
Method Article
追赶:用于批量 ATAC-Seq 和 ChIP-Seq 数据的高通量上游管道
摘要
ATAC-seq 和 ChIP-seq 允许详细研究基因调控;但是,处理这些数据类型具有挑战性,并且研究小组之间通常不一致。我们提出了 CATCH-UP:一种易于使用的计算管道,允许对新的和已发布的 ATAC/ChIP-seq 数据集进行标准化和可重复的数据处理和分析。
摘要
转座酶可及染色质可及染色质 (ATAC) 和染色质免疫沉淀 (ChIP) 检测与下一代测序 (NGS) 相结合,彻底改变了基因调控的研究。这些技术生成的高维数据集的分析缺乏标准化,这使得难以实现可重复性,从而导致已发布、处理的数据出现差异。这个问题的部分原因是可用于分析这些类型数据的生物信息学工具多种多样。其次,需要按顺序使用许多不同的生物信息学工具才能将原始数据转换为完全处理和可解释的输出,这些工具需要不同级别的计算技能。此外,在数据处理过程中,有许多质量控制选项并未统一采用。我们通过转座酶可及染色质测序 (ATAC-seq) 和染色质免疫沉淀测序 (ChIP-seq) 上游管道 (CATCH-UP) 的完整分析来解决这些问题,这是一种易于使用的基于 Python 的管道,用于分析从原始 fastq 文件到可视化的大人物轨迹和峰值调用的批量 ChIP-seq 和 ATAC-seq 数据集。此管道易于安装和运行,只需最少的计算知识。该管道是模块化的、可扩展的,并且可以在各种计算基础设施上并行化,从而可以轻松报告方法,从而对新的或已发布的数据集进行可重复的分析。
引言
基因表达必须受到严格调节,细胞才能建立和维持其正确的生物学功能。众所周知,异常基因表达是许多疾病发病机制的基础,因此,了解基因调控的机制1 是很大的研究兴趣。基因表达由启动子和增强子等调节元件促进。在它们的序列中,这些元件包含转录因子 (TF) 结合位点,当转录因子 (TF) 结合位点激活时,为 TF 结合提供了一个平台。TF 在这些位点的结合导致核小体的置换,导致 DNA 可及性增加,随后对转录机制的允许性增加。由于这种可及性增加,这些 DNA 区域对核酸酶和转座酶(如 DNase 和 Tn5)更敏感,这是一种生化特性,研究人员已利用这种生化特性来研究转录调控 2,3。
DNase-seq 和 ATAC-seq 使研究人员能够绘制整个基因组中开放染色质区域、TF 结合位点和核小体定位。在这两种技术中,ATAC-seq 在过去十年中越来越受欢迎,因为它具有简单的两步方案和低细胞数量要求(50,000 个细胞,而 DNase-seq 每个重复 100 万个细胞)。虽然 ATAC-seq 概述了细胞群中一般染色质的概况,但它在很大程度上与哪些特定蛋白质结合基因组无关 4,5。为了确定特定蛋白质与基因组相互作用的位置,金标准技术是染色质免疫沉淀 (ChIP)-seq。ChIP-seq 包括化学固定细胞中的蛋白质-DNA 相互作用,然后使用目标蛋白质特异性抗体进行免疫沉淀(“pull-down”),以选择与目标蛋白质 (POI) 结合的 DNA 片段。这些 DNA 片段可以测序,以揭示特定蛋白质(如 TF)或包含特定组蛋白修饰的位点的基因组结合位置1。通过结合 ATAC-seq 和 ChIP-seq 数据集,可以得出细胞群的监管环境的详细图片。
分析所需的基本工作流程如下:在与参考基因组比对(“映射”)之前,必须对原始测序读数进行质量控制。然后可以过滤成功映射的 reads 以去除低质量 reads 和 PCR 重复。为了可视化这些映射和过滤的 reads,有必要计算这些 reads 在整个基因组中的 “覆盖率”。这将生成一个文件,该文件可以作为“轨道”上传到基因组浏览器,例如多位点视图 (MLV) 或 UCSC 基因组浏览器6,7。这些覆盖轨迹的峰值识别或“峰值调用”通常使用 LanceOtron 或 MACS2 等工具实现 8,9。最后,通过分析峰位置、形状和大小,可以在样品或生物条件之间进行比较。这些数据集的分析和集成是一个复杂的多步骤过程,其中可以实施生物信息学工具的不同组合。不同版本的工具可能彼此不兼容,并且可能会更改数据处理的输出。实施数据处理的不同部分所需的计算能力和用户熟练程度也多种多样,如 nf-core10、panpipes11、genpipes12、PEPATAC13 或 ChIP-AP14 管道所示。
总体而言,这导致分析和分析报告不一致,这反过来又导致生物信息学知识有限的任何人的可重复性、可访问性和便利性不佳。我们使用 CATCH-UP(完整的 ATAC-seq 和 ChIP-seq 上游管道)解决了所有这些问题,这是一种易于使用、灵活且模块化的管道,用于处理 ChIP-seq 和 ATAC/DNase-seq 数据。CATCH-UP 的实施需要最少的生物信息学经验;它可以在各种计算基础设施上运行,并支持在研究小组内部和研究小组之间进行可重复的数据分析。
CATCH-UP 是一个基于 Python 的 Snakemake 管道,旨在标准化 ChIP-seq 和 ATAC-seq 数据的分析。它以原始测序数据(fastq.gz 文件)作为输入,并以峰 (.bed) 文件的形式生成输出,提供每个步骤的相应结果。我们提供了一个 yaml 格式的配置文件(config.yaml),用户可以在其中编辑每个分析步骤的参数。在 snakemake 中实现的管理系统允许使用不同的计算基础设施(例如服务器、集群、云系统或个人计算机),如果用户提供大量数据,则可以并行使用。
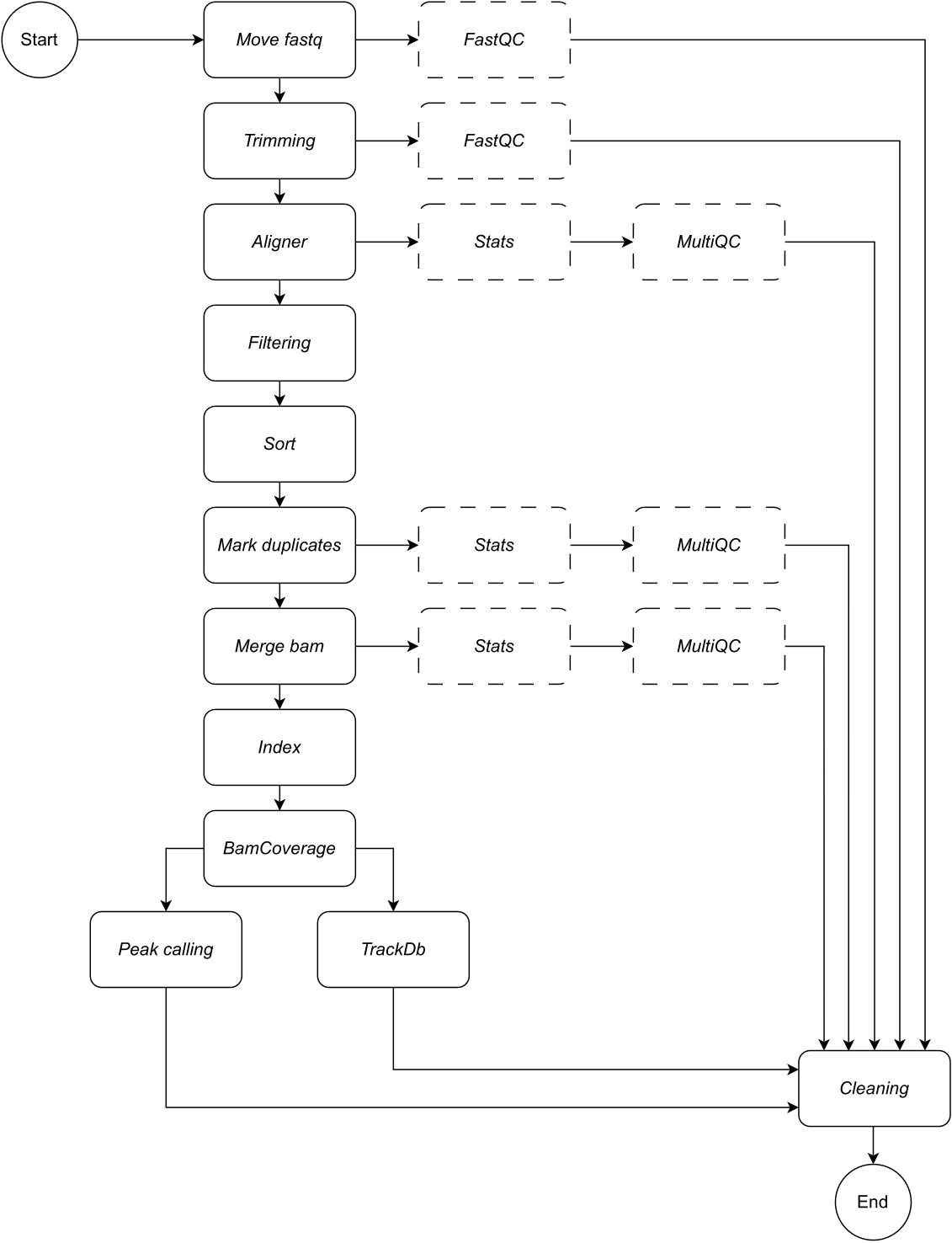
下面,我们提供了工作流每个步骤的详细说明(参见 图 1 的工作流图示)。此说明对于按照协议部分中的分步作至关重要:
Move fastq: pipeline 的第一步是将原始 fastq 文件复制到命名的分析目录中。这将使原始数据保持不变,以避免损坏或修改原始数据文件。
连接:如果原始测序数据包含多个泳道,则需要在分析之前执行此步骤来连接泳道。默认情况下,管道将所有 fastq 文件作为单个样本处理。必须在配置文件中定义此串联步骤。
Trimming:可选的数据清理步骤。这允许使用 trimmomatic15 修剪低质量的读长或接头序列。用户可以提供适配器序列的自定义 fasta 文件;Adapter 目录中提供了一个示例。可以在配置文件中定义其他 trimming 参数。默认情况下,工作流会跳过此规则。
Aligner:对齐时,默认应用 Bowtie216 ;也可以指定其他对齐工具,例如 BWA-MEM217 。选择 Bowtie2 比对工具为默认工具,因为它特别擅长将相对较短的读数比对到相对较大的基因组,因此非常适合将 ChIP-seq 和 ATAC-seq 数据与哺乳动物基因组比对。为了避免任何中间文件,将 aligner 通过管道传输到 samtools view 中,以将 bam 文件保存在输出中。对于此规则,用户必须指定首选的基因组构建,以在其上映射读数,例如 hg19/hg38(人类)、mm10/mm39(小鼠)。
筛选:保留正确映射的读取,并筛选掉质量低的读取。默认值:samtools 视图,带参数:-bShuF 4 -f 3 -q 30。
Sort:对齐的读取按最左侧坐标的顺序排序。默认值:samtools sort (snakemake wrapper),参数为:-m 4G。
Mark duplicates:识别并标记所有重复的读取。用户可以通过更改配置文件参数来决定删除它们。默认值:Picard MarkDuplicates(snakemake 包装器),参数:--REMOVE_DUPLICATES False 标记并保留重复项。
合并 bam:如果测序数据由重复或样本组成,用户可能希望合并为单个 bam。在这种情况下,用户可以选择合并 bams 或在整个分析过程中将 bam 文件分开。如果用户选择合并 bams(使用 samtools merge),则必须为合并的 bams 指定一个通用前缀。
Index:此步骤对排序后的坐标进行索引。默认值:samtools 索引(snakemake 包装器),使用 samtools 指定的默认参数。
BamCoverage:此规则从对齐的读取创建一个大型覆盖率跟踪。应用 deepTools 的 bamCoverage 工具,覆盖率计算为每个 bin 的读取次数,其中 bin 表示指定大小的窗口。在此管道中,bamCoverage 应用时,将以下参数设置为默认值:-bs 1 -normalizeUsing RPKM -extendReads。
峰值调用:LanceOtron8 被选为该管道的默认峰值调用器。与传统的峰值调用器不同,传统的峰值调用器主要基于统计测试,而 LanceOtron 是一种基于深度学习的峰值调用器,它结合了基因组富集测量和统计测试,并已被证明优于行业标准的峰值调用器 MACS29。为了使 bigwigs 与 LanceOtron 兼容,必须按碱基对计算覆盖率,并对 RPKM 进行标准化;这反映在 BamCoverage 步骤的默认设置中。可以选择 MACS2 作为替代峰值调用方。将监控新峰值调用方的释放,并在适用时纳入其中,以维护和优化此分析管道的性能。
TrackDb:这将创建 bigwig 文件的键值对关联,以便在 MLV6 或 UCSC Genome Browser18 平台等工具中加载和可视化它们。
除了输出数据之外,管道的每个步骤都会输出一个日志文件,并提供适当的质量控制检查,以便用户可以跟踪分析进度。FastQC19 适用于原始和修剪(如果选择)测序数据(步骤 1 - 移动 fastq 和 2 - 修剪)。Samtools stats 和 MultiQC20 用于收集、生成和可视化输出中 bam 文件的质量控制报告,步骤为 3 - 对齐器,6 - 标记重复项,和 7 - 合并 bam。 有关上述步骤中应用的每个工具的更多信息,请参阅 表 1。
研究方案
1. 运行 CATCH-UP 管道
- 从 https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline 克隆 UpStreamPipeline 存储库:
导航到所选的工作目录,复制以下代码并在命令行上运行:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - 在使用以下命令下载的 UpStreamPipeline 文件夹中导航: cd UpStreamPipeline
- 安装 anaconda 发行版(如有必要):
- 使用 conda 命令检查系统上是否已安装 anaconda。如果该命令未显示任何 conda 发行版的任何路径,请从 https://github.com/conda-forge/miniforge#mambaforge 下载 mambaforge 并为系统选择正确的发行版和版本。例如,对于 Linux 用户,请使用 wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh。请访问此网页了解不同的作系统: https://github.com/conda-forge/miniforge/。
- 使用 sh Mambaforge-Linux-x86_64.sh 运行安装程序,然后通过运行 conda init 在系统中初始化 conda。
- 安装并激活上游 conda 环境(上游 conda 环境的要求如 表 2 中列出):
- 使用命令 mamba env create - file=envs/upstream.yml 安装环境。
- 使用命令 conda activate upstream 激活环境。
- 成功安装上游 conda 环境后,使用命令 conda activate upstream 激活环境,然后使用 cd genetics/CATCH-UP 导航到 CATCH-UP 文件夹。
- 编辑配置文件,可以使用命令 cd /config/analysis.yaml 在 config 文件夹中找到该文件,并使用文本编辑器根据分析规范对其进行相应修改。按照逐行说明编辑文件本身中的每个参数。该文件将在分析后保留,并用于记录运行参数以帮助重现。
- 在文本编辑器(例如,Mac 的 TextEdit 或 Windows 的记事本)中打开并编辑以下三个文件:
- 编辑 1_fastqfile_home_dir.txt 文件以包含所有要分析的 fastq 文件的列表。
注意:必须排除读取号码和分机(例如, _R1/_R2 和 .fastq.gz)。例如,如果一个项目包含以下 fastq 文件列表:
Sample1_conditionA_L001_R1 .fastq .广州
Sample1_conditionA_L001_R2 .fastq .广州
Sample1_conditionA_L002_R1 .fastq .广州
Sample1_conditionA_L002_R2 .fastq .广州
Sample1_conditionB_L001_R1 .fastq .广州
Sample1_conditionB_L001_R2 .fastq .广州
Sample1_conditionB_L002_R1 .fastq .广州
Sample1_conditionB_L002_R2 .fastq .广州
Sample2_conditionA_L001_R1 .fastq .广州
Sample2_conditionA_L001_R2 .fastq .广州
Sample2_conditionA_L002_R1 .fastq .广州
Sample2_conditionA_L002_R2 .fastq .广州
Sample2_conditionB_L001_R1 .fastq .广州
Sample2_conditionB_L001_R2 .fastq .广州
Sample2_conditionB_L002_R1 .fastq .广州
Sample2_conditionB_L002_R2 .fastq .广州
在这种情况下, 1_fastqfile_home_dir.txt如下:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - 如果原始数据包含需要串联的测序泳道,请编辑 file_concat.txt 文件2_fastq以定义要串联的文件名的前缀。如果没有要连接的排序通道,则不要编辑 2_fastqfile_concat.txt。确保 2_fastqfile_concat.txt的每一行都包含一个示例前缀,如下所示:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - 如果需要合并不同样本的数据,则使用要合并的文件名的前缀 编辑3_merge_bams.txt 文件。确保每行都包含一个示例前缀,如下所示:
样本 1
示例 2
图 2 显示了如何汇总这三个文件的摘要。该方案可应用于单端或双端测序数据。除非另有指定,否则管道默认为 paired-end analysis;这可以在配置文件中修改(请参阅步骤 1.6)。
- 编辑 1_fastqfile_home_dir.txt 文件以包含所有要分析的 fastq 文件的列表。
- 编辑完所有必需的文件后,使用 snakemake 运行 CATCH-UP,如下所示: snakemake --configfile=config/analysis_name.yaml all --cores 4。
注意:有关更详细的说明和文档,请参阅 UpStreamPipeline GitHub 存储库中的 CATCH-UP 文件夹, 可在此处获取。这包括有关正确修改配置文件的详细文档,从更改测序数据文件的路径和存储结果到编辑每个步骤的参数。
结果
CATCH-UP 管道为每个步骤生成结果、日志和质量控制 (QC) 输出。在配置文件中,用户可以选择保留或删除输出文件以减少所需的存储内存。所有输出都解释如下:
00. fastq_home_dir:配置文件、 fastqfile_home_dir.txt 和 merge_bams.txt 被复制到此文件夹中以供参考和重现。
01. 读取:将 FastQ 文件复制到此文件夹中,以避免在工作流过程中更改原始数据,如果指定,可以连接泳道。
02. 修剪:如果指定,则修剪读取和适配器的 FastQ 文件。
03. Aligner:与选定的基因组进行比对。
04. 过滤:质量控制过滤。
05. sorted: BAM 文件的排序。
06. duplicates:标记重复项。
07. merge:如果在 config.yaml 中指定了,则合并 BAM 文件。
08. bam_coverages:报道的 bigwig 文件。
09. peak_calling:LanceOtron peak calling 输出的 bed 文件。
10. 跟踪:生成一个格式化的文本文件,如果需要,可以在 Genome Browser 上使用。
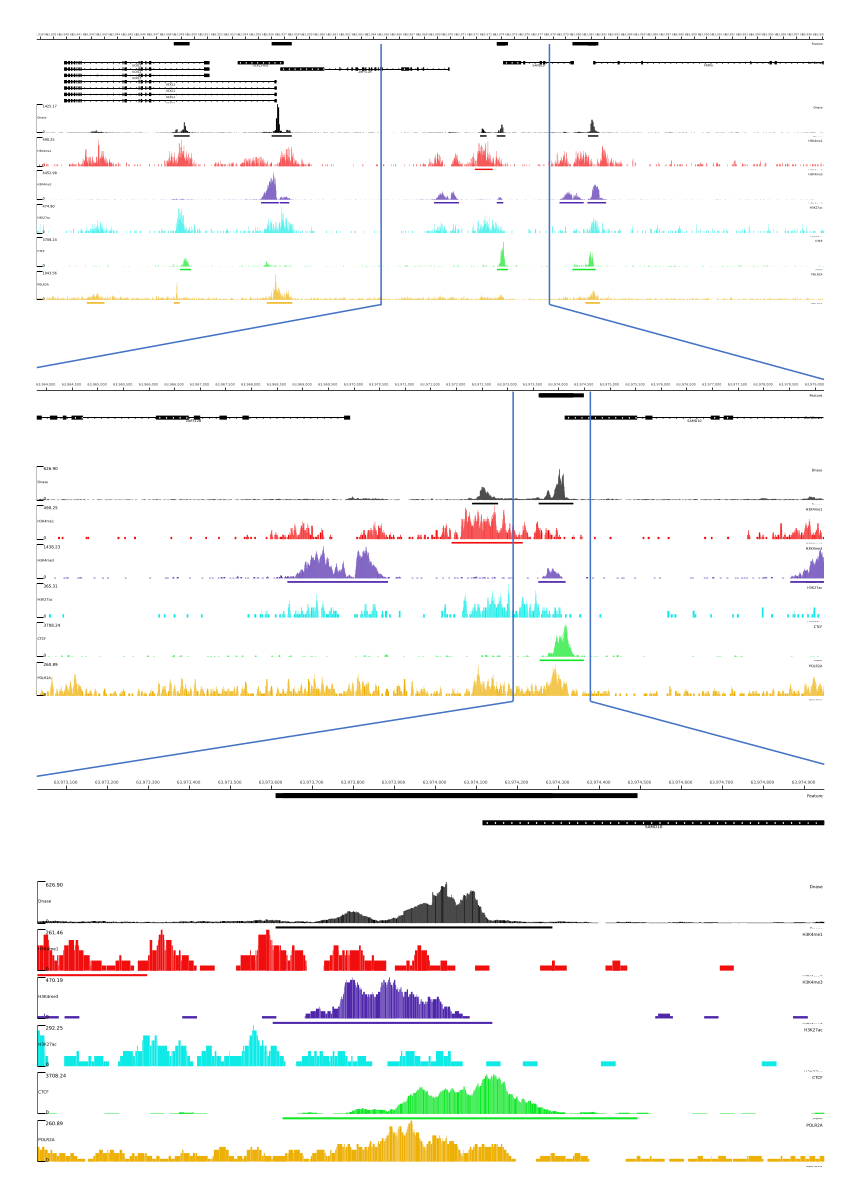
对于 01、02、03、06 和 07 输出,提供了 QC 指标和 HTML 文件。此外,在 图 3 中,我们提供了一个使用 CATCH-UP 处理数据的示例,通过 MLV 平台可视化最终输出。

图 1:CATCH-UP 的工作流程。 给定 fastq 文件列表,CATCH-UP 会并行处理所有上游步骤中的所有样本。 请单击此处查看此图的较大版本。
{kind=link}

图 2:说明如何正确修改 1_fastqfile_home_dir.txt、2_fastqfile_concat.txt 和 3_merge_bams.txt 才能运行 CATCH-UP 的说明性表示。请单击此处查看此图的较大版本。
{kind=link}

图 3:CATCH-UP 管道的示例输出。 原始测序数据 (fastq 文件) 从 ENCODE21 下载。CATCH-UP 管道用于处理 DNase-seq 和 5 种类型的 ChIP-seq (H3K4me1、H3K4me3、H3K27ac、CTCF 和 POLR2A) 的 fastq 文件。将 Bigwig 输出文件上传到 Multi Locus View,用于可视化和鉴定基因组调控元件。 请单击此处查看此图的较大版本。
{kind=link}
表 1:文档资源。 下表显示了 CATCH-UP 工作流中涉及的工具、其文档的链接以及相应的参考资料。 请点击此处下载此表格。
表 2:上游 conda 环境的通道和依赖项要求列表。请点击此处下载此表格。
表 3:用于测试 CATCH-UP 的作系统。 Ubuntu 在高性能集群和本地计算机上进行了测试。 请点击此处下载此表格。
讨论
NGS 技术在生成基因组数据方面的采用和利用的增加与用于分析这些数据的生物信息学工具的开发增加相匹配。数据分析的每个步骤都可以应用多种工具,也可以在每个工具中指定许多不同的参数 6,8,9,15,16,17,18,19,20,22,23,24 .这使得可以应用的分析策略组合非常多样化,每种策略都可能产生结果的变化。为了准确比较实验,生物信息学分析的标准化是必不可少的。从历史上看,NGS 数据由湿实验室科学家生成,数据由生物信息学家分析。
NGS 数据分析可分为“上游”和“下游”流程,其中上游包括从测序机输出的原始数据到研究人员可目视解释的格式的必要步骤。下游分析包括针对研究问题和实验设计定制的其他步骤。因此,上游管道是可推广的,并且适合标准化,以提高科学可重复性。另一方面,下游管道是定制的,取决于生物学问题,并且需要研究人员的洞察力,这使得它们不太适合标准化。我们创建了一个用户友好的上游管道,使湿实验室科学家能够可重复地分析自己的数据,而无需任何生物信息学知识。在这里,我们介绍了 CATCH-UP,这是一个使用 snakemake 框架构建的管道,旨在既用户友好,又可以解决 ChIP-seq 和 ATAC-seq 数据分析中的可重复性问题。此管道已构建用于处理 ChIP-seq 或 ATAC-seq 数据。用户下载 CATCH-UP 后,必须先定义分析参数和样品命名,然后才能使用一行代码在命令行上运行管道。配置文件本身和 CATCH-UP GitHub 存储库中的分步指南中提供了有关如何自定义 ChIP-seq 或 ATAC-seq 分析分析参数的简单分步说明。
目前已有用于 ChIP-seq 或 ATAC-seq 数据的分析流程,例如 PEPATAC 和 ChIP-AP。虽然这些工具具有优势,例如将上游和下游分析整合到单个工作流程中或使用图形用户界面 (GUI),但这些工具针对的是具有中等计算培训水平的生物信息学家和科学家13,14。CATCH-UP 旨在解决两个问题:使没有生物信息学培训的湿实验室科学家能够进行自己的上游分析,并通过促进跨实验室的轻松报告和精确重现性来实现上游分析的标准化。CATCH-UP 有意仅限于上游分析,但输出与下游分析工具兼容,例如用于统计比较数据集或推断转录因子结合的工具25,26。
执行可重复上游分析所需的所有关键步骤都在 CATCH-UP 管道中预定义,以确保稳定性。此管道的详细性质允许用户逐步跟踪管道的输出,这对于故障排除和启用可复制的分析工作流程都非常有用。鉴于 NGS 技术的快速发展性质,该流程的模块化特性是有益的,因为它提供了轻松适应的能力,以整合工具版本更新的发布和新工具的实施。CATCH-UP 已成功针对以下作系统进行测试:Ubuntu、CentOS、macOS (Intel CPU) 和 Windows(表 3)。该流程旨在通过并行化工作流程来处理包含数十个样品的大型实验,使其能够适应不同的实验设计。总体而言,在 ChIP-seq 和 ATAC-seq 数据的分析中实施 CATCH-UP 可实现用户友好、可重现且适应性强的分析工作流程。
披露声明
J.R.H. 是 Nucleome Therapeutics 的联合创始人兼董事,并为该公司提供咨询。
致谢
JRH 得到了 Wellcome Trust(225220/Z/22/Z 和 106130/Z/14/Z)和 MRC (MC_UU_00029/3) 的资助。MB得到了Wellcome Trust赠款(225220/Z/22/Z)的支持。E.R.G 得到了土耳其共和国国家教育部国家教育部出国研究生教育 (YLSY) 奖学金的选择和安置支持。E.G. 得到了 Wellcome 基因组医学和统计学博士计划 (108861/Z/15/Z) 的支持。S.G.R. 得到了医学研究委员会 (MRC) 资助 (MC_UU_00029/3) 的支持。
材料
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
参考文献
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。