
Method Article
اللحاق بالركب: خط أنابيب عالي الإنتاجية لبيانات ATAC-Seq و ChIP-Seq المجمعة
In This Article
Summary
يسمح ATAC-seq و ChIP-seq بالتحقيق التفصيلي لتنظيم الجينات. ومع ذلك ، فإن معالجة أنواع البيانات هذه تمثل تحديا وغالبا ما تكون غير متسقة بين مجموعات البحث. نقدم CATCH-UP: خط أنابيب حسابي سهل الاستخدام يسمح بمعالجة البيانات الموحدة والقابلة للتكرار وتحليلها لمجموعات بيانات ATAC / ChIP-seq الجديدة والمنشورة.
Abstract
أحدث اختبار الكروماتين الذي يمكن الوصول إليه عبر الترانسبوزاز (ATAC) والترسيب المناعي للكروماتين (ChIP) ، إلى جانب تسلسل الجيل التالي (NGS) ، ثورة في دراسة تنظيم الجينات. أدى الافتقار إلى التوحيد القياسي في تحليل مجموعات البيانات عالية الأبعاد الناتجة عن هذه التقنيات إلى صعوبة تحقيق التكرار ، مما أدى إلى تناقضات في البيانات المنشورة والمعالجة. يرجع جزء من هذه المشكلة إلى مجموعة متنوعة من أدوات المعلوماتية الحيوية المتاحة لتحليل هذه الأنواع من البيانات. ثانيا ، هناك حاجة إلى عدد من أدوات المعلوماتية الحيوية المختلفة بالتتابع لتحويل البيانات الأولية إلى مخرجات معالجة بالكامل وقابلة للتفسير ، وتتطلب هذه الأدوات مستويات متفاوتة من المهارات الحسابية. علاوة على ذلك ، هناك العديد من الخيارات لمراقبة الجودة التي لا يتم استخدامها بشكل موحد أثناء معالجة البيانات. نعالج هذه المشكلات من خلال فحص كامل لتسلسل الكروماتين الذي يمكن الوصول إليه بواسطة الترانسبوزاز (ATAC-seq) وتسلسل الترسيب المناعي للكروماتين (ChIP-seq) خط أنابيب المنبع (CATCH-UP) ، وهو خط أنابيب سهل الاستخدام قائم على Python لتحليل مجموعات بيانات ChIP-seq و ATAC-seq المجمعة من ملفات fastq الأولية إلى المسارات الكبيرة القابلة للتصور ومكالمات الذروة. خط الأنابيب هذا سهل التثبيت والتشغيل ، ويتطلب الحد الأدنى من المعرفة الحسابية. خط الأنابيب معياري وقابل للتطوير ومتوازي على مختلف البنى التحتية للحوسبة ، مما يسمح بإعداد تقارير سهلة عن المنهجية لتمكين التحليل القابل للتكرار لمجموعات البيانات الجديدة أو المنشورة.
Introduction
يجب تنظيم التعبير الجيني بإحكام حتى تتمكن الخلايا من إنشاء وظيفتها البيولوجية الصحيحة والحفاظ عليها. من المعروف أن التعبير الجيني الشاذ يكمن وراء التسبب في العديد من الأمراض ، وبالتالي ، يكمن قدر كبير من الاهتمام البحثي في فهم آليات تنظيم الجينات1. يتم تسهيل التعبير الجيني من خلال العناصر التنظيمية مثل المحفزات والمعززات. ضمن تسلسلها ، تحتوي هذه العناصر على مواقع ربط عامل النسخ (TF) ، والتي ، عند نشاطها ، توفر منصة لربط TF. يؤدي ارتباط TFs في هذه المواقع إلى إزاحة النيوكليوسومات ، مما يؤدي إلى زيادة إمكانية الوصول إلى الحمض النووي وزيادة لاحقة في السماح بآلية النسخ. نتيجة لهذا الوصول المتزايد ، تكون مناطق الحمض النووي هذه أكثر حساسية للنوكليازات والينقولات مثل DNase و Tn5 ، وهي خاصية كيميائية حيوية تم استغلالها من قبل الباحثين الذين يحققون في تنظيم النسخ2،3.
يسمح DNase-seq و ATAC-seq للباحثين برسم خرائط لمناطق الكروماتين المفتوح ومواقع ربط TF وتحديد المواقع النووية عبر الجينوم. من بين هاتين التقنيتين ، نمت شعبية ATAC-seq على مدار العقد الماضي بسبب البروتوكول البسيط المكون من خطوتين ومتطلبات عدد الخلايا المنخفض (50,000 خلية مقارنة ب 1 مليون لكل تكرار ل DNase-seq). بينما يقدم ATAC-seq نظرة عامة على المشهد العام للكروماتين في مجموعة من الخلايا ، إلا أنه لا أدري إلى حد كبير الذي ترتبط به بروتينات معينة بالجينوم4،5. من أجل تحديد المواقع التي يتفاعل فيها بروتين معين مع الجينوم ، فإن التقنية القياسية الذهبية هي Chromatin Immunoprecipitation (ChIP)-seq. يتضمن ChIP-seq التثبيت الكيميائي لتفاعلات البروتين والحمض النووي في الخلية ، متبوعا بالترسيب المناعي ("المنسدل") باستخدام جسم مضاد خاص بالبروتين محل الاهتمام لاختيار شظايا الحمض النووي المرتبطة بالبروتين محل الاهتمام (POI). يمكن تسلسل شظايا الحمض النووي هذه للكشف عن مواقع الارتباط الجينومي لبروتينات معينة مثل TFs ، أو المواقع التي تحتوي على تعديلات هيستون محددة1. من خلال الجمع بين مجموعات بيانات ATAC-seq و ChIP-seq ، يمكن اشتقاق صورة مفصلة للمشهد التنظيمي لمجموعة من الخلايا.
سير العمل الأساسي المطلوب للتحليل هو كما يلي: يجب التحكم في جودة قراءات التسلسل الخام قبل المحاذاة مع الجينوم المرجعي ("رسم الخرائط"). يمكن بعد ذلك تصفية القراءات المعينة بنجاح لإزالة كل من القراءات منخفضة الجودة وتكرارات PCR. من أجل تصور هذه القراءات المعينة والمفلترة ، من الضروري حساب "تغطية" هذه القراءات عبر الجينوم. يؤدي هذا إلى إنشاء ملف يمكن تحميله إلى متصفح الجينوم مثل عرض متعدد المواقع (MLV) أو متصفح الجينوم UCSC ك "مسار"6،7. عادة ما يتم تحديد الذروة ، أو "استدعاء الذروة" لمسارات التغطية هذه باستخدام أدوات مثل LanceOtron أو MACS2 8,9. أخيرا ، من خلال تحليل موقع الذروة والشكل والحجم ، يمكن إجراء مقارنات بين العينات أو الظروف البيولوجية. يعد تحليل مجموعات البيانات هذه ودمجها عملية معقدة متعددة الخطوات يمكن من خلالها تنفيذ مجموعات مختلفة من أدوات المعلوماتية الحيوية. قد تكون الإصدارات المختلفة من الأدوات غير متوافقة مع بعضها البعض وقد تغير إخراج معالجة البيانات. هناك أيضا مجموعة متنوعة في القوة الحسابية وكفاءة المستخدم المطلوبة لتنفيذ أجزاء مختلفة من معالجة البيانات كما هو موضح في خطوط أنابيبnf-core 10 أوpanpipes 11 أو genpipes12 أو PEPATAC13 أو ChIP-AP14.
بشكل عام ، أدى هذا إلى تناقضات في كل من تحليل التحليل والإبلاغ عنه ، مما أدى بدوره إلى ضعف قابلية التكرار وإمكانية الوصول والراحة لأي شخص لديه معرفة محدودة بالمعلوماتية الحيوية. نعالج كل هذه المشكلات من خلال CATCH-UP (خط أنابيب ATAC-seq و ChIP-seq الكامل ، وهو خط أنابيب سهل الاستخدام ومرن ومعياري لمعالجة بيانات ChIP-seq و ATAC / DNase- seq. يتطلب تنفيذ اللحاق بالركب الحد الأدنى من الخبرة في المعلوماتية الحيوية. يمكن تشغيله على بنى تحتية مختلفة للحوسبة ويتيح تحليل البيانات القابلة للتكرار داخل مجموعات البحث وعبرها.
CATCH-UP هو خط أنابيب Snakemake قائم على Python تم تصميمه لتوحيد تحليل بيانات ChIP-seq و ATAC-seq. يأخذ بيانات التسلسل الأولية (ملفات fastq.gz) كمدخلات ويولد مخرجات في شكل ملفات الذروة (.bed) التي توفر النتيجة المعنية لكل خطوة. نحن نقدم ملف تكوين بتنسيق yaml (config.yaml) ، حيث يمكن للمستخدم تحرير معلمات كل خطوة تحليل. يتيح نظام الإدارة المنفذة داخل snakemake استخدام بنى تحتية مختلفة للحوسبة (مثل الخوادم أو المجموعات أو الأنظمة السحابية أو أجهزة الكمبيوتر الشخصية) وبالتوازي إذا كان المستخدم يوفر كمية كبيرة من البيانات.
أدناه ، نقدم وصفا تفصيليا لكل خطوة من خطوات سير العمل (انظر الشكل 1 للحصول على توضيح سير العمل). هذا التفسير ضروري لاتباع خطوة بخطوة في قسم البروتوكول:
نقل fastq: تتمثل الخطوة الأولى في المسار في نسخ ملفات fastq الأولية إلى دليل التحليل المسمى. هذا يترك البيانات الأصلية كما هي لتجنب إتلاف أو تعديل ملفات البيانات الأولية.
التسلسل: إذا كانت بيانات التسلسل الأولية تحتوي على ممرات متعددة ، فهذه الخطوة مطلوبة لتسلسل الممرات قبل التحليل. بشكل افتراضي، يعالج المسار جميع ملفات fastq كعينات فردية. يجب تحديد خطوة التسلسل هذه في ملف التكوين.
التشذيب: خطوة تنظيف البيانات الاختيارية. يسمح ذلك بقص القراءات منخفضة الجودة أو تسلسلات المحول باستخدام trimmomatic15. يمكن للمستخدم توفير ملفات fasta مخصصة لتسلسلات المحول. يتم توفير مثال في دليل المحول. يمكن تحديد معلمات التشذيب الإضافية في ملف التكوين. بشكل افتراضي، يتخطى سير العمل هذه القاعدة.
التقويم: للمحاذاة ، يتم تطبيق Bowtie216 افتراضيا ؛ يمكن أيضا تحديد أدوات المحاذاة البديلة مثل BWA-MEM217 . يتم تحديد أداة محاذاة Bowtie2 كإعداد افتراضي لأنها بارعة بشكل خاص في محاذاة القراءات القصيرة نسبيا مع الجينومات الكبيرة نسبيا ، وبالتالي فهي مناسبة تماما لمحاذاة بيانات ChIP-seq و ATAC-seq مع جينومات الثدييات. لتجنب أي ملفات وسيطة ، يتم إدخال المحاذاة في عرض samtools لحفظ ملف bam في الإخراج. بالنسبة لهذه القاعدة ، يجب على المستخدم تحديد بناء الجينوم المفضل الذي يتم تعيين القراءات عليه على سبيل المثال ، hg19 / hg38 (إنسان) ، mm10 / mm39 (الماوس).
التصفية: يتم الاحتفاظ بالقراءات المعينة بشكل صحيح ، ويتم تصفية القراءات ذات الجودة المنخفضة. الافتراضي: عرض samtools ، مع المعلمات: -bShuF 4 -f 3 -q 30.
الفرز: يتم فرز القراءات المحاذاة بترتيب الإحداثيات في أقصى اليسار. الافتراضي: فرز samtools (غلاف صنع الثعابين) ، مع المعلمة: -m 4G.
وضع علامة على التكرارات: يتم تحديد جميع عمليات القراءة المكررة ووضع علامة عليها. يمكن للمستخدم أن يقرر إزالتها عن طريق تغيير معلمة ملف التكوين. الافتراضي: Picard MarkDuplicates (غلاف snakemake) ، مع المعلمة: --REMOVE_DUPLICATES False للإبلاغ عن التكرارات والاحتفاظ بها.
دمج بام: إذا كانت بيانات التسلسل تتكون من نسخ متماثلة أو عينات ، فقد يرغب المستخدم في الدمج في بام واحد. في هذه الحالة ، يمكن للمستخدم اختيار دمج bams أو الاحتفاظ بملفات bam منفصلة طوال التحليل. إذا اختار المستخدم دمج bams (استخدام دمج samtools) ، فيجب تحديد بادئة مشتركة ل bams المدمجة.
الفهرس: تقوم هذه الخطوة بفهرسة الإحداثيات التي تم فرزها. الافتراضي: فهرس samtools (غلاف snakemake) ، باستخدام المعلمات الافتراضية المحددة بواسطة samtools.
BamCoverage: تنشئ هذه القاعدة مسار تغطية كبير من القراءات المحاذاة. يتم تطبيق أداة bamCoverage من deepTools ، ويتم حساب التغطية على أنها عدد القراءات لكل سلة ، حيث تمثل الحاوية نافذة بحجم محدد. في هذا المسار ، يتم تطبيق bamCoverage مع تعيين المعلمات التالية كافتراضي: -bs 1 -normalizeUsing RPKM -extendReads.
ذروة الاتصال: تم تحديد LanceOtron8 كمستدعي الذروة الافتراضي لخط الأنابيب هذا. على عكس المتصلين التقليديين بالذروة ، والتي تعتمد في الغالب على الاختبار الإحصائي ، فإن LanceOtron عبارة عن متصل ذروة قائم على التعلم العميق ، والذي يتضمن قياسات الإثراء الجيني والاختبارات الإحصائية وقد ثبت أنه يتفوق على متصل الذروة القياسي في الصناعة ، MACS29. لكي تكون الشخصيات البارزة متوافقة مع LanceOtron ، يجب حساب التغطية لكل زوج أساسي ، وتطبيع RPKM ؛ ينعكس هذا في الإعدادات الافتراضية لخطوة BamCoverage. يمكن تحديد MACS2 كمتصل ذو ذروة بديل. ستتم مراقبة إصدار المتصلين الجدد بالذروة ودمجها حسب الاقتضاء من أجل الحفاظ على أداء خط أنابيب التحليل هذا وتحسينه.
TrackDb: يؤدي هذا إلى إنشاء اقتران زوج من المفتاح والقيمة للملفات الكبيرة من أجل تحميلها وتصورها في أدوات مثل الأنظمة الأساسية MLV6 أو UCSC Genome Browser18 .
بالإضافة إلى بيانات الإخراج، تقوم كل خطوة من خطوات المسار بإخراج ملف سجل، ويتم توفير فحوصات مراقبة الجودة المناسبة حتى يتمكن المستخدم من تتبع تقدم التحليل. يتم تطبيق FastQC19 على بيانات التسلسل الأولية والمقتطعة (إذا تم تحديدها) (الخطوات 1 - نقل fastq و 2- التشذيب). تستخدم إحصائيات Samtools بالإضافة إلى MultiQC20 لجمع وإنتاج وتصور تقارير مراقبة الجودة على ملفات bam في الإخراج في الخطوات 3 - Aligner و 6 - وضع علامة على التكرارات و 7 - دمج bam. لمزيد من المعلومات حول كل أداة من الأدوات المطبقة في الخطوات أعلاه ، انظر الجدول 1.
Protocol
1. تشغيل خط أنابيب اللحاق بالركب
- استنساخ مستودع UpStreamPipeline من https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
انتقل إلى دليل العمل المختار ، وانسخ التعليمات البرمجية التالية وقم بتشغيلها على سطر الأوامر:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - انتقل داخل مجلد UpStreamPipeline الذي تم تنزيله باستخدام الأمر: cd UpStreamPipeline
- قم بتثبيت توزيع anaconda (إذا لزم الأمر):
- تحقق مما إذا كان anaconda مثبتا بالفعل على النظام باستخدام الأمر which conda. إذا لم يظهر الأمر أي مسار لأي توزيعة conda ، فقم بتنزيل mambaforge من https://github.com/conda-forge/miniforge#mambaforge وحدد التوزيع والإصدار المناسبين للنظام. على سبيل المثال ، بالنسبة لمستخدمي Linux ، استخدم wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. قم بزيارة صفحة الويب هذه لأنظمة التشغيل المختلفة: https://github.com/conda-forge/miniforge/.
- قم بتشغيل المثبت باستخدام sh Mambaforge-Linux-x86_64.sh ، وتهيئة conda في النظام عن طريق تشغيل conda init.
- تثبيت وتنشيط بيئة conda الأولية (متطلبات بيئة conda الأولية مدرجة في الجدول 2):
- قم بتثبيت البيئة باستخدام الأمر mamba env create - file=envs/upstream.yml.
- قم بتنشيط البيئة باستخدام الأمر conda activate upstream.
- بمجرد تثبيت بيئة conda الأولية بنجاح، قم بتنشيط البيئة باستخدام الأمر conda activate upstream وانتقل إلى المجلد CATCH-UP باستخدام cd genetics/CATCH-UP.
- قم بتحرير ملف التكوين ، والذي يمكن العثور عليه داخل مجلد التكوين باستخدام الأمر cd /config/analysis.yaml ، وقم بتعديله وفقا لمواصفات التحليل باستخدام محرر نصوص. اتبع التعليمات سطرا بسطر لتحرير كل معلمة داخل الملف نفسه. سيتم الاحتفاظ بهذا الملف بعد التحليل والعمل على توثيق معلمات التشغيل للمساعدة في التكرار.
- افتح الملفات الثلاثة التالية وقم بتحريرها في محرر نصوص (على سبيل المثال ، TextEdit لنظام التشغيل Mac أو Notepad لنظام التشغيل Windows):
- قم بتحرير 1_fastqfile_home_dir.txt الملف ليحتوي على قائمة بجميع ملفات fastq المراد تحليلها.
ملاحظة: يجب استبعاد أرقام القراءة والملحقات (على سبيل المثال، _R1/_R2 و .fastq.gz). على سبيل المثال، إذا كان المشروع يحتوي على قائمة ملفات fastq هذه:
Sample1_conditionA_L001_R1 . فاستق . جي جي
Sample1_conditionA_L001_R2 . فاستق . جي جي
Sample1_conditionA_L002_R1 . فاستق . جي جي
Sample1_conditionA_L002_R2 . فاستق . جي جي
Sample1_conditionB_L001_R1 . فاستق . جي جي
Sample1_conditionB_L001_R2 . فاستق . جي جي
Sample1_conditionB_L002_R1 . فاستق . جي جي
Sample1_conditionB_L002_R2 . فاستق . جي جي
Sample2_conditionA_L001_R1 . فاستق . جي جي
Sample2_conditionA_L001_R2 . فاستق . جي جي
Sample2_conditionA_L002_R1 . فاستق . جي جي
Sample2_conditionA_L002_R2 . فاستق . جي جي
Sample2_conditionB_L001_R1 . فاستق . جي جي
Sample2_conditionB_L001_R2 . فاستق . جي جي
Sample2_conditionB_L002_R1 . فاستق . جي جي
Sample2_conditionB_L002_R2 . فاستق . جي جي
في هذه الحالة ، يكون 1_fastqfile_home_dir.txt كما يلي:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - إذا كانت البيانات الأولية تحتوي على ممرات تسلسل تتطلب التسلسل، فقم بتحرير ملف 2_fastqfile_concat.txt لتحديد بادئة أسماء الملفات المراد تسلسلها. إذا لم تكن هناك ممرات تسلسل للتسلسل ، فلا تقم بتحرير 2_fastqfile_concat.txt. تأكد من أن كل سطر من 2_fastq le_concat.txtfi يحتوي على بادئة عينة واحدة على النحو التالي:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - إذا كان دمج بيانات العينات المختلفة مطلوبا ، فقم بتحرير 3_merge_bams.txt الملف ببادئة أسماء الملفات المراد دمجها. تأكد من أن كل سطر يحتوي على بادئة عينة واحدة على النحو التالي:
عينة 1
عينة 2
يوضح الشكل 2 ملخصا لكيفية تلخيص هذه الملفات الثلاثة. يمكن تطبيق البروتوكول على بيانات التسلسل الفردية أو المزدوجة. يتم تعيين المسار افتراضيا إلى تحليل الطرف المقترن ما لم ينص على خلاف ذلك. يمكن تعديل ذلك في ملف التكوين (راجع الخطوة 1.6).
- قم بتحرير 1_fastqfile_home_dir.txt الملف ليحتوي على قائمة بجميع ملفات fastq المراد تحليلها.
- بمجرد تحرير جميع الملفات المطلوبة ، استخدم snakemake لتشغيل CATCH-UP على النحو التالي: snakemake --configfile=config/analysis_name.yaml all --cores 4.
ملاحظة: لمزيد من الإرشادات والوثائق التفصيلية، راجع مجلد CATCH-UP داخل مستودع UpStreamPipeline GitHub، المتوفر هنا. يتضمن ذلك وثائق مفصلة حول تعديل ملف التكوين بشكل صحيح ، من تغيير مسارات تسلسل ملفات البيانات وتخزين النتائج إلى تحرير المعلمات لكل خطوة.
النتائج
ينتج خط أنابيب CATCH-UP نتيجة وسجل وإخراج مراقبة الجودة (QC) لكل خطوة. داخل ملف التكوين ، يمكن للمستخدم اختيار إما الاحتفاظ بملفات الإخراج أو إزالتها لتقليل ذاكرة التخزين المطلوبة. يتم شرح جميع المخرجات على النحو التالي:
00. fastq_home_dir: يتم نسخ ملف التكوين و FastQFile_home_dir.txt و merge_bams.txt إلى هذا المجلد للرجوع إليها وقابلية التكرار.
01. يقرأ: يتم نسخ ملفات FastQ إلى هذا المجلد لتجنب التغييرات في البيانات الأولية الأصلية أثناء عملية سير العمل ، ويمكن تسلسل الممرات إذا تم تحديدها.
02. التشذيب: ملفات FastQ مع قراءة ومحولات مقطوعة إذا تم تحديدها.
03. التقويم: المحاذاة مع الجينوم المحدد.
04. FILTERING: تصفية مراقبة الجودة.
05. فرز: فرز ملفات BAM.
06. التكرارات: وضع علامة على التكرارات.
07. دمج: دمج ملفات BAM إذا تم تحديد ذلك في config.yaml.
08. bam_coverages: ملف bigwig من التغطية.
09. peak_calling: ملف سرير لإخراج استدعاء الذروة LanceOtron.
10. المسار: ينتج ملفا نصيا منسقا جاهزا للاستخدام في متصفح الجينوم إذا لزم الأمر.
بالنسبة للمخرجات 01 و 02 و 03 و 06 و 07 ، يتم توفير مقاييس مراقبة الجودة وملفات HTML. بالإضافة إلى ذلك ، في الشكل 3 ، نقدم مثالا على البيانات المعالجة باستخدام CATCH-UP ، وتصور المخرجات النهائية من خلال منصة MLV.

الشكل 1: سير عمل اللحاق بالركب. بالنظر إلى قائمة ملفات fastq ، يعالج CATCH-UP بالتوازي جميع العينات من خلال جميع خطوات المنبع. الرجاء النقر هنا لعرض نسخة أكبر من هذا الرقم.
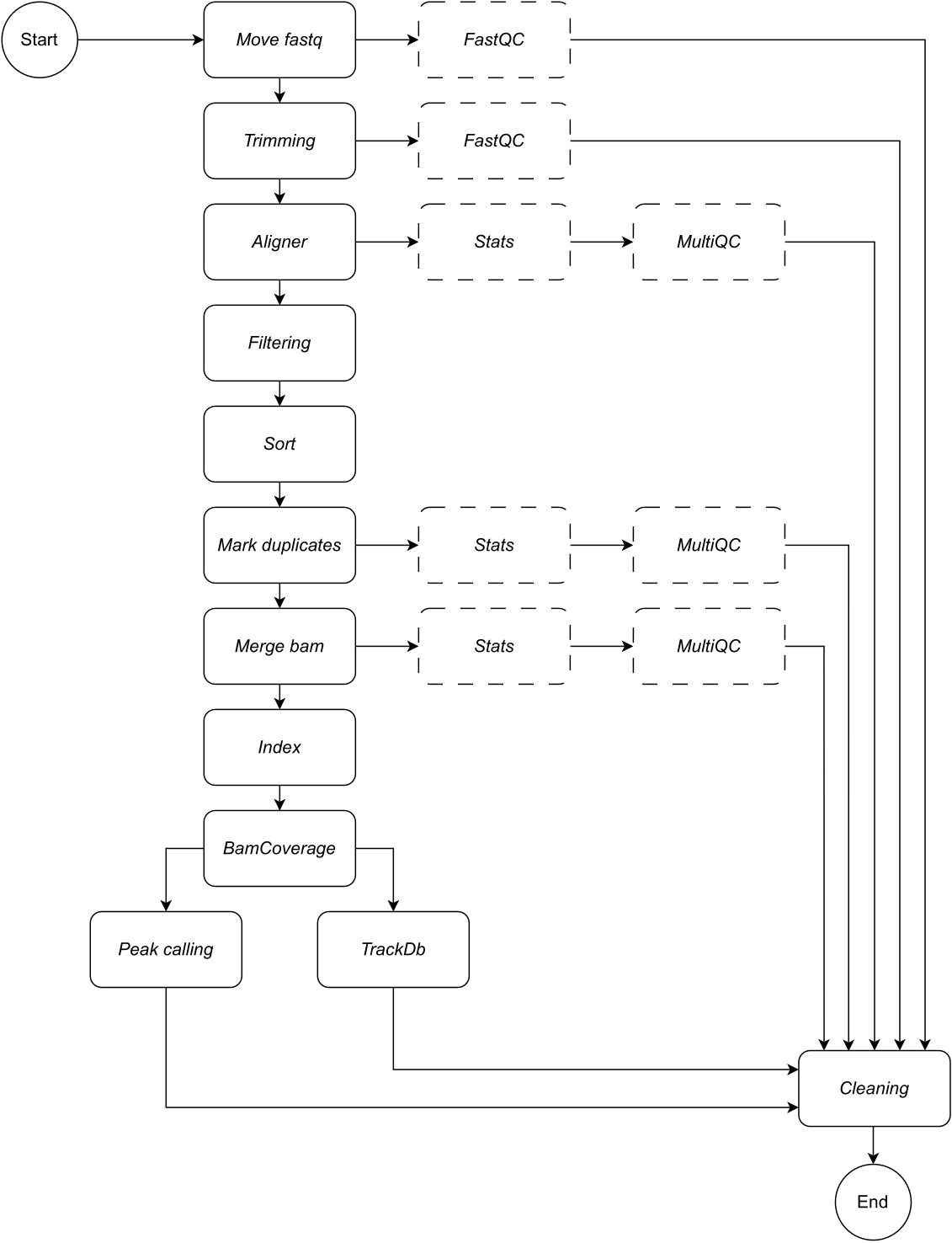{kind=link}

الشكل 2: تمثيل توضيحي يشرح كيف يجب تعديل 1_fastqfile_home_dir.txt و 2_fastqfile_concat.txt و 3_merge_bams.txt بشكل صحيحمن أجل تشغيل اللحاق بالركب. الرجاء النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}

الشكل 3: مثال على الإخراج من خط أنابيب CATCH-UP. تم تنزيل بيانات التسلسل الأولية (ملفات fastq) من ENCODE21. تم استخدام خط أنابيب CATCH-UP لمعالجة ملفات fastq ل DNase-seq و 5 أنواع من ChIP-seq (H3K4me1 و H3K4me3 و H3K27ac و CTCF و POLR2A). تم تحميل ملفات إخراج Bigwig إلى Multi Locus View لتصور وتحديد العناصر التنظيمية الجينومية. الرجاء النقر هنا لعرض نسخة أكبر من هذا الرقم.
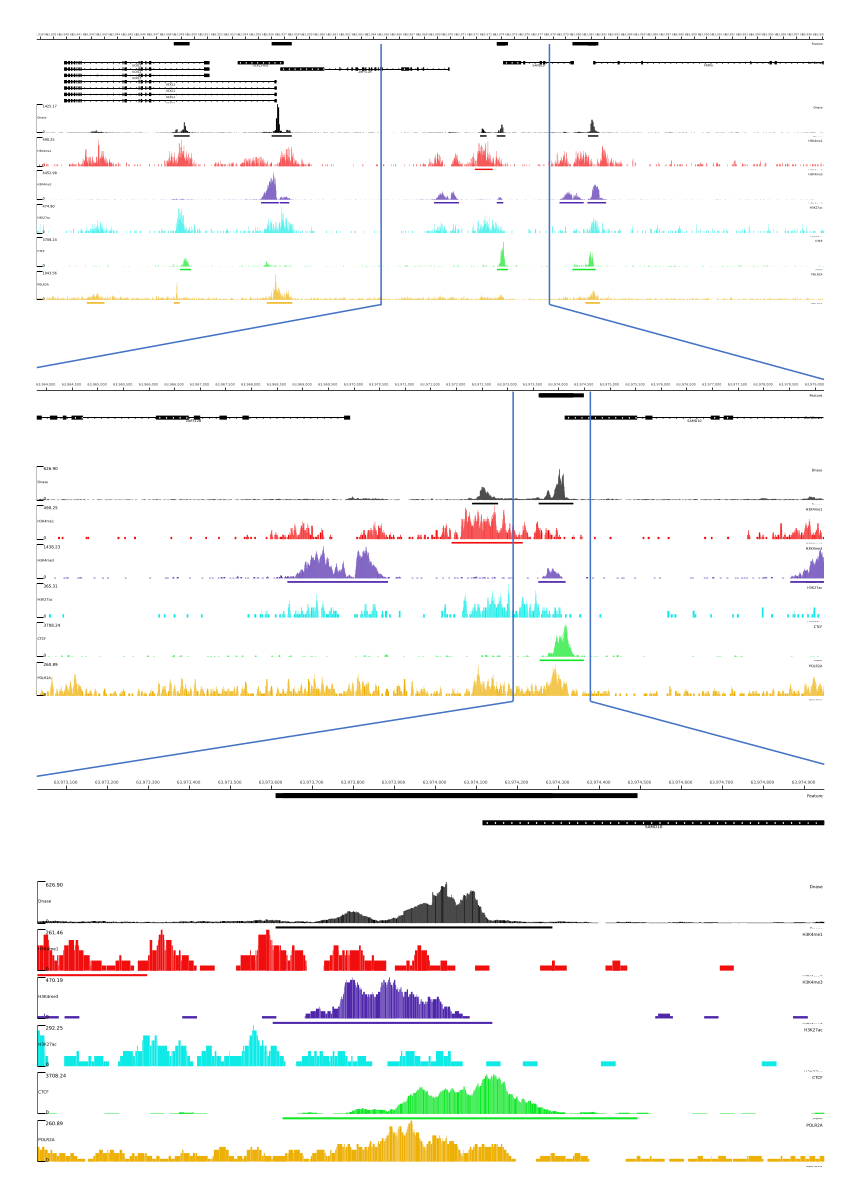{kind=link}
الجدول 1: موارد الوثائق. يعرض هذا الجدول الأدوات المتضمنة في سير عمل CATCH-UP ورابط وثائقها والمراجع ذات الصلة. الرجاء النقر هنا لتنزيل هذا الجدول.
الجدول 2: قائمة متطلبات القناة والتبعية لبيئة conda الأولية. الرجاء النقر هنا لتنزيل هذا الجدول.
الجدول 3: أنظمة التشغيل المستخدمة لاختبار اللحاق بالركب. تم اختبار Ubuntu على مجموعة عالية الأداء وجهاز محلي. الرجاء النقر هنا لتنزيل هذا الجدول.
Discussion
وقد قابلت زيادة امتصاص واستخدام تقنيات NGS لتوليد البيانات الجينومية زيادة في تطوير أدوات المعلوماتية الحيوية لتحليل هذه البيانات. هناك العديد من الأدوات التي يمكن تطبيقها لكل خطوة من خطوات تحليل البيانات ، بالإضافة إلى العديد من المعلمات المختلفة التي يمكن تحديدها داخل كل أداة6،8،9،15،16،17،18،19،20،22،23،24. هذا يجعل مجموعة متنوعة إلى حد كبير من استراتيجيات التحليل التي يمكن تطبيقها ، والتي يمكن لكل منها أن ينتج اختلافات في النتيجة. من أجل المقارنة بدقة عبر التجارب ، يعد توحيد تحليل المعلوماتية الحيوية أمرا ضروريا. تاريخيا ، يتم إنشاء بيانات NGS بواسطة علماء المختبرات الرطبة ، ويتم تحليل البيانات بواسطة علماء المعلوماتية الحيوية.
يمكن تقسيم تحليل بيانات NGS إلى خطوط أنابيب "المنبع" و "المصب" ، حيث يتضمن المنبع الخطوات اللازمة للانتقال من إخراج البيانات الأولية من آلة التسلسل إلى تنسيق قابل للتفسير بصريا من قبل الباحث. يتضمن التحليل النهائي خطوات إضافية مخصصة لسؤال البحث والتصميم التجريبي. لذلك ، فإن خطوط أنابيب المنبع قابلة للتعميم وقابلة للتوحيد القياسي لتحسين قابلية التكاثر العلمي. من ناحية أخرى ، فإن خطوط الأنابيب النهائية مخصصة ، وتعتمد على السؤال البيولوجي ، وتتطلب نظرة ثاقبة من المحقق ، مما يجعلها أقل ملاءمة للتوحيد القياسي. لقد أنشأنا خط أنابيب سهل الاستخدام يسمح لعلماء المختبرات الرطبة بتحليل بياناتهم الخاصة بشكل متكرر دون الحاجة إلى أي معرفة مسبقة بالمعلوماتية الحيوية. هنا ، نقدم CATCH-UP ، وهو خط أنابيب تم إنشاؤه باستخدام إطار عمل snakemake ومصمم ليكون سهل الاستخدام ولمكافحة مشكلة التكرار في تحليل بيانات ChIP-seq و ATAC-seq. تم إنشاء خط الأنابيب هذا للتعامل مع بيانات ChIP-seq أو ATAC-seq. بمجرد قيام المستخدم بتنزيل CATCH-UP، يجب أولا تحديد معلمات التحليل وتسمية العينة قبل تشغيل المسار على سطر الأوامر باستخدام سطر واحد من التعليمات البرمجية. يتم توفير إرشادات بسيطة خطوة بخطوة حول كيفية تخصيص معلمات التحليل إما لتحليل ChIP-seq أو ATAC-seq داخل ملف التكوين نفسه وفي دليلنا المفصل خطوة بخطوة في مستودع CATCH-UP GitHub.
توجد خطوط أنابيب تحليل موجودة لبيانات ChIP-seq أو ATAC-seq ، مثل PEPATAC و ChIP-AP. في حين أن خطوط الأنابيب هذه لها مزايا مثل دمج كل من تحليلات المنبع والمصب في سير عمل واحد أو استخدام واجهة مستخدم رسومية (GUI) ، فإن هذه الأدوات تستهدف علماء المعلوماتية الحيوية والعلماء بمستوى معتدل من التدريب الحسابي13،14. تم تصميم CATCH-UP لحل مشكلتين: تمكين علماء المختبرات الرطبة الذين لم يتلقوا تدريبا في مجال المعلوماتية الحيوية من إجراء تحليلاتهم الأولية وتمكين توحيد تحليل المنبع من خلال تسهيل إعداد التقارير السهلة والاستنساخ الدقيق عبر المختبرات. يقتصر CATCH-UP عن قصد على التحليل الأولي ، لكن المخرجات متوافقة مع أدوات التحليل النهائية مثل تلك المستخدمة لمقارنة مجموعات البيانات إحصائيا أو استنتاج ربط عاملالنسخ 25،26.
يتم تحديد جميع الخطوات الحاسمة اللازمة لإجراء تحليل المنبع القابل للتكرار مسبقا داخل خط أنابيب CATCH-UP لضمان المتانة. تسمح الطبيعة المطولة لمسار الأنابيب هذا للمستخدم بمتابعة إخراج المسار خطوة بخطوة، وهو أمر مفيد لكل من استكشاف الأخطاء وإصلاحها وتمكين تكرار سير العمل التحليلي. نظرا للطبيعة سريعة التطور لتقنيات NGS ، فإن الطبيعة المعيارية لخط الأنابيب هذا مفيدة لأنها توفر القدرة على التكيف بسهولة لدمج كل من إصدار تحديثات إصدار الأداة وتنفيذ أدوات جديدة. تم اختبار CATCH-UP بنجاح لأنظمة التشغيل التالية: Ubuntu و CentOS و macOS (Intel CPU) و Windows (الجدول 3). تم بناء خط الأنابيب للتعامل مع التجارب الكبيرة التي تحتوي على عشرات العينات من خلال موازاة سير العمل ، مما يجعله قابلا للتكيف مع التصميمات التجريبية المختلفة. بشكل عام ، يتيح تنفيذ CATCH-UP في تحليل بيانات ChIP-seq و ATAC-seq سير عمل تحليل سهل الاستخدام وقابل للتكرار وقابل للتكيف بدرجة كبيرة.
Disclosures
JRH هو مؤسس مشارك ومدير Nucleome Therapeutics ويقدم الاستشارات للشركة.
Acknowledgements
تم دعم JRH بمنح من Wellcome Trust (225220 / Z / 22 / Z و 106130 / Z / 14 / Z) و MRC (MC_UU_00029/3). تم دعم M.B. من خلال منحة Wellcome Trust (225220 / Z / 22 / Z). تم دعم E.R.G من قبل وزارة التعليم الوطني لاختيار وتنسيب المرشحين المرسلين إلى الخارج للحصول على منحة التعليم العالي (YLSY) ، وزارة التعليم الوطني في جمهورية تركيا. تم دعم E.G. من قبل برنامج الدكتوراه في الطب الجينومي والإحصاء في ويلكوم (108861/Z/15/Z). تم دعم S.G.R. من قبل منحة مجلس البحوث الطبية (MRC) (MC_UU_00029/3).
Materials
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
References
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623(2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521(2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037(2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101(2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537(2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008(2021).
- Picard Toolkit. , http://broadinstitute.github.io/picard/ (2019).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved