
Method Article
A Human Cerebral Organoid Model of Neural Cell Transplantation
In This Article
Summary
Here, we describe a protocol for the transplantation and tracking of labeled neural cells into human cerebral organoids.
Abstract
The advancement of cell transplantation approaches requires model systems that allow an accurate assessment of transplanted cell functional potency. For the central nervous system, although xenotransplantation remains state-of-the-art, such models are technically challenging, limited in throughput, and expensive. Moreover, the environmental signals present do not perfectly cross-react with human cells. This paper presents an inexpensive, accessible, and high-throughput-compatible model for the transplantation and tracking of human neural cells into human cerebral organoids. These organoids can be easily generated from human induced pluripotent stem cells using commercial kits and contain the key cell types of the cerebrum.
We first demonstrate this transplant protocol with the injection of EGFP-labeled human iPSC-derived neural progenitor cells (NPCs) into these organoids. We next discuss considerations for tracking the growth of these cells in the organoid by live-cell fluorescence microscopy and demonstrate the tracking of transplanted EGFP-labeled NPCs in an organoid over a 4 month period. Finally, we present a protocol for the sectioning, cyclic immunofluorescent staining, and imaging of the transplanted cells in their local context. The organoid transplantation model presented here allows the long-term (at least 4 months) tracking of transplanted human cells directly in a human microenvironment with an inexpensive and simple-to-perform protocol. It, thus, represents a useful model both for neural cell therapies (transplants) and, likely, for modeling central nervous system (CNS) tumors in a more microenvironmentally accurate manner.
Introduction
The human brain is a complex organ composed of multiple cell types of the neural and glial lineages. Together, these form a sophisticated network that gives rise to cognition. There is significant interest in the transplantation of cells into this system as a treatment for a wide variety of neurological disorders, including traumatic brain injury (TBI)1,2, neurodegenerative disorders3,4,5,6,7, and stroke8. One major limitation in the advancement of such strategies, however, is the relative paucity of available preclinical models to determine the expected transplantation outcomes. The most used models currently are in vitro culture methods to determine cell potential and xenotransplantation into mice. While cell culture methods can assess differentiation and self-renewal potential9, these are performed under optimal growth conditions that do not mimic the microenvironment the cells would encounter in a transplant context. Moreover, the way in which the cells are grown can influence their behavior10.
Mouse brains contain all the cells of the microenvironment and are, thus, extremely powerful model systems for transplantation11. There are, however, important differences between the mouse and human cortex12,13, and not all growth factors cross-react between species. Primate models are a closer alternative that better mimic the human system and have also yielded important preclinical results14. Even these more closely related relatives, however, retain important differences in their cellular makeup15. While both of these model systems provide valuable insights into cell behavior during transplant and incorporate the surgical elements of an eventual therapy, they remain imperfect. They are also costly and technically challenging (i.e., one must perform brain surgery on the animals), thus limiting the possible throughput. Moreover, there are a plethora of ethical issues associated with transplanting human brain cells into animals16. Brain slice cultures allow one brain to be cut and used for multiple treatments, thus removing some of the limitations of animal transplants; however, these have limited lifespans (weeks), are still animal-derived, and (being a thin slice) do not have sufficient volume/surface integrity to mimic the injection of cells17. Thus, there remains an important gap between strictly cell culture/potential models and in vivo transplantation.
Cerebral organoids are an in vitro model containing the main neural cell types present in the brain and can be generated in high numbers from human induced pluripotent stem cells (iPSCs)18,19. Such organoids thus provide a cellular context, which could allow the assessment of the functional capacity of a test cell of interest in the transplant setting. Indeed, a recent study demonstrated that neural progenitor cells (NPCs) transplanted into human cerebral organoids survive, proliferate, and differentiate similarly to NPCs transplanted into the brain of a non-obese diabetic severe combined immunodeficient gamma (NSG) mouse20. Cerebral organoids thus represent a cruelty-free, long-lived (>6 months), cost-effective system that captures the cell types of the human brain. As such, they could represent an ideal transplant recipient for the early-stage testing of the regenerative capacity of neural cells.
This paper presents a protocol for the transplantation and subsequent tracking of labeled human NPCs into human cerebral organoids (Figure 1). This begins with the injection of GFP-labeled NPCs into mature (2-4 months old) cerebral organoids18. The transplanted cells are then followed by live-cell fluorescence microscopy over a 4 month period. During this time, we show both the persistence of cells at the injection site but also migration to distal regions of the organoid. At the endpoint, we demonstrate the antigen retrieval, staining, and imaging of histological sections derived from these organoids, including a protocol for the quenching of existing AlexaFluor-based dyes to allow additional staining and imaging rounds, based on previous work21. This protocol could, thus, be useful in the measurement of the differentiation capacity of cells in a transplant setting, graft durability, cell expansion in situ, and cell migration from the site of the transplant. We anticipate that this will be useful both for regenerative medicine/cell therapy applications, as well as tumor modeling by engrafting tumor cells into relevant region-specific organoids.
Protocol
NOTE: See the Table of Materials for details related to all the materials, reagents, and equipment used in this protocol.
1. Fluorophore labeling of cells by lentiviral transduction
- Thaw an aliquot of the solubilized basement membrane matrix of choice on ice.
- While the aliquot is thawing, add sterile water to fill in the gaps between the wells in a 24-well plate, along with all the outer wells.
NOTE: This leaves eight wells open in the middle for use and ensures that the cells remain in high humidity, thus minimizing evaporation-induced variability. - Dilute the solubilized basement membrane matrix 1:100 in ice-cold DMEM/F12 medium (a final concentration of 0.089 mg/mL was obtained with the lot used here).
- Add 300 µL of the diluted solubilized basement membrane matrix to one of the middle (open) wells of the prepared 24-well plate per infection to be performed. Be sure to evenly cover the entire bottom of the well, and add more, if needed, to ensure an even coating.
- Incubate the plate for 30 min at 37 °C to allow a layer to solidify along the well bottom. Verify at the end of this incubation that the liquid is still fully covering the well bottom and has not evaporated in the center. Evaporation is a sign that the humidity is too low; if this happens, repeat the coating.
NOTE: The plates can be coated the evening before and left in diluted solubilized basement membrane matrix without evident ill effects; however, the risk of evaporation is greatly increased, so they must be carefully inspected before use. - Remove the medium from one well of confluent iPSC-derived NPCs growing in a well plate.
NOTE: For this paper, iPSC-derived neural progenitors produced using a commercial kit as per the manufacturer's instructions between passage 3 and passage 10 were used. - Gently add 1 mL of PBS to the side of the well, and rock the plate to ensure even washing.
- Remove the PBS, and replace with 300 µL of the proteolytic-collagenolytic enzyme mix, again rocking the plate to ensure to fully coat the well bottom. Incubate for 5 min at 37 °C. Verify that the cells are detaching from the plate surface by light microscopy.
- With a P1,000 pipette, add 700 µL of DMEM/F12, and detach the cells by spraying this mixture across all the surfaces of the bottom of the plate. Pipette up and down to ensure the cell aggregates are broken up into a uniform single-cell suspension. Take up the cells, and add to 9 mL of DMEM/F12 medium in a conical 15 mL tube.
- Centrifuge at 300 × g for 5 min.
- During this time, prepare enough medium supplemented with 10 µM Y-27632 for 500 µL per well to be plated, plus 1 mL for resuspension.
NOTE: Y-27632 is necessary to ensure high survival of the NPCs after passage as single cells. - Remove the supernatant, and resuspend the cells in 1 mL of Y-27632-supplemented medium, pipetting up and down gently several times to break up the cell aggregates.
- Take out a few microliters of cells, and dilute them (1:5 to 1:10) in trypan blue.
- Count the viable (trypan blue-negative) cells on a hemocytometer. Adjust the trypan blue dilution to get at least 50 viable cells per quadrant of the hemocytometer to ensure the counts are accurate. Calculate the volume of cell suspension needed for 100,000 cells to be present in each well.
- Mix the cells gently up and down by pipetting, and take up the calculated volume (for 100,000 cells per well). Add this to the Y-27632-supplemented medium (from step 1.11) such that the final volume is 500 µL per well to be plated.
- Remove the diluted solubilized basement membrane matrix from the previously coated wells (assuming the 30 min or longer incubation has been completed).
- Mix the cells gently by pipetting, and add 500 µL of the cell suspension to each well.
- To evenly distribute the cells, move the plate in a large "+" sign motion with gentle linear sweeps (forward-backward, stop, back-forth) immediately prior to placing into a cell culture incubator.
- Incubate overnight (16-24 h) at 37 °C and 5% CO2.
- Remove the medium, and add the titrated lentivirus. If single integration events are required, aim for a multiplicity of infection of 0.3 infectious units per cell (ideally titrated on the cells of interest), and top up to 500 µL in medium (without Y-27632).
NOTE: The lentivirus of interest can be purchased pre-titrated or produced in-house as done previously22. Here, an in-house-produced GFP lentivirus with puromycin resistance was used to generate and select the NPCs expressing EGFP. If the lentivirus has not been titrated on the cells of interest and the copy number is critical, it is recommended that several volumes be added to a couple of wells and that the well with ~30% EGFP+ cells 48-72 h be used later. If the copy number is not critical, excess virus can be added with the caveat that insertional mutagenesis may result in anomalous behavior in some clones. CAUTION: Lentiviral vectors are a level 2-3 biological hazard (depending on their contents). Follow local biosafety regulations for handling the lentivirus, the decontamination of surfaces and plasticware, and liquid waste. - Return the cells to the incubator, and incubate them for 24 h at 37 °C and 5% CO2.
- Remove the cells from the incubator, and remove the supernatant.
CAUTION: As the supernatant from the cells still contains lentiviral particles at this point, continue to follow local biosafety regulations for its handling and disposal. - Rinse the cells with 2 x 500 µL of PBS by gently adding PBS to the wall of the well; then, remove from the well corner to wash away any remaining lentivirus.
CAUTION: As the supernatant from the cells still contains lentiviral particles at this point, continue to follow local biosafety regulations for its handling and disposal. - Change to 500 µL of fresh medium (without Y-27632), and continue to grow at 37 °C and 5% CO2 overnight.
- Use live-cell fluorescence microscopy to verify the proportion of cells that express EGFP and select the well with which to proceed.
- If the lentiviral vector of choice contains a selection cassette (antibiotic resistance gene such as puromycin resistance), add the selection agent (here, 1 µg/mL puromycin) to the medium at a previously validated dose that kills sensitive but not resistant cells. Otherwise, use fluorescence-activated cell sorting to isolate the EGFP+ cells at the time of the first split.
- From now onward, maintain the cells with daily monitoring for confluency, and perform complete medium changes daily.
- When confluency is reached, prepare one or more wells of a fresh 6-well coated plate (with 1 mL of diluted solubilized basement membrane matrix; see steps 1.3-1.5), and harvest the cells as described in steps 1.6-1.15.
NOTE: If no selection agent is included, sort the cells by flow cytometry at this stage. Ensure that the cells are sorted with a sufficiently wide nozzle and low pressure to minimize stress, as well as that 10 µM Y-27632 is included at every step in the sorting process. - Plate the cells to a final density of 200,000 cells/cm2.
- Maintain the daily monitoring and complete medium changes daily.
NOTE: When the cells again reach confluency, they are now ready for use. Alternatively, a stock can now be frozen down for future use as follows. - Split, harvest, and count the cells as described in steps 1.6-1.15.
- Pre-label a set of cryovials with the relevant information (e.g., passage, EGFP%, line).
- Resuspend at 400,000 cells/mL in medium + 10% DMSO.
NOTE: DMSO is toxic to cells when not frozen, so minimize the time spent at room temperature in the presence of DMSO. - Add 1 mL (400,000 cells) of the mixture from step 1.33 per cryovial, and place in a cell-freezing container.
- Transfer the container to a −80 °C freezer overnight.
- The following day, transfer the cells into liquid nitrogen for long-term storage.
2. Labeled cell injection into the brain organoid
NOTE: For this paper, cerebral organoids were produced using a commercial kit as per the manufacturer's instructions. This can be replaced with the cerebral organoid of interest. The materials used in this part of the protocol must be prechilled to avoid the gelling of the solubilized basement membrane matrix at above 4 °C.
- Place the insulin syringe(s), tips, and tubes that are going to be in contact with the solubilized basement membrane matrix at −20 °C to let them cool.
- Thaw a suitable-size aliquot of solubilized basement membrane matrix on ice depending on the number of injections that are going to be performed (~2 µL/injection). This takes approximately 30 min.
- While the aliquot is thawing, split, harvest, and count the cells as described in steps 1.6-1.15.
- Calculate the volumes needed per injection. If doing multiple injections, place the total volume of cells in a 1.5 mL tube, and add up to 1 mL of DMEM/F12.
NOTE: A volume that allows for a few extra injections should be used to account for losses/pipetting errors. - Place the cells into ice while the next steps are being prepared.
- After thawing, dilute the solubilized basement membrane matrix to a final concentration of 3 mg/mL in ice-cold DMEM/F12.
- Take the single-cell suspension from the ice, and spin it down at 300 × g for 5 min at 4 °C.
NOTE: If a refrigerated centrifuge is not available, spinning at room temperature generally appears to be tolerated. - Gently remove the medium completely without disturbing the cell pellet. Resuspend the cell pellet by gentle pipetting in the diluted solubilized basement membrane matrix (3 mg/mL) to obtain a final volume of 2 µL per injection to be performed, and immediately place it back on ice until use.
NOTE: The resuspension of the cells needs to be performed slowly and with prechilled tips to avoid the formation of bubbles and gelling. - Transfer the prechilled syringe(s) from the −20 °C freezer into an ice bucket. Keep them there until use.
- Remove the plate with the brain organoids from the incubator. Use a wide-bore tip to transfer the organoid to be injected into a 35 mm dish. Remove all the medium possible without damaging the organoid to stabilize it and facilitate the injection.
NOTE: It is important to use wide-bore tips to avoid destroying the organoid. If they are not available, cut the end of a regular P1,000 tip with sterile scissors. - Place the 35 mm dish containing the organoid under a dissecting microscope to help guide and facilitate the injection.
NOTE: If a dissecting microscope is not available in a sterile area, it is possible to perform the injection without one, albeit with somewhat reduced control. Another alternative is the use of a magnification loop or glasses. - Once the organoid(s) are ready to be injected, gently resuspend the cells with a chilled P20 pipette tip, and move 2 µL (containing the desired number of cells to be injected) onto a prechilled sterile glass slide.
NOTE: For multiple injections, additional 2 µL volumes can be added across the glass slide. Keep it on ice, and take care to avoid evaporation. - Take a prechilled insulin syringe from the ice, and slowly draw up the 2 µL of cell suspension with the bevel of the needle facing down to allow all the cells/medium to be taken up.
NOTE: Make sure to go extremely slowly to avoid drawing in any air. - Open the lid of the dish containing the organoid, and focus the microscope on it. Hold the dish with one hand. Place the bevel of the needle up, and with the other hand, inject the cells slowly into the organoid surface.
NOTE: It is important to inject slowly to avoid damaging the tissue. - After the injection, place the lid back into the dish, and let the injected organoid sit for 1-2 min.
NOTE: This allows the diluted solubilized basement membrane matrix to gel, holding the cells in place. If the medium is added too early, it can wash away the cells sitting at the surface. - Gently add 500 µL of the organoid medium, and transfer the organoid with a wide-bore tip into a well of a 24-well plate. Incubate the organoid at 37 °C and 5% CO2 overnight. Perform complete medium changes every second day.
NOTE: It is important to include a negative control organoid (mock injected), which will be used for imaging adjustments. To do this, inject an organoid as previously described with diluted solubilized basement membrane matrix (3 mg/mL) to obtain a final volume of 2 µL without cells.
3. Graft tracking by live-cell fluorescence imaging
NOTE: Use a fluorescence microscope that can excite the fluorophore of interest and has the filter set needed to detect its fluorescence. As mentioned before, the NPCs used here were EGFP+, with an excitation peak of 488 nm and an emission peak of ~510 nm.
- Load a negative control organoid (non-injected or mock injected), and set the illumination intensity and exposure time such that autofluorescence is minimal. For the instrument used here, the illumination intensity was set between 1 and 2 and the exposure time between 80 ms and 100 ms.
NOTE: As many excitation wavelengths (particularly lower wavelengths) can be toxic to cells, it is recommended to keep the intensity low and to avoid looking for too long to prevent damage to the organoid. - Load the positive control organoid, and raise the exposure time if necessary to ensure that labeled cells are clearly visible. Reposition the organoid with a wide-bore pipette tip to find the injection site (EGFP+ region).
- Remove the medium completely from the negative control organoid. Load the negative control organoid, and image it with the selected settings (for the instrument used here, the illumination intensity was set between 1 and 2 and the exposure time between 80 ms and 100 ms). When the imaging is complete, immediately add fresh medium to prevent the organoid from drying out.
NOTE: Medium removal is critical for obtaining a good image as it fixes the height and orientation of the organoids, which are otherwise floating and constantly moving, precluding high-quality imaging. - Repeat the medium removal/imaging with each of the test organoids.
- Return the organoids to normal incubation/medium changes (as in step 2.16), and repeat the imaging at desired intervals. Here, the organoids were imaged at week 1, week 9, and week 16 post injection to follow and test the survival, proliferation, differentiation, and migration of the injected EGFP+ NPCs.
4. Histology and immunofluorescence
- At the endpoint, transfer the organoid to a histology cassette labeled with the ID of the organoid and other necessary identifiers.
- Fill the cassette with 10% formalin, and close it. Keep it at room temperature for 24 h.
- Transfer the closed cassette with the organoid to a tissue process, and set the following protocol: ethanol 70% 10 min, ethanol 80% 20 min, ethanol 95% 30 min, ethanol 100% 30 min, ethanol 100% 40 min, ethanol 100% 50 min, toluene 30 min, toluene 40 min, toluene 50 min, paraffin 25 min at 59 °C, paraffin 35 min at 59 °C, paraffin 40 min at 59 °C, paraffin 50 min at 59 °C.
NOTE: The above steps are done under a vacuum at room temperature unless otherwise specified. This protocol replaces the water with gradients of alcohol up to 100%. The intermediate step with toluene is a transition between the alcohol and paraffin, as both are soluble in it. CAUTION: Toluene and paraffin are considered hazardous materials as they can be highly flammable liquids and can cause damage by inhalation or contact with the skin or eyes. Make sure to store them tightly closed in a secure space. Wear proper personal protective equipment when handling them. Dispose of waste according to local chemical hazard regulations. - Transfer the closed cassette with the organoid into a paraffin bath in the embedding station.
- Take the cassette from the bath, open it, and transfer the organoid into a steel base mold. Place the mold under the paraffin dispenser, and cover it completely with paraffin.
- Place the empty cassette on the top of the steel base mold. Here, add more paraffin on top of the cassette.
- Transfer the steel base mold with the cassette on top into ice, and let it cool for 5 min. Remove the steel mold, and keep the cassette with the organoid embedded in a paraffin block.
- Transfer the cassette into a microtome to cut sections. Cut sections as desired.
NOTE: Here, sections of 15 µm thickness were made throughout the organoid. - Drop each section onto the surface of a water bath set at 45 °C, and pick it up on a glass microscope slide.
- Let the slides dry in an incubator at 42 °C overnight.
NOTE: The slides can now be stored at room temperature until the time of staining. - Place the slide in a Coplin slide staining jar filled with toluene for 2 min. Repeat this step once more.
CAUTION: Toluene is considered a hazardous material as it can be a highly flammable liquid and can cause damage by inhalation or contact with the skin or eyes. Make sure to store it tightly closed in a secure space. Wear proper personal protective equipment when handling it. Dispose of waste according to local chemical hazard regulations. - Transfer to a glass Coplin slide staining jar filled with EtOH 100% for 2 min. Repeat this step once more.
- Transfer to a glass Coplin slide staining jar filled with distilled water for 2 min. Repeat this step once more.
- For antigen retrieval, prepare citrate buffer (10 mM citric acid, 0.05% Tween 20, pH 6.0).
NOTE: Citrate buffer can be stored at room temperature for up to 3 months or at 4 °C for longer storage. - Set a water bath between 95 °C and 100 °C.
CAUTION: This water bath can cause burns if one is not careful. Take all necessary precautions to avoid direct contact with the bath or its components. - In the water bath, float a plastic container that can fit the slides inside. Ensure the plastic does not touch the bottom of the bath (assuming it is a bottom-heating bath) to avoid melting. Pour the citrate buffer inside the plastic container, and let it reach 95-100 °C.
- Once the buffer reaches the desired temperature, place the slides inside the buffer, and loosely cover the container with a lid. Leave the slides inside the container in the water bath for 30-40 min.
- Remove the plastic container from the water bath, and let it cool at room temperature for 20 min more.
- Wash the slides for 3 x 2 min with PBS. Remove the PBS with a paper tissue without touching the sample or overdrying it.
- Prepare the permeabilization buffer (90 mL of PBS + 0.1% Tween-20), and fill a glass slide staining jar with it. Immerse the slides in the permeabilization buffer, and incubate for 10 min.
- Wash the slides for 3 x 2 min with PBS.
- Prepare enough staining mix to cover each sample (~25 µL).
NOTE: See Table 1 for the antibodies and the final concentrations used here. - Add 25 µL of staining mix to cover each organoid slice. Incubate at room temperature for 1 h or overnight at 4 °C in the dark.
NOTE: Ensure that the samples remain at a high humidity to prevent the evaporation of the staining mix. If a container to keep the slides humid is not available, put them inside a box with some wet tissues on the corner. - Wash the slides for 3 x 2 min with PBS.
- Rinse once with distilled water inside a glass slide staining jar to remove the salt.
- Add 10 µL of liquid mountant + 4',6-diamidino-2-phenylindole (DAPI) to each of the samples.
NOTE: The slides can be sealed if a second round of staining is not performed. Here, the slides were not covered to allow access for quenching/restaining as coverslip removal can damage the tissue. - Image the slides using the selected fluorescence microscope.
NOTE: Make sure to have positive and negative controls for each antibody to allow the proper setting of the intensities and exposure times (for the instrument used here, the illumination intensity was set between 5 and 6 and the exposure time between 2,000 ms and 3,000 ms), though this can vary depending on the antibody. Additionally, ensure the microscope has adequate detectors for each channel used. Be aware that as the samples are not covered, they need to be placed face up while imaging them. - After imaging, prepare fresh 2x quenching buffer (9% H2O2 + 50 mM NaOH in PBS).
NOTE: The quenching solution must be prepared fresh immediately before using it. The reaction is sensitive to the activity of the H2O2, which will decrease over time. CAUTION: H2O2 is an irritant, and NaOH is caustic. These should be treated with appropriate safety equipment, and skin contact should be avoided. Ensure they are disposed of in accordance with local chemical safety policies. - Fill the glass Coplin slide staining jar halfway with 2x quenching buffer (45 mL), add 45 mL PBS, and put the slides inside. Incubate overnight at 4 °C.
- Check by fluorescence microscopy that the fluorophores were effectively quenched.
- Repeat the staining and imaging as above.
NOTE: The quenching and restaining can be repeated for additional rounds as needed, though the risk of damage increases with each additional round.
5. Image registration
- Open FIJI.
- Make a folder with the DAPI images from round 1 and round 2. Change the names to clarify which is which, if needed.
- Make an empty folder for the output of the image registration.
- Click on Plugins | Registration | Register Virtual Stack Slices.
- Under Source directory, select the folder containing the DAPI images from each round.
- Under Output directory, select the folder made for the registration output.
- Set the Feature extraction model to Rigid and the Registration model dropdown menu to Rigid - translate+rotate to fix the registration, only allowing the image to be moved and rotated for alignment rather than be deformed.
NOTE: Should deformation be expected, the feature extraction model and registration model could be adjusted to Affine or other models as desired, though this can result in undesired transformations. Occasionally a slice may be damaged between rounds, which can prevent successful registration. - Check the Save transforms box to save the transformation parameters, allowing them to be applied to the other channels.
- Click on OK.
- Select the location to save the transform file (defaults to the input directory), and click on Open.
- Select the DAPI image to serve as a reference for the transforms. The DAPI image from each of the other rounds will be aligned to this one.
NOTE: When complete, the registered images will appear as a stack (and in the output folder). - Use the slide bar on the bottom to flip back and forth between the images to verify that the image registration was successful (the nuclei are in the same place in both images, at least for the overlapping areas).
- Verify that an .xml file has been created for each round of the input images.
NOTE: These contain the translation parameters needed for the files from that round. - Make a folder containing all the images to be registered (each channel of each round). Note the order of the files.
- Make a folder for the transformation parameters, and make one copy of the .xml file for each round per channel to be registered. Double-check that the file order (by name) is the same between the images to be registered and the transformation parameter copies, as the next step will go through and transform each image in the order of the transformation parameter files.
- Click on Plugins | Transform | Transform Virtual Stack Slices.
- Under Source directory, select the folder containing the images to be registered.
- Under Output directory, select the folder made for the registration output.
- Under Transforms directory, select the folder containing the .xml files with one copy per image file to be transformed. Again, make sure the order matches the files (i.e., one copy of the round 1 .xml for each round 1 channel, one copy of the round 2 .xml for each round 2 channel, etc.).
- Click on OK.
NOTE: When complete, the registered images from that round will appear as a stack (and in the output folder). - Use the slide bar on the bottom to flip back and forth between the channels. Verify that all the channels were transformed in the same way. If not, this is likely due to a wrong .xml file in the transforms directory; in this case, fix the file and repeat.
NOTE: The brightness range displayed will be based on one channel, but this does not affect the actual saved images, just the display. - Observe that the registered images are all successfully aligned across rounds and ready for overlays/downstream analysis.
NOTE: There are more efficient programmatic ways to accomplish registration for large batches. The method presented here is just an easy one that does not require programming.
Results
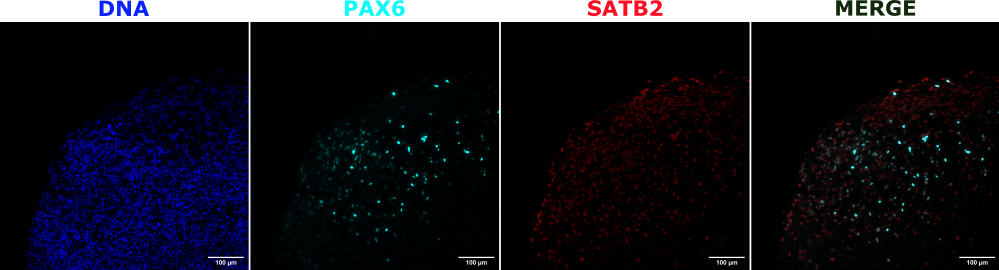
As a validation of the cerebral organoid identity, histological sections of a mature (2 month old) cerebral organoid were stained for PAX6 (a marker of dorsal NPCs23) and SATB2 (a marker of mature, postmitotic, upper-layer neurons24). As expected, PAX6+ cells were present at the interior of the organoid, and SATB2+ cells were present in the upper layers (Figure 2). These results support that the cerebral organoids used were indeed dorsal forebrain as specified in the differentiation kit. To establish the dose-dependence of the cerebral organoid transplant system, 2 month old cerebral organoids were injected with increasing numbers of EGFP+ iPSC-derived NPCs. A clear dose-dependence of GFP fluorescence on input cell number was present, with consistent EGFP+ cell patch detection at 10,000 cells and above (Figure 3). The persistence and migration of the transplanted NPCs were next assessed by following the transplanted organoids over time. For this, 50,000 iPSC-derived EGFP+ NPCs were transplanted into 2-3 month old cerebral organoids generated from the same iPSC line. The injected organoids and controls were imaged for EGFP positivity at indicated timepoints over the next 3-4 months. In this transplantation series, we observed persistence of the injected site throughout the 4 month tracking period (Figure 4A). Additional EGFP+ cell patches appeared by 9 days post transplantation and persisted until the study endpoint (3-4 months depending on the organoid), indicating the migration of the cells and integration at their new sites (Figure 4A). At a higher magnification, clear neural morphology was observable with long projections into the organoid (Figure 4B), confirming the integration of the injected cells.
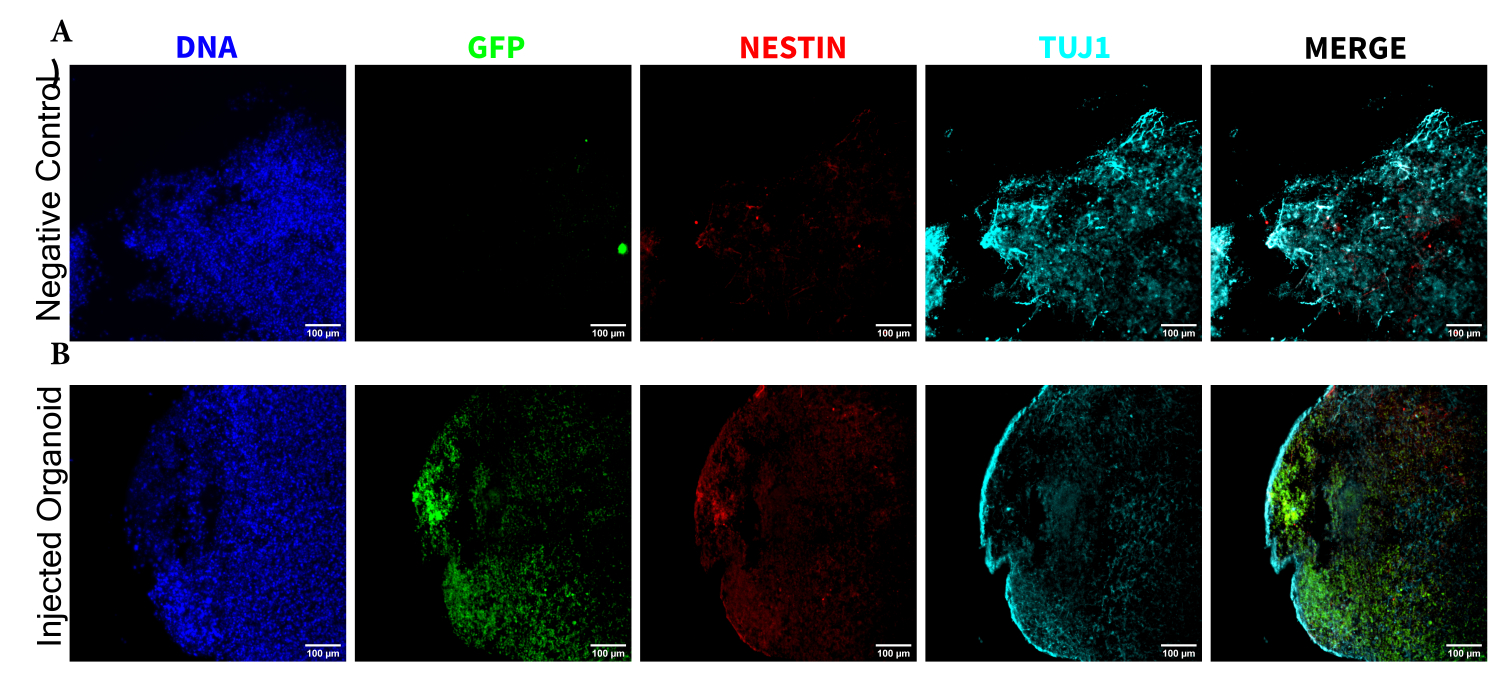
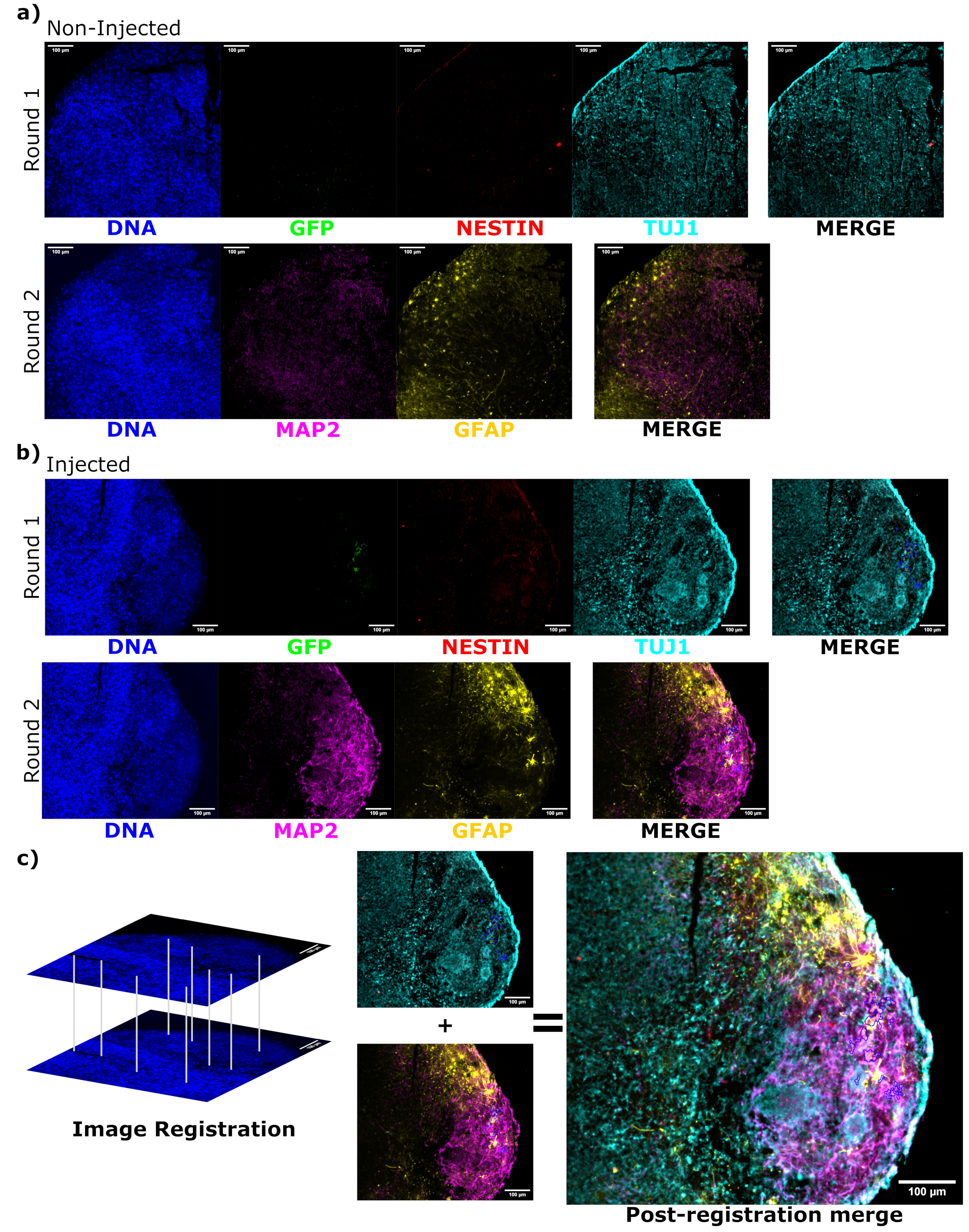
To determine the differentiation status of the injected cells late post injection, the 4 month tracked organoid and its control were fixed, paraffin-embedded, cut to 15 µm thick slices, and mounted onto glass slides. The slices were then processed and stained in either a single round of fluorescent staining (EGFP, TUJ1, NESTIN) or for two consecutive staining cycles to add additional markers (MAP2, GFAP). The initial single-round staining confirmed the presence of EGFP+ cells at the injection site, including a mixture of cells retaining NPC status (NESTIN+TUJ1−) and those that had differentiated towards a neural fate (NESTIN−TUJ1+) (Figure 5). For both the control and injected organoids, very few NESTIN+ NPCs were observed (most, though not all being EGFP+ transplanted NPCs at the site of injection), with a majority of TUJ1+ immature-mature neurons (Figure 5). The two-round staining gave more detail, revealing mature neurons (NESTIN−TUJ1+MAP2+GFAP−) around most of the outer region of the organoid, with areas of immature (NESTIN−TUJ1+MAP2−GFAP−) neurons toward the middle (Figure 6A,B). Astrocytes (NESTIN−TUJ1−MAP2−GFAP+) were present in both the injected and control organoids and were interspersed around the outer edges (Figure 6A,B). The slice for which the two-round staining was performed in the injected organoid showed a small satellite colony of EGFP+ cells far from the injection site that had adopted the phenotype of mature neurons (Figure 6B,C). Some of these appeared to be in close proximity to astrocytes; however, there were no EGFP+ cells with complete overlap to the GFAP staining, suggesting they were adjacent rather than generating the astrocytes themselves (Figure 6B,C).

Figure 1: Transplantation model of labeled cells into cerebral organoids. Schematic overview of the generation of labeled cells by lentiviral transduction, their transplantation into cerebral organoids, and tracking by live-cell imaging and immunofluorescence. Abbreviation: GFP = green fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Immunofluorescence of histological sections showing the architecture of early and late organoids. A 2 month old cerebral organoid was fixed, paraffin-embedded, sliced, and stained with PAX6, SATB2, and DAPI. An unstained section was used to set the exposure and integration time to avoid a false-positive signal from autofluorescence. PAX6+ cells were present at the interior of the organoid, while SATB2+ cells were present in the upper layers. Z-stack images were taken every 4.2 µm through the whole 15 µm tissue section. Optical sections were combined using the focus stacking option in the Gen5 software with default options. Scale bars = 100 µm. Abbreviations: PAX6 = paired box 6 protein; SATB2 = special AT-rich sequence-binding protein 2; DAPI = 4',6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Dose-dependent engraftment of NPCs into cerebral organoids. The organoids were transplanted with 0 (negative control), 1,000, 5,000, 10,000, or 50,000 GFP+ iPSC-derived NPCs. At 1 week post transplantation, the organoids were imaged on a Cytation 5 with a GFP filter cube. The negative control was used to set the exposure and integration time to minimize autofluorescence. Darker colors indicate more EGFP fluorescence relative to the negative control. Scale bars = 100 µm. These are 4x images of the whole organoids. After imaging, rolling-ball background subtraction with a pixel radius of 50 was performed prior to display to correct for the variable background intensity across the organoids. Injection sites identified as the regions of highest engraftment are indicated with a red arrow where engraftment was present. Abbreviations: NPCs = neural progenitor cells; GFP = green fluorescent protein; EGFP = enhanced GFP; iPSC = induced pluripotent stem cell. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Tracking of transplanted cell growth, migration, and persistence with fluorescence live-cell imaging. (A) Control and transplanted (50,000 GFP+ iPSC-derived NPCs) organoids were followed by fluorescence live-cell imaging over the course of 2-4 months from two independent transplant sets. Negative control organoids were used to set the exposure and integration time to minimize the autofluorescence at each time point. EGFP images were captured using a GFP filter cube on the Cytation 5 at indicated times post transplant. Darker colors indicate more EGFP fluorescence relative to the negative control for that time point. Rolling-ball background subtraction with a pixel radius of 50 was performed prior to display to correct for the variable background intensity across the organoids. The organoids were placed in approximately the same orientation at each time point, and the images were rotated for consistency of the display and to clearly show the transplanted cell growth. The injection sites identified as the regions of highest engraftment at the earliest time point are indicated with a red arrow. An example negative control organoid is shown in the bottom of the figure. (B) Example 20x images are shown from engrafted organoids at week 1 and week 15 post transplantation. The local contrast was enhanced prior to display using FIJI to ensure neurite visibility. Scale bars = 100 µm. Abbreviations: NPCs = neural progenitor cells; GFP = green fluorescent protein; EGFP = enhanced GFP; iPSC = induced pluripotent stem cell. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Immunofluorescence of the histological sections revealing the persistence of the transplanted NPCs at the injection site along with migration and neural differentiation. Single-channel fluorescence images from (A) non-injected and (B) transplanted organoids. The overlayed image on the right shows the three channels of interest (NESTIN, TUJ1, and EGFP), but does not include DAPI. The display minimums were set to just exclude the signal from the negative cells (determined for EGFP from the non-injected control and for other channels based on known marker combinations). The display maximums were based on the highest signal observed for that antibody in any cell. The display ranges were kept constant between the non-injected and transplanted organoids to allow for direct comparison. Scale bars = 100 µm. Abbreviations: NPCs = neural progenitor cells; DAPI = 4',6-diamidino-2-phenylindole; TUJ1 = beta-III tubulin; EGFP = enhanced green fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Assessment of engrafted cell differentiation state and localization using cyclic immunofluorescence of the histological sections. Single-channel fluorescence images from the non-injected, (A) age-matched, and (B) transplanted organoids are shown for the first and second round of staining as indicated. The display minimums were set to just exclude the signal from the negative cells (determined for EGFP from the non-injected control and for other channels based on known marker combinations). The display maximums were based on the highest signal observed for that antibody in any cell. The display ranges (and of course, the imaging parameters) were kept constant between the control and injected organoids to allow for direct comparison. Scale bars = 100 µm. An overlayed image (excluding DAPI) is shown for each staining round for each organoid on the right. (B) For the injected organoid where image registration was performed, all images are cropped to the region observed in both the staining rounds. (B,C) For the injected organoid, the EGFP+ cell regions are outlined in blue. A diagram of how DAPI is used to match the features during image registration is shown in (C), followed by an overall merge of the registered image. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole; TUJ1 = beta-III tubulin; EGFP = enhanced green fluorescent protein; MAP2 = microtubule-associated protein 2; GFAP = glial fibrillary acidic protein. Please click here to view a larger version of this figure.
{kind=link}
| Clone | Fluorophore | Concentration | |
| Anti-NESTIN | 10C2 | AlexaFluor 594 | 1 in 2,000 |
| Anti-TUBB3 | TUJ1 | AlexaFluor 647 | 1 in 2,000 |
| Anti-GFP | FM264G | AlexaFluor 488 | 1 in 200 |
| Anti-GFAP | SMI 25 | AlexaFluor 594 | 1 in 500 |
| Anti-MAP2 | SMI 52 | AlexaFluor 488 | 1 in 1,000 |
| Anti-PAX6 | O18-1330 | AlexaFluor 647 | 1 in 100 |
| Anti-SATB2 | EPNCIR130A | AlexaFluor 594 | 1 in 500 |
Table 1: Antibody concentrations for staining. Abbreviations: TUBB3 = beta-tubulin III; GFP = green fluorescent protein; GFAP = glial fibrillary acidic protein; MAP2 = microtubule-associated protein 2; PAX6 = paired box 6 protein; SATB2 = special AT-rich sequence-binding protein 2.
Discussion
Given the significant interest in cell therapeutic approaches for the treatment of CNS injuries/neurodegenerative disorders1,2,3,4,5,6,7,8, models of cell function in a transplant setting are gaining importance. This paper presents a method for the transplantation of labeled, human NPCs into human cerebral organoids, along with their live-cell tracking and end-point assessment by histology and immunofluorescent staining. Importantly, we showed that the transplanted cells were capable of migration, differentiation, and long-term (4 month) persistence in the organoid setting. Such long-term persistence is a marked increase over the maintainability of brain slice cultures17. This system is, thus, appropriate for examining many of the behaviors one would need to assess in a potential therapeutic setting, such as survival, proliferation, and differentiation. Indeed, an orthogonal study recently demonstrated that transplanted NPCs behaved similarly in cerebral organoids compared to NPCs transplanted into NSG mouse brains20, thus confirming the utility of organoids as a transplant recipient. As this is an in vitro system, it is also straightforward to add cytokines or drugs of interest. This could be used to better understand the effects of specific environments such as inflammation and immunosuppressants on the transplanted cells to further mimic what they might encounter in a therapeutic setting. The cyclic immunofluorescence protocol we demonstrated (based on previous research21) further extends the power of this approach, allowing a wide array of lineage- and, potentially, disease-specific markers to be simultaneously assessed in a single section, and, thus, allowing accurate tracking of the transplanted cells and their impact on the tissue. Of course, other endpoint assessment methods could be used instead depending on the goals of the analysis. For example, tissue clearing with 3D reconstruction could be used if cell morphology is of primary interest, or dissociation followed by flow cytometry could be used if the quantification of specific cell types is the end goal. We expect this method to be easily extensible to other cell types such as CNS tumors, potentially allowing their study in a microenvironmentally relevant context. Similarly, the organoids used as recipients could be exchanged for disease-model organoids25,26,27, potentially allowing for the modeling of transplantation approaches for these conditions.
As with all models, the one presented here also has its own limitations. For one, iPSC-derived organoids are developmentally immature19 and, thus, have important differences compared to the aging brain, in which many neurodegenerative diseases manifest. Cerebral organoids are also non-uniform in development19, thus precluding consistent injection into the same exact physiological niche. Moreover, while they contain the cell types of the relevant brain regions18, 19, they lack the endothelial, microglial, and immune components, which are also important in the in vivo setting14. This limits the study of how the host will react to the cell transplantation. Techniques are currently coming online to add vascular28 and microglial29 cells, as well as to increase the organoid consistency and regionalization18, thus improving the modeling power of the organoid transplantation system. They would, however, require further testing and optimization beyond what is presented here. While this protocol is inexpensive and does not require specialized equipment, there remain a number of important technical considerations-the injection depth, for example. This is both due to the fact that organoids are not perfused and, thus, often have a necrotic center if they grow too large19 and that light cannot penetrate through the organoid core for live-cell tracking. Thus, the cells that have been injected too deep and colonies that have migrated inside may be missed. While this can be ameliorated by the use of longer-wavelength fluorophores with better tissue penetrance30, depending on the organoid size and detection apparatus, this will likely remain a consideration. Finally, as brain organoids are in a state of development, the transplantation timing is another key consideration, as the environment will likely differ depending on the developmental stage of the organoid into which it is injected. While this can be controlled to some extent by ensuring a consistent organoid age at time of injection, it is, without doubt, a factor that needs consideration.
This protocol is inexpensive, simple, animal-free, and does not require specialized equipment, thus making transplantation modeling accessible to a wider variety of labs. With the rapid pace of advancement both in neural cell therapeutics and organoid model systems, we anticipate that the organoid transplantation protocol presented here will be a useful model for a range of diseases and therapeutic approaches.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Funds for this work were provided through the IRIC Philanthropic funds from the Marcelle and Jean Coutu foundation and from the Fonds de recherche du Québec - Santé (FRQS #295647). D.J.H.F.K. has salary support from FRQS in the form of a Chercheurs-boursiers Junior 1 fellowship (#283502). M.I.I.R. was supported by an IRIC Doctoral Award from the Institut of Research in Immunology and Cancer, a Bourse de passage accélère de la maitrise au doctorat from the University of Montreal, and Bourse de Mérite aux cycles supérieurs.
Materials
| Name | Company | Catalog Number | Comments |
| Accutase | StemCell Technologies | 7920 | proteolytic-collagenolytic enzyme mix |
| Alexa Fluor 488 anti-GFP Antibody | BioLegend | 338008 | |
| Alexa Fluor 488 anti-MAP2 (clone SMI 52) | BioLegend | 801804 | |
| Alexa Fluor 594 anti-GFAP Antibody (clone SMI 25) | BioLegend | 837510 | |
| Alexa Fluor 594 anti-Nestin (clone 10C2) | BioLegend | 656804 | |
| Alexa Fluor 647 anti-Tubulin β 3 (TUBB3) (clone TUJ1) | BioLegend | 801209 | |
| Citric Acid Monohydrate | Fisher Chemical | A104-500 | |
| Cytation 5 Cell Imaging Multimode Reader | Biotek | - | |
| Denaturated Ethyl Alcohol (Anhydrous) | ChapTec | - | |
| DMEM F12/Glutamax | Thermo | 10565018 | |
| Dymethil Sulfoxide (DMSO), Sterile | BioShop | DMS666.100 | |
| FIJI 1.53c | - | - | |
| Formalin solution, neutral buffered, 10% | Sigma | HT501128-4L | |
| Gen5 | - | - | |
| HistoCore Arcadia H | Leica Biosystems | - | |
| Matrigel Growth Factor Reduced (GFR) | Corning | 356231 | Phenol Red-free, LDEV-free |
| MX35 microtome blade | Epredia | 3053835 | |
| NaOH | Sigma | 655104 | |
| PBS (-Ca -Mg) | Sigma | D8537 | |
| Puromycin Dihydrochloride | Thermo | A1113803 | |
| ROCK inhibitor Y-27632 | Abcam | ab120129 | |
| Simport Scientific Stainless-Steel Base Molds | Fisher Scientific | 22-038-209 | |
| Simport Scientific UNISETTE Biopsy Processing/Embedding Cassette | Fisher Scientific | 36-101-9255 | |
| STEMdiff Forebrain Neuron Differentiation Kit | StemCell Technologies | 8600 | |
| STEMdiff Neural Progenitor Medium | StemCell Technologies | 5833 | |
| STEMdiff SMADi Neural Induction Kit | StemCell Technologies | 8581 | |
| Thermo Scientific Shandon Finesse ME Microtome | Thermo Scientific | - | |
| Tissue Prep | Fisher Scientific | T555 | |
| Tissue-Tek VIP 6 AI Tissue Processor | Sakura Finetek | - | |
| Toluene (histological) | ChapTec | - | |
| Trypan blue; 0.4% (wt/vol) | Thermo | 15250061 | |
| Tween 20 | BioShop | TWN510.100 |
References
- Spurlock, M. S., et al. Amelioration of penetrating ballistic-like brain injury induced cognitive deficits after neuronal differentiation of transplanted human neural stem cells. Journal of Neurotrauma. 34 (11), 1981 (2017).
- Zhou, Y., Shao, A., Xu, W., Wu, H., Deng, Y. Advance of stem cell treatment for traumatic brain injury. Frontiers in Cellular Neuroscience. 13, 301 (2019).
- Hayashi, Y., Lin, H. -. T., Lee, C. -. C., Tsai, K. -. J. Effects of neural stem cell transplantation in Alzheimer's disease models. Journal of Biomedical Science. 27 (1), 29 (2020).
- Kefalopoulou, Z., et al. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: Two case reports. JAMA Neurology. 71 (1), 83-87 (2014).
- Li, W., et al. Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proceedings of the National Academy of Sciences of the United States of America. 113 (23), 6544-6549 (2016).
- Parmar, M., Grealish, S., Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nature Reviews Neuroscience. 21 (2), 103-115 (2020).
- Takahashi, J. iPS cell-based therapy for Parkinson's disease: A Kyoto trial. Regenerative Therapy. 13, 18-22 (2020).
- Krause, M., Phan, T. G., Ma, H., Sobey, C. G., Lim, R. Cell-based therapies for stroke: Are we there yet. Frontiers in Neurology. 10, 656 (2019).
- Coles-Takabe, B. L. K., et al. Don't look: Growing clonal versus nonclonal neural stem cell colonies. Stem Cells. 26 (11), 2938-2944 (2008).
- Duval, K., et al. Modeling physiological events in 2D vs. 3D cell culture. Physiology. 32 (4), 266-277 (2017).
- Jakel, R. J., Schneider, B. L., Svendsen, C. N. Using human neural stem cells to model neurological disease. Nature Reviews Genetics. 5 (2), 136-144 (2004).
- Hodge, R. D., et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 573 (7772), 61-68 (2019).
- Li, J., et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nature Communications. 12 (1), 3958 (2021).
- Morizane, A., et al. MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nature Communications. 8 (1), 385 (2017).
- Khrameeva, E., et al. Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Research. 30 (5), 776-789 (2020).
- Powell, K. Hybrid brains: The ethics of transplanting human neurons into animals. Nature. 608 (7921), 22-25 (2022).
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience. 305, 86-98 (2015).
- Birey, F., et al. Assembly of functionally integrated human forebrain spheroids. Nature. 545 (7652), 54-59 (2017).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- García-Delgado, A. B., et al. Brain organoids to evaluate cellular therapies. Animals. 12 (22), (2022).
- Lin, J. -. R., Fallahi-Sichani, M., Sorger, P. K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nature Communications. 6 (1), 8390 (2015).
- Knapp, D. J. H. F., et al. Single-cell analysis identifies a CD33+ subset of human cord blood cells with high regenerative potential. Nature Cell Biology. 20 (6), 710-720 (2018).
- Georgala, P. A., Carr, C. B., Price, D. J. The role of Pax6 in forebrain development. Developmental Neurobiology. 71 (8), 690-709 (2011).
- Britanova, O., et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 57 (3), 378-392 (2008).
- Kim, H., et al. Modeling G2019S-LRRK2 sporadic Parkinson's disease in 3D midbrain organoids. Stem Cell Reports. 12 (3), 518-531 (2019).
- Smits, L. M., et al. Modeling Parkinson's disease in midbrain-like organoids. NPJ Parkinson's Disease. 5, 5 (2019).
- Jin, M., et al. Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer's disease. Cell Stem Cell. 29 (7), 1135.e8-1153.e8 (2022).
- Sun, X. -. Y., et al. Generation of vascularized brain organoids to study neurovascular interactions. eLife. 11, e76707 (2022).
- Popova, G., et al. Human microglia states are conserved across experimental models and regulate neural stem cell responses in chimeric organoids. Cell Stem Cell. 28 (12), 2153.e6-2166.e6 (2021).
- Wang, S., Li, B., Zhang, F. Molecular fluorophores for deep-tissue bioimaging. ACS Central Science. 6 (8), 1302-1316 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved