Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine. 19 (11), 1423-1437 (2013).
- Velaei, K., Samadi, N., Barazvan, B., Soleimani Rad, J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast. 30, 92-100 (2016).
- Cheng, J. D., Weiner, L. M., et al. Tumors and their microenvironments: tilling the soil. Commentary re: A. M. Scott et al., A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clinical Cancer Research. 9 (5), 1690-1695 (2003).
- Hui, L., Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Letters. 368 (1), 7-13 (2015).
- Balkwill, F. R., Capasso, M., Hagemann, T. The tumor microenvironment at a glance. Journal of Cell Science. 125, 5591-5596 (2012).
- Chen, X., Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nature Review. Drug Discovery. 18 (2), 99-115 (2019).
- Franco, O. E., Shaw, A. K., Strand, D. W., Hayward, S. W. Cancer associated fibroblasts in cancer pathogenesis. Seminars in Cell & Developmental Biology. 21 (1), 33-39 (2010).
- LeBleu, V. S., Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Disease Models & Mechanisms. 11, (2018).
- Li, X. -Y., Hu, S. -Q., Xiao, L. The cancer-associated fibroblasts and drug resistance. European Review for Medical and Pharmacological Sciences. 19 (11), 2112-2119 (2015).
- Luo, H., Tu, G., Liu, Z., Liu, M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Letters. 361 (2), 155-163 (2015).
- Truffi, M., Sorrentino, L., Corsi, F. Fibroblasts in the tumor microenvironment. Advances in Experimental Medicine and Biology. 1234, 15-29 (2020).
- Sugimoto, H., Mundel, T. M., Kieran, M. W., Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biology & Therapy. 5 (12), 1640-1646 (2006).
- Costa, A., et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 33 (3), 463-479 (2018).
- Ishii, G., Ochiai, A., Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Advances Drug Delivery Reviews. 99, Part B 186-196 (2016).
- Chen, Q., et al. Remodeling the tumor microenvironment with emerging nanotherapeutics. Trends in Pharmacological Sciences. 39 (1), 59-74 (2018).
- Truffi, M., et al. Nano-strategies to target breast cancer-associated fibroblasts: rearranging the tumor microenvironment to achieve antitumor efficacy. International Journal of Molecular Sciences. 20 (6), 1263(2019).
- Tong, R., Langer, R. Nanomedicines Targeting the Tumor Microenvironment. Cancer Journal. 21 (4), 314-321 (2015).
- Kaps, L., Schuppan, D. Targeting cancer associated fibroblasts in liver fibrosis and liver cancer using nanocarriers. Cells. 9 (9), 2027(2020).
- Miao, L., et al. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Research. 77 (3), 719-731 (2017).
- Kelly, T., Huang, Y., Simms, A. E., Mazur, A. Fibroblast activation protein-α: a key modulator of the microenvironment in multiple pathologies. International Review of Cell and Molecular Biology. 297, 83-116 (2012).
- Brennen, W. N., Isaacs, J. T., Denmeade, S. R. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Molecular Cancer Therapeutics. 11 (2), 257-266 (2012).
- Juillerat-Jeanneret, L., Tafelmeyer, P., Golshayan, D. Fibroblast activation protein-α in fibrogenic disorders and cancer: more than a prolyl-specific peptidase. Expert Opinion on Therapeutic Targets. 21, 977-991 (2017).
- Falvo, E., et al. Antibody-drug conjugates: targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale. 5 (24), 12278-12285 (2013).
- Truffi, M., et al. Ferritin nanocages: A biological platform for drug delivery, imaging and theranostics in cancer. Pharmacological Research. 107, 57-65 (2016).
- Cai, Y., et al. Enhanced magnetic resonance imaging and staining of cancer cells using ferrimagnetic H-ferritin nanoparticles with increasing core size. International Journal of Nanomedicine. 10, 2619-2634 (2015).
- Heger, Z., Skalickova, S., Zitka, O., Adam, V., Kizek, R. Apoferritin applications in nanomedicine. Nanomedicine. 9 (14), 2233-2245 (2014).
- Mazzucchelli, S., et al. Nanometronomic treatment of 4T1 breast cancer with nanocaged doxorubicin prevents drug resistance and circumvents cardiotoxicity. Oncotarget. 8 (5), 8383-8396 (2017).
- Yu, Q., et al. Targeting cancer-associated fibroblasts by dual-responsive lipid-albumin nanoparticles to enhance drug perfusion for pancreatic tumor therapy. Journal of Controlled Release. 321, 564-575 (2020).
- Mertens, J. C., et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Research. 73 (2), 897-907 (2013).
- Kovács, D., et al. Core-shell nanoparticles suppress metastasis and modify the tumour-supportive activity of cancer-associated fibroblasts. Journal of Nanobiotechnology. 18, 18(2020).
- Sharon, Y., Alon, L., Glanz, S., Servais, C., Erez, N. Isolation of normal and cancer-associated fibroblasts from fresh tissues by Fluorescence Activated Cell Sorting (FACS). Journal of Visualized Experiments. (71), e4425(2013).
- Sitia, L., et al. Selective targeting of cancer-associated fibroblasts by engineered H-ferritin nanocages loaded with navitoclax. Cells. 10 (2), 328(2021).
- Bellini, M., et al. Protein nanocages for self-triggered nuclear delivery of DNA-targeted chemotherapeutics in cancer cells. Journal of Controlled Release. 196, 184-196 (2014).
- Sahai, E., et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer. 20, 174-186 (2020).
- Dvořánková, B., Lacina, L., Smetana, K. Isolation of normal fibroblasts and their cancer-associated counterparts (CAFs) for biomedical research. Methods in Molecular Biology. 1879, 393-406 (2018).
- Calvo, F., Hooper, S., Sahai, E. Isolation and immortalization of fibroblasts from different tumoral stages. Bio-protocol. 4 (7), 1097(2014).
- Sha, M., et al. Isolation of cancer-associated fibroblasts and its promotion to the progression of intrahepatic cholangiocarcinoma. Cancer Medicine. 7 (9), 4665-4677 (2018).
- Hwang, R. F., et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Research. 68 (3), 918-926 (2008).
- Zhen, Z., et al. Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic T cell infiltration and tumor control. Nano Letters. 17 (2), 862-869 (2017).
- Ji, T., et al. Peptide assembly integration of fibroblast-targeting and cell-penetration features for enhanced antitumor drug delivery. Advances Materials. 27 (11), 1865-1873 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved