
Method Article
CATCH-UP: צנרת תפוקה גבוהה במעלה הזרם עבור נתוני ATAC-Seq ו-ChIP-Seq בתפזורת
In This Article
Summary
ATAC-seq ו-ChIP-seq מאפשרים חקירה מפורטת של ויסות גנים; עם זאת, עיבוד סוגי נתונים אלה מאתגר ולעתים קרובות אינו עקבי בין קבוצות מחקר. אנו מציגים את CATCH-UP: צינור חישובי קל לשימוש המאפשר עיבוד וניתוח נתונים סטנדרטיים וניתנים לשחזור של מערכי נתונים חדשים ופורסמו של ATAC/ChIP-seq.
Abstract
בדיקה לכרומטין נגיש לטרנספוזאז (ATAC) וכרומטין אימונו-משקעים (ChIP), יחד עם ריצוף הדור הבא (NGS), חוללו מהפכה בחקר ויסות הגנים. היעדר סטנדרטיזציה בניתוח מערכי הנתונים הממדיים ביותר שנוצרו על ידי טכניקות אלה הקשה על השגת השחזור, מה שהוביל לאי התאמות בנתונים המעובדים שפורסמו. חלק מבעיה זו נובע מהמגוון המגוון של כלים ביואינפורמטיים הזמינים לניתוח נתונים מסוג זה. שנית, מספר כלים ביואינפורמטיים שונים נדרשים ברצף כדי להמיר נתונים גולמיים לפלט מעובד וניתן לפירוש מלא, וכלים אלה דורשים רמות שונות של מיומנויות חישוביות. יתר על כן, ישנן אפשרויות רבות לבקרת איכות שאינן מופעלות באופן אחיד במהלך עיבוד הנתונים. אנו מטפלים בבעיות אלה באמצעות בדיקה מלאה לריצוף כרומטין נגיש לטרנספוזאז (ATAC-seq) ורצף כרומטין אימונו-משקעים (ChIP-seq) במעלה הזרם (CATCH-UP), צינור קל לשימוש מבוסס Python לניתוח מערכי נתונים של ChIP-seq ו-ATAC-seq בתפזורת מקבצי fastq גולמיים ועד רצועות גדולות ושיחות פסגות הניתנות להמחשה. צינור זה פשוט להתקנה ולהפעלה, ודורש ידע חישובי מינימלי. הצינור הוא מודולרי, ניתן להרחבה ומקביל בתשתיות מחשוב שונות, ומאפשר דיווח קל על מתודולוגיה כדי לאפשר ניתוח ניתן לשחזור של מערכי נתונים חדשים או שפורסמו.
Introduction
ביטוי גנים חייב להיות מווסת היטב כדי שהתאים יוכלו לבסס ולשמור על תפקודם הביולוגי הנכון. ידוע היטב כי ביטוי גנים חריג עומד בבסיס הפתוגנזה של מחלות רבות, ולכן עניין מחקרי רב טמון בהבנת מנגנוני הבקרה של גנים1. ביטוי גנים מקל על ידי אלמנטים רגולטוריים כגון מקדמים ומשפרים. בתוך הרצף שלהם, אלמנטים אלה מכילים אתרי קשירה של גורם שעתוק (TF), אשר, כאשר הם פעילים, מספקים פלטפורמה לקשירת TF. קשירת TFs באתרים אלה מביאה לתזוזה של נוקלאוזומים, וכתוצאה מכך לעלייה בנגישות ה-DNA ולעלייה לאחר מכן במותר למנגנון השעתוק. כתוצאה מהנגישות המוגברת הזו, אזורים אלה של ה-DNA רגישים יותר לנוקלאזות וטרנספוזות כגון DNase ו-Tn5, תכונה ביוכימית שנוצלה על ידי חוקרים החוקרים ויסות שעתוק 2,3.
DNase-seq ו-ATAC-seq מאפשרים לחוקרים למפות אזורים של כרומטין פתוח, אתרי קשירת TF ומיקום נוקלאוזומי על פני הגנום. מבין שתי הטכניקות הללו, ATAC-seq גדלה בפופולריות בעשור האחרון בשל הפרוטוקול הדו-שלבי הפשוט והדרישה למספר תאים נמוך (50,000 תאים בהשוואה למיליון לכל שכפול עבור DNase-seq). בעוד ש-ATAC-seq מספק סקירה כללית של נוף הכרומטין הכללי באוכלוסיית תאים, הוא אגנוסטי במידה רבה לאילו חלבונים ספציפיים נקשרים לגנום 4,5. על מנת לזהות את המיקומים שבהם חלבון ספציפי מקיים אינטראקציה עם הגנום, טכניקת תקן הזהב היא Chromatin Immunoprecipitation (ChIP)-seq. ChIP-seq כרוך בקיבוע כימי של אינטראקציות חלבון-DNA בתא, ואחריו משקעים חיסוניים ("משיכה כלפי מטה") באמצעות נוגדן ספציפי לחלבון המעניין כדי לבחור מקטעי DNA הקשורים לחלבון המעניין (POI). ניתן לרצף את מקטעי ה-DNA הללו כדי לחשוף את מיקומי הקישור הגנומי של חלבונים ספציפיים כגון TFs, או אתרים המכילים שינויים ספציפיים בהיסטון1. על ידי שילוב מערכי נתונים של ATAC-seq ו-ChIP-seq, ניתן לגזור תמונה מפורטת של הנוף הרגולטורי עבור אוכלוסיית תאים.
זרימת העבודה הבסיסית הנדרשת לניתוח היא כדלקמן: קריאות ריצוף גולמיות חייבות להיות מבוקרות איכות לפני יישור לגנום ייחוס ("מיפוי"). לאחר מכן ניתן לסנן את הקריאות שמופו בהצלחה כדי להסיר גם קריאות באיכות נמוכה וגם כפילויות PCR. על מנת לדמיין את הקריאות הממופות והמסוננות הללו, יש צורך לחשב את "הכיסוי" של קריאות אלה על פני הגנום. זה יוצר קובץ שניתן להעלות לדפדפן גנום כגון תצוגת ריבוי מיקומים (MLV) או לדפדפן הגנום של UCSC כ"מסלול"6,7. זיהוי שיא, או "קריאת שיא" של מסלולי כיסוי אלה מושג בדרך כלל באמצעות כלים כגון LanceOtron או MACS2 8,9. לבסוף, באמצעות ניתוח מיקום שיא, ניתן לבצע השוואות בין דגימות או תנאים ביולוגיים. ניתוח ואינטגרציה של מערכי נתונים אלה הוא תהליך מורכב ורב-שלבי בו ניתן ליישם שילובים שונים של כלים ביואינפורמטיים. גרסאות שונות של הכלים עשויות להיות לא תואמות זו לזו ועשויות לשנות את הפלט של עיבוד הנתונים. קיים גם מגוון רחב בכוח החישוב ובמיומנות המשתמש הנדרשים ליישום חלקים שונים של עיבוד נתונים כפי שמוצג בצינורות nf-core10, panpipes11, genpipes12, PEPATAC13 או ChIP-AP14.
בסך הכל, זה הוביל לחוסר עקביות הן בניתוח והן בדיווח של הניתוח, מה שהוביל, בתורו, לשחזור, נגישות ונוחות לקויים עבור כל מי שיש לו ידע מוגבל בביואינפורמטיקה. אנו מטפלים בכל הבעיות הללו עם CATCH-UP (צינור ATAC-seq ו-ChIP-seq מלא במעלה הזרם), צינור קל לשימוש, גמיש ומודולרי לעיבוד נתוני ChIP-seq ו-ATAC/DNase-seq. יישום CATCH-UP דורש ניסיון מינימלי בביואינפורמטיקה; ניתן להפעיל אותו על תשתיות מחשוב שונות ומאפשר ניתוח נתונים הניתן לשחזור בתוך ובין קבוצות מחקר.
CATCH-UP הוא צינור Snakemake מבוסס Python שנבנה כדי לתקנן את הניתוח של נתוני ChIP-seq ו-ATAC-seq. הוא לוקח נתוני רצף גולמיים (fastq.gz קבצים) כקלט ומייצר פלט בצורה של קבצי שיא (.bed) המספקים את התוצאה המתאימה לכל שלב. אנו מספקים קובץ תצורה בפורמט yaml (config.yaml), בו המשתמש יכול לערוך את הפרמטרים של כל שלב ניתוח. מערכת הניהול המיושמת בתוך snakemake מאפשרת שימוש בתשתיות מחשוב שונות (כגון שרתים, אשכולות, מערכות ענן או מחשבים אישיים) ובמקביל אם המשתמש מספק כמות גדולה של נתונים.
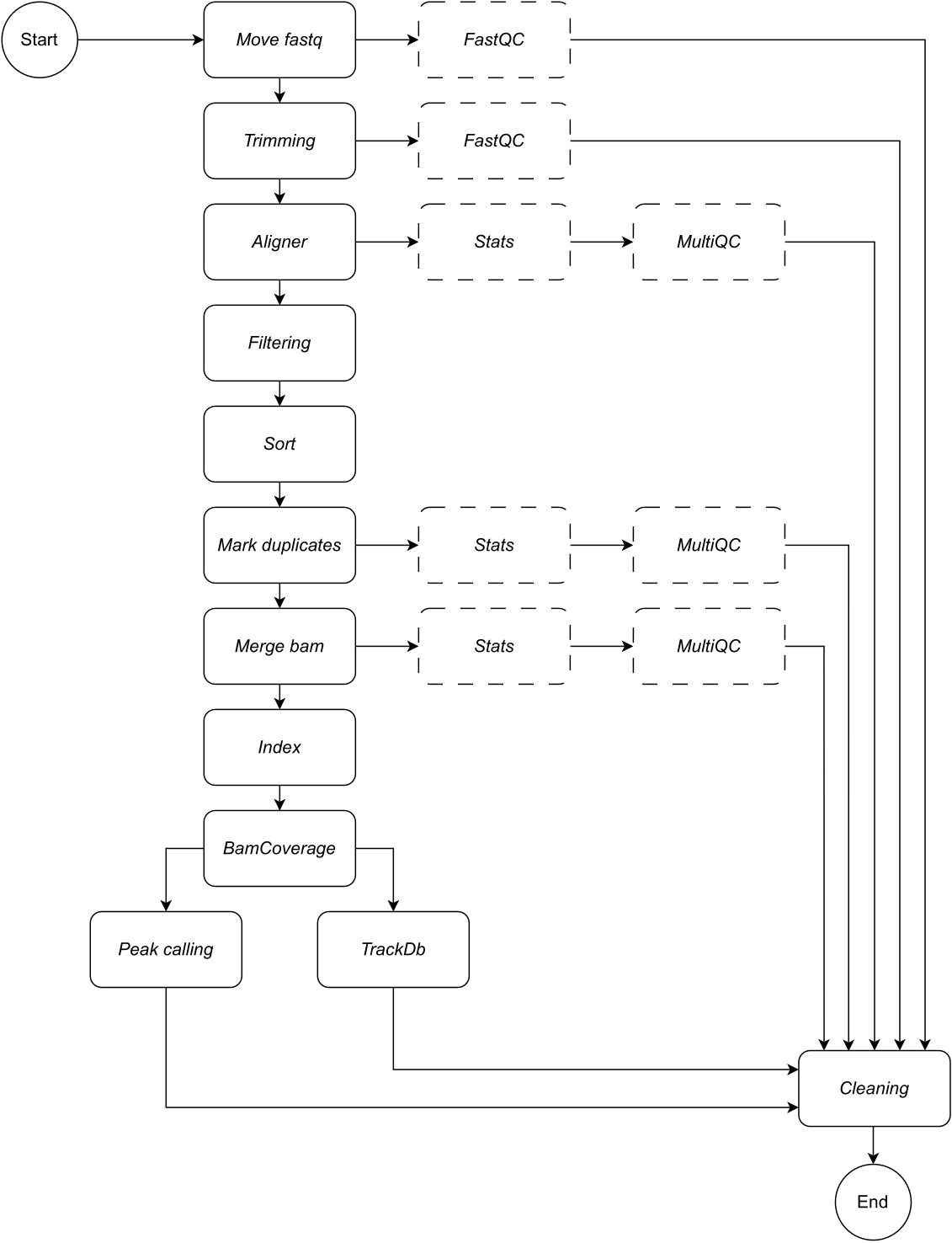
להלן, אנו מספקים תיאור מפורט של כל שלב בזרימת העבודה (ראה איור 1 לאיור זרימת העבודה). הסבר זה חיוני על מנת לעקוב אחר שלב אחר שלב בסעיף הפרוטוקול:
העבר fastq: השלב הראשון של הצינור הוא להעתיק את קבצי ה-fastq הגולמיים לספריית הניתוח בעלת השם. זה משאיר את הנתונים המקוריים ללא שינוי כדי למנוע השחתה או שינוי של קבצי הנתונים הגולמיים.
שרשור: אם נתוני ריצוף גולמיים מכילים מספר נתיבים, שלב זה נדרש כדי לשרשר את הנתיבים לפני הניתוח. כברירת מחדל, הצינור מטפל בכל קבצי ה-fastq כדוגמאות בודדות. יש להגדיר שלב שרשור זה בקובץ התצורה.
חיתוך: שלב ניקוי נתונים אופציונלי. זה מאפשר חיתוך של קריאות באיכות נמוכה או רצפי מתאמים באמצעות trimmomatic15. המשתמש יכול לספק קבצי fasta מותאמים אישית של רצפי מתאמים; דוגמה לכך מסופקת בספריית המתאם. ניתן להגדיר פרמטרי חיתוך נוספים בקובץ התצורה. כברירת מחדל, זרימת העבודה מדלגת על כלל זה.
קשתית: ליישור, Bowtie216 מוחל כברירת מחדל; ניתן לציין גם כלי יישור חלופיים כגון BWA-MEM217 . כלי היישור Bowtie2 נבחר כברירת מחדל מכיוון שהוא מיומן במיוחד ביישור קריאות קצרות יחסית לגנומים גדולים יחסית ולכן הוא מתאים היטב ליישור נתוני ChIP-seq ו-ATAC-seq לגנומים של יונקים. כדי להימנע מקבצי ביניים, היישור מועבר לתצוגת samtools כדי לשמור את קובץ ה-bam בפלט. עבור כלל זה, על המשתמש לציין את מבנה הגנום המועדף עליו למפות את הקריאות, למשל, hg19/hg38 (אנושי), mm10/mm39 (עכבר).
סינון: קריאות ממופות כהלכה נשמרות, וקריאות באיכות נמוכה מסוננות החוצה. ברירת מחדל: תצוגת samtools, עם פרמטרים: -bShuF 4 -f 3 -q 30.
מיון: קריאות מיושרות ממוינות לפי סדר הקואורדינטה השמאלית ביותר. ברירת מחדל: מיון samtools (עטיפת נחש), עם פרמטר: -m 4G.
סימון כפילויות: כל הקריאות הכפולות מזוהות ומסומנות בדגל. המשתמש יכול להחליט להסיר אותם על ידי שינוי פרמטר קובץ התצורה. ברירת מחדל: Picard MarkDuplicates (עטיפת snakemake), עם פרמטר: --REMOVE_DUPLICATES False כדי לסמן ולשמור כפילויות.
מיזוג bam: אם נתוני הרצף מורכבים משכפולים או דוגמאות, ייתכן שהמשתמש ירצה להתמזג ל-bam יחיד. במקרה זה, המשתמש יכול לבחור למזג את ה-bams או לשמור על קבצי bam נפרדים לאורך הניתוח. אם המשתמש בוחר למזג bams (תוך שימוש במיזוג samtools), יש לציין קידומת משותפת ל-bams הממוזגים.
אינדקס: שלב זה יוצר אינדקס של הקואורדינטות הממויינות. ברירת מחדל: אינדקס samtools (עטיפת snakemake), תוך שימוש בפרמטרי ברירת מחדל שצוינו על ידי samtools.
BamCoverage: כלל זה יוצר מסלול כיסוי גדול מקריאות מיושרות. הכלי bamCoverage מבית deepTools מוחל, והכיסוי מחושב כמספר הקריאות לכל סל, שבו הסל מייצג חלון בגודל מוגדר. בקו צינור זה, bamCoverage מוחל עם הפרמטרים הבאים המוגדרים כברירת מחדל: -bs 1 -normalizeUsing RPKM -extendReads.
שיחות שיא: LanceOtron8 נבחר כברירת המחדל עבור צינור זה. בניגוד למתקשרים שיא מסורתיים, המבוססים ברובם על בדיקות סטטיסטיות, LanceOtron הוא מתקשר שיא מבוסס למידה עמוקה, המשלב מדידות העשרה גנומיות ובדיקות סטטיסטיות והוכח כעולה על מתקשר השיא הסטנדרטי בתעשייה, MACS29. כדי ש-bigwigs יהיו תואמים ל-LanceOtron, יש לחשב את הכיסוי לכל זוג בסיסים, ולנרמל RPKM; הדבר בא לידי ביטוי בהגדרות ברירת המחדל עבור השלב BamCoverage. ניתן לבחור ב-MACS2 כמתקשר שיא חלופי. שחרורם של מתקשרי שיא חדשים ינוטר וישולב לפי העניין על מנת לשמור ולייעל את הביצועים של צינור ניתוח זה.
TrackDb: זה יוצר אסוציאציה של צמד מפתח-ערך של קבצי bigwig על מנת לטעון ולדמיין אותם בכלים כמו פלטפורמות MLV6 או UCSC Genome Browser18 .
בנוסף לנתוני הפלט, כל שלב בצנרת מוציא קובץ יומן, ובדיקות בקרת איכות מתאימות מסופקות כדי שהמשתמש יוכל לעקוב אחר התקדמות הניתוח. FastQC19 מוחל על נתוני רצף גולמיים וחתוכים (אם נבחר) (שלבים 1 - Move fastq ו- 2- חיתוך). נתונים סטטיסטיים של Samtools בתוספת MultiQC20 משמשים לאיסוף, הפקה והדמיה של דוחות בקרת איכות על קבצי bam בפלט בשלבים 3 - Aligner, 6 - סימון כפילויות ו-7 - מיזוג bam. למידע נוסף על כל אחד מהכלים המיושמים בשלבים לעיל, ראה טבלה 1.
Protocol
1. הפעלת צינור CATCH-UP
- שכפל את מאגר UpStreamPipeline מ-https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
נווט לספריית העבודה שנבחרה, העתק את הקוד הבא והפעל בשורת הפקודה:
שיבוט git git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - נווט בתוך תיקיית UpStreamPipeline שהורדת באמצעות הפקודה: cd UpStreamPipeline
- התקן את הפצת האנקונדה (במידת הצורך):
- בדוק אם אנקונדה כבר מותקנת במערכת באמצעות הפקודה שקונדה. אם הפקודה אינה מציגה נתיב כלשהו להפצת conda כלשהי, הורד את mambaforge מ-https://github.com/conda-forge/miniforge#mambaforge ובחר את ההפצה והגרסה הנכונות עבור המערכת. לדוגמה, עבור משתמשי לינוקס, השתמש ב-wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. בקר בדף אינטרנט זה עבור מערכות הפעלה שונות: https://github.com/conda-forge/miniforge/.
- הפעל את תוכנית ההתקנה באמצעות sh Mambaforge-Linux-x86_64.sh, ואתחל את conda במערכת על ידי הפעלת conda init.
- התקן והפעל את סביבת conda במעלה הזרם (הדרישות של סביבת conda במעלה הזרם מפורטות בטבלה 2):
- התקן את הסביבה באמצעות הפקודה mamba env create - file=envs/upstream.yml.
- הפעל את הסביבה באמצעות הפקודה conda activate upstream.
- לאחר התקנת סביבת conda במעלה הזרם בהצלחה, הפעל את הסביבה באמצעות הפקודה conda הפעל במעלה הזרם ונווט לתיקיית CATCH-UP באמצעות cd genetics/CATCH-UP.
- ערוך את קובץ התצורה, שניתן למצוא בתוך תיקיית התצורה באמצעות הפקודה cd /config/analysis.yaml, ושנה אותו בהתאם למפרט הניתוח באמצעות עורך טקסט. בצע את ההוראות שורה אחר שורה כדי לערוך כל פרמטר בקובץ עצמו. קובץ זה יישמר לאחר הניתוח ויפעל לתיעוד פרמטרי הריצה כדי לסייע בשחזור.
- פתח וערוך את שלושת הקבצים הבאים בעורך טקסט (לדוגמה, TextEdit עבור Mac או Notepad עבור Windows):
- ערוך קובץ 1_fastqfile_home_dir.txt כך שיכיל רשימה של כל קבצי ה-fastq שיש לנתח.
הערה: יש לא לכלול מספרי קריאה וסיומות (לדוגמה, _R1/_R2 ו- .fastq.gz). לדוגמה, אם פרויקט מכיל רשימה זו של קבצי fastq:
Sample1_conditionA_L001_R1 . fastq . ג'י.ז.
Sample1_conditionA_L001_R2 . fastq . ג'י.ז.
Sample1_conditionA_L002_R1 . fastq . ג'י.ז.
Sample1_conditionA_L002_R2 . fastq . ג'י.ז.
Sample1_conditionB_L001_R1 . fastq . ג'י.ז.
Sample1_conditionB_L001_R2 . fastq . ג'י.ז.
Sample1_conditionB_L002_R1 . fastq . ג'י.ז.
Sample1_conditionB_L002_R2 . fastq . ג'י.ז.
Sample2_conditionA_L001_R1 . fastq . ג'י.ז.
Sample2_conditionA_L001_R2 . fastq . ג'י.ז.
Sample2_conditionA_L002_R1 . fastq . ג'י.ז.
Sample2_conditionA_L002_R2 . fastq . ג'י.ז.
Sample2_conditionB_L001_R1 . fastq . ג'י.ז.
Sample2_conditionB_L001_R2 . fastq . ג'י.ז.
Sample2_conditionB_L002_R1 . fastq . ג'י.ז.
Sample2_conditionB_L002_R2 . fastq . ג'י.ז.
במקרה זה, 1_fastqfile_home_dir.txt הוא כדלקמן:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - אם נתונים גולמיים מכילים נתיבי רצף הדורשים שרשור, ערוך קובץ2_fastq file_concat.txt כדי להגדיר את הקידומת של שמות הקבצים שיש לשרשר. אם אין נתיבי רצף לשרשור, אל תערוך 2_fastqfile_concat.txt. ודא שכל שורה של 2_fastqfile_concat.txt מכילה קידומת אחת לדוגמה כדלקמן:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - אם נדרש מיזוג נתונים של דוגמאות שונות, ערוך קובץ 3_merge_bams.txt עם הקידומת של שמות הקבצים למיזוג. ודא שכל שורה מכילה קידומת לדוגמה אחת כדלקמן:
דוגמה1
דוגמא 2
איור 2 מציג סיכום של אופן סיכום שלושת הקבצים הללו. ניתן להחיל את הפרוטוקול על נתוני ריצוף יחידים או מזווגים. ברירת המחדל של קו הצינור היא ניתוח קצה מזווג אלא אם צוין אחרת; ניתן לשנות זאת בקובץ התצורה (ראה שלב 1.6).
- ערוך קובץ 1_fastqfile_home_dir.txt כך שיכיל רשימה של כל קבצי ה-fastq שיש לנתח.
- לאחר עריכת כל הקבצים הנדרשים, השתמש ב-snakemake כדי להריץ את CATCH-UP באופן הבא: snakemake --configfile=config/analysis_name.yaml כל --cores 4.
הערה: להוראות ותיעוד מפורטים יותר, עיין בתיקיית CATCH-UP בתוך מאגר UpStreamPipeline GitHub, הזמין כאן. זה כולל תיעוד מפורט על שינוי נכון של קובץ התצורה, החל משינוי נתיבים לרצף קבצי נתונים ואחסון תוצאות ועד לעריכת פרמטרים עבור כל שלב.
תוצאות
צינור ה-CATCH-UP מייצר תוצאה, יומן ופלט בקרת איכות (QC) עבור כל שלב. בתוך קובץ התצורה, המשתמש יכול לבחור לשמור או להסיר קבצי פלט כדי להפחית את זיכרון האחסון הנדרש. כל התפוקות מוסברות כדלקמן:
00. fastq_home_dir: קובץ תצורה, le_home_dir.txt FastQFi ו-merge_bams.txt מועתקים לתיקיה זו לצורך עיון ושחזור.
01. קורא: קבצי FastQ מועתקים לתיקיה זו כדי למנוע שינויים בנתונים הגולמיים המקוריים במהלך תהליך זרימת העבודה, ניתן לשרשר נתיבים אם צוין.
02. חיתוך: קבצי FastQ עם קריאה ומתאמים חתוכים אם צוין.
03. מיישר: יישור כנגד הגנום הנבחר.
04. פילטרינג: סינון בקרת איכות.
05. ממוין: מיון קבצי BAM.
06. כפילויות: סימון כפילויות.
07. מיזוג: מיזוג קבצי BAM אם זה צוין ב-config.yaml.
08. bam_coverages: קובץ גדול של הסיקור.
09. peak_calling: קובץ מיטה של פלט שיחות שיא של LanceOtron.
10. מסלול: מייצר קובץ טקסט מעוצב מוכן לשימוש בדפדפן הגנום במידת הצורך.
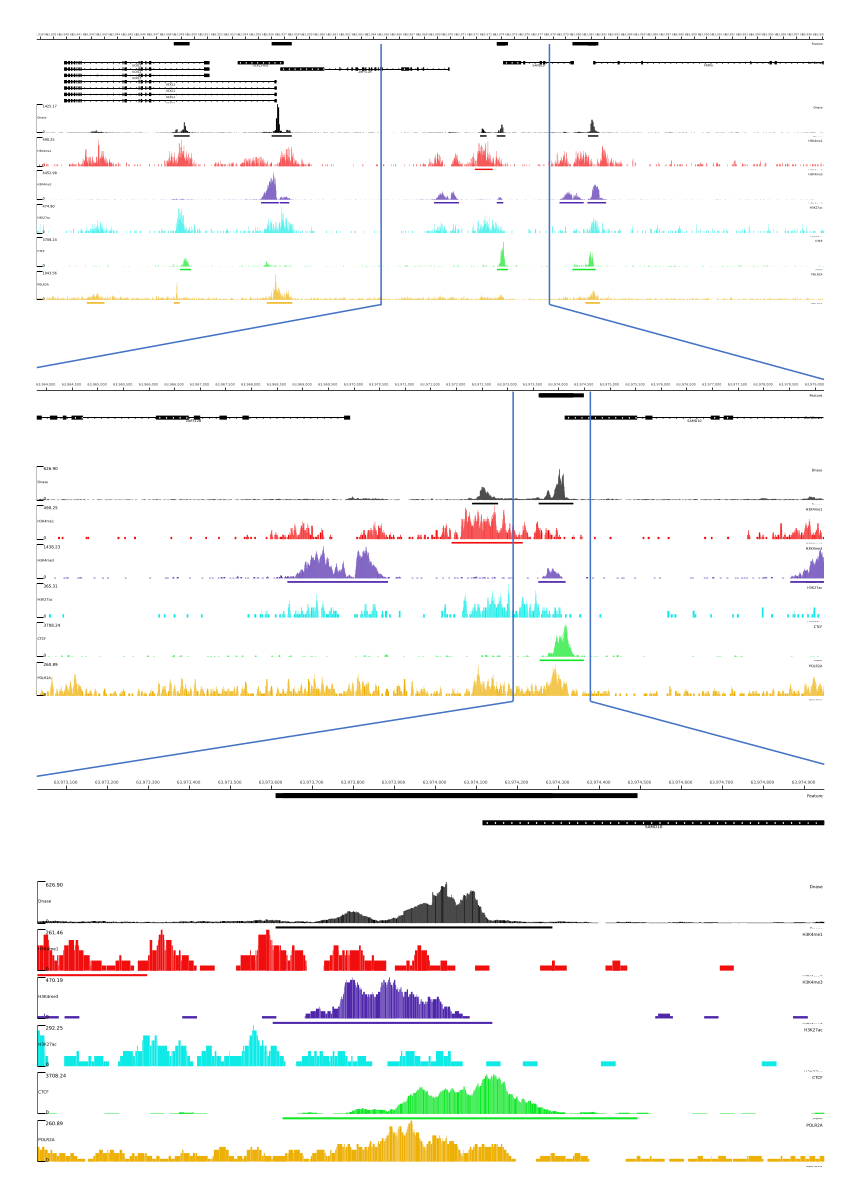
עבור יציאות 01, 02, 03, 06 ו-07, מסופקים מדדי QC וקבצי HTML. בנוסף, באיור 3, אנו מספקים דוגמה לנתונים מעובדים באמצעות CATCH-UP, המדמיינים את הפלט הסופי באמצעות פלטפורמת MLV.

איור 1: זרימת עבודה של CATCH-UP. בהינתן רשימה של קבצי fastq, CATCH-UP מעבד במקביל את כל הדגימות דרך כל השלבים במעלה הזרם. אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}

איור 2: ייצוג אילוסטרטיבי המסביר כיצד 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt ו-3_merge_bams.txt חייבים להיות מודיים בצורה נכונהעל מנת להריץ CATCH-UP. אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}

איור 3: פלט לדוגמה מצינור CATCH-UP. נתוני ריצוף גולמיים (קבצי fastq) הורדו מ-ENCODE21. צינור CATCH-UP שימש לעיבוד קבצי fastq עבור DNase-seq ו-5 סוגים של ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF ו-POLR2A). קבצי פלט של Bigwig הועלו ל-Multi Locus View לצורך הדמיה וזיהוי של אלמנטים רגולטוריים גנומיים. אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}
טבלה 1: משאבי תיעוד. טבלה זו מציגה את הכלים המעורבים בזרימת העבודה של CATCH-UP, את הקישור לתיעוד שלהם ואת ההפניות המתאימות. אנא לחץ כאן להורדת טבלה זו.
טבלה 2: רשימת דרישות הערוץ והתלות עבור סביבת conda במעלה הזרם. אנא לחץ כאן להורדת טבלה זו.
טבלה 3: מערכות הפעלה המשמשות לבדיקת CATCH-UP. אובונטו נבדקה על אשכול בעל ביצועים גבוהים ומכונה מקומית. אנא לחץ כאן להורדת טבלה זו.
Discussion
הקליטה והניצול המוגברים של טכניקות NGS ליצירת נתונים גנומיים הותאמו לעלייה בפיתוח כלים ביואינפורמטיים לניתוח נתונים אלה. ישנם מספר כלים שניתן ליישם עבור כל שלב בניתוח הנתונים, כמו גם פרמטרים רבים ושונים שניתן לציין בתוך כל כלי 6,8,9,15,16,17,18,19,20,22,23,24 . זה יוצר שילוב מגוון מאוד של אסטרטגיות ניתוח שניתן ליישם, שכל אחת מהן יכולה לייצר שינויים בתוצאה. על מנת להשוות במדויק בין ניסויים, סטנדרטיזציה של ניתוח ביואינפורמטי היא חיונית. מבחינה היסטורית, נתוני NGS נוצרים על ידי מדעני מעבדה רטובה, והנתונים מנותחים על ידי ביואינפורמטיקאים.
ניתן לחלק את ניתוח נתוני NGS לצינורות "במעלה הזרם" ו"במורד הזרם", כאשר במעלה הזרם כולל את השלבים הדרושים כדי לעבור מפלט נתונים גולמיים ממכונת ריצוף לפורמט הניתן לפירוש חזותי על ידי חוקר. ניתוח במורד הזרם כולל שלבים נוספים המותאמים לשאלת המחקר ולתכנון הניסוי. לפיכך, צינורות במעלה הזרם ניתנים להכללה וניתנים לסטנדרטיזציה לשיפור השחזור המדעי. צינורות במורד הזרם, לעומת זאת, מותאמים אישית, תלויים בשאלה הביולוגית, ודורשים תובנה מהחוקר, מה שהופך אותם לפחות מתאימים לסטנדרטיזציה. יצרנו צינור ידידותי למשתמש במעלה הזרם המאפשר למדעני מעבדה רטובה לנתח את הנתונים שלהם באופן שחזורי ללא צורך בידע מוקדם בביואינפורמטיקה. כאן, אנו מציגים את CATCH-UP, צינור שנבנה באמצעות מסגרת Snakemake ונועד להיות גם ידידותי למשתמש וגם להילחם בסוגיית השחזור בניתוח נתוני ChIP-seq ו-ATAC-seq. צינור זה נבנה כדי לטפל בנתוני ChIP-seq או ATAC-seq. לאחר שהמשתמש הוריד את CATCH-UP, יש להגדיר תחילה את פרמטרי הניתוח ושמות הדגימה לפני הפעלת הצינור בשורת הפקודה באמצעות שורת קוד אחת. הוראות פשוטות שלב אחר שלב כיצד להתאים אישית את פרמטרי הניתוח עבור ניתוח ChIP-seq או ATAC-seq מסופקות בתוך קובץ התצורה עצמו ובמדריך שלנו שלב אחר שלב במאגר CATCH-UP GitHub.
ישנם צינורות ניתוח קיימים עבור נתוני ChIP-seq או ATAC-seq, כגון PEPATAC ו-ChIP-AP. בעוד שלצינורות אלה יש יתרונות כגון שילוב של ניתוחים במעלה הזרם ובמורד הזרם בזרימת עבודה אחת או שימוש בממשק משתמש גרפי (GUI), כלים אלה מיועדים לביואינפורמטיקאים ומדענים עם רמה בינונית של הכשרה חישובית13,14. CATCH-UP תוכנן לפתור שתי בעיות: לאפשר למדעני מעבדה רטובה ללא הכשרה ביואינפורמטית לבצע ניתוח משלהם במעלה הזרם ולאפשר סטנדרטיזציה של ניתוח במעלה הזרם על ידי הקלה על דיווח קל ושחזור מדויק בין מעבדות. CATCH-UP מוגבל בכוונה לניתוח במעלה הזרם, אך התפוקות תואמות לכלי ניתוח במורד הזרם כגון אלה המשמשים להשוואה סטטיסטית של מערכי נתונים או להסקת קשירת גורם שעתוק25,26.
כל השלבים הקריטיים הדרושים לביצוע ניתוח במעלה הזרם הניתן לשכפול מוגדרים מראש בתוך צינור ה-CATCH-UP כדי להבטיח חוסן. האופי המילולי של קו צינור זה מאפשר למשתמש לעקוב אחר פלט הצינור שלב אחר שלב, דבר שימושי הן לפתרון בעיות והן לאפשר שכפול זרימת העבודה האנליטית. בהתחשב באופי המתפתח במהירות של טכניקות NGS, האופי המודולרי של צינור זה מועיל מכיוון שהוא מספק את היכולת להתאים בקלות לשילוב הן שחרור עדכוני גרסת כלים והן יישום כלים חדשים. CATCH-UP נבדק בהצלחה עבור מערכות ההפעלה הבאות: Ubuntu, CentOS, macOS (Intel CPU) ו-Windows (טבלה 3). הצינור נבנה כדי לטפל בניסויים גדולים המכילים עשרות דגימות על ידי הקבלה לזרימת העבודה, מה שהופך אותו להתאמה לעיצובים ניסיוניים שונים. בסך הכל, יישום CATCH-UP בניתוח נתוני ChIP-seq ו-ATAC-seq מאפשר זרימת עבודה ידידותית למשתמש, ניתנת לשחזור וניתנת להתאמה גבוהה.
Disclosures
J.R.H. הוא מייסד שותף ומנהל של Nucleome Therapeutics ומספק ייעוץ לחברה.
Acknowledgements
J.R.H. נתמך על ידי מענקים מקרן וולקאם (225220/Z/22/Z ו-106130/Z/14/Z) ו-MRC (MC_UU_00029/3). M.B. נתמך על ידי מענק Wellcome Trust (225220/Z/22/Z). E.R.G נתמך על ידי משרד החינוך הלאומי לבחירה והשמה של מועמדים שנשלחו לחו"ל למלגת השכלה מתקדמת (YLSY), משרד החינוך הלאומי של הרפובליקה של טורקיה. E.G. נתמך על ידי תוכנית הדוקטורט לרפואה גנומית וסטטיסטיקה של Wellcome (108861/Z/15/Z). S.G.R. נתמך על ידי מענק המועצה למחקר רפואי (MRC) (MC_UU_00029/3).
Materials
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
References
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved