
Method Article
CATCH-UP:ATAC-SeqおよびChIP-Seqのバルクデータのためのハイスループットアップストリームパイプライン
要約
ATAC-seqおよびChIP-seqは、遺伝子制御の詳細な調査を可能にします。しかし、これらのデータタイプの処理は困難であり、多くの場合、研究グループ間で一貫性がありません。CATCH-UPは、新規および公開されたATAC/ChIP-seqデータセットの標準化された再現性のあるデータ処理と分析を可能にする、使いやすい計算パイプラインです。
要約
トランスポゼースアクセス性クロマチン(ATAC)およびクロマチン免疫沈降(ChIP)のアッセイは、次世代シーケンシング(NGS)と相まって、遺伝子制御の研究に革命をもたらしました。これらの手法によって生成された高次元データセットの分析における標準化の欠如は、再現性の達成を困難にし、公開および処理されたデータに不一致をもたらしています。この問題の一部は、これらのタイプのデータの分析に利用できるさまざまなバイオインフォマティクスツールによるものです。次に、生データを完全に処理され解釈可能な出力に変換するには、さまざまなバイオインフォマティクスツールが順次必要であり、これらのツールにはさまざまなレベルの計算スキルが必要です。さらに、品質管理には、データ処理中に一様に採用されていない多くのオプションがあります。私たちは、トランスポザーゼアクセス可能なクロマチンシーケンシング(ATAC-seq)およびクロマチン免疫沈降シーケンシング(ChIP-seq)アップストリームパイプライン(CATCH-UP)の完全なアッセイ、つまり、生のfastqファイルから視覚化可能なbigwigトラックおよびピークコールまでのバルクChIP-seqおよびATAC-seqデータセットの解析のための使いやすいPythonベースのパイプラインで、これらの問題に対処します。このパイプラインはインストールと実行が簡単で、必要な計算知識は最小限です。このパイプラインはモジュール式でスケーラブルで、さまざまなコンピューティングインフラストラクチャで並列化できるため、新規または公開されたデータセットの再現性のある分析を可能にする方法論のレポート作成が容易になります。
概要
細胞が正しい生物学的機能を確立し維持するためには、遺伝子発現を厳密に制御する必要があります。多くの疾患の病因の根底には遺伝子の異常な発現があることはよく知られており、遺伝子制御のメカニズムの解明には大きな研究関心が集まっています1。遺伝子発現は、プロモーターやエンハンサーなどの調節要素によって促進されます。それらの配列内には、これらの要素には転写因子(TF)結合部位が含まれており、活性するとTF結合のプラットフォームが提供されます。これらの部位でのTFの結合は、ヌクレオソームの置換をもたらし、DNAアクセシビリティの増加とそれに続く転写機構への許容性の増大をもたらす。このアクセス可能性の向上の結果として、DNAのこれらの領域は、DNaseやTn5などのヌクレアーゼやトランスポゼーゼに対してより敏感になり、転写制御を研究する研究者によって利用されている生化学的特性です2,3。
DNase-seqおよびATAC-seqにより、研究者はゲノム全体のオープンクロマチン、TF結合部位、およびヌクレオソーム位置の領域をマッピングできます。これら2つの技術のうち、ATAC-seqは、シンプルな2ステップのプロトコルと必要な細胞数が少ない(DNase-seqの複製あたり100万個に対して50,000個の細胞)ため、過去10年間で人気が高まっています。ATAC-seqは、細胞集団における一般的なクロマチンの状況を概観することができますが、特定のタンパク質がゲノムに結合しているかどうかは大きく左右されません4,5。特定のタンパク質がゲノムと相互作用している場所を特定するためのゴールドスタンダード技術は、Chromatin Immunoprecipitation (ChIP)-seqです。ChIP-seqでは、細胞内のタンパク質-DNA相互作用を化学的に固定した後、目的のタンパク質に特異的な抗体を用いて免疫沈降(プルダウン)を行い、目的のタンパク質(POI)に結合したDNA断片を選択します。これらのDNA断片を配列決定して、TFなどの特定のタンパク質のゲノム結合位置、または特定のヒストン修飾を含む部位を明らかにすることができます1。ATAC-seqとChIP-seqのデータセットを組み合わせることで、細胞集団の制御状況を詳細に把握することができます。
解析に必要な基本的なワークフローは次のとおりです:生のシーケンシングリードは、参照ゲノムにアラインメントする前に品質管理する必要があります(「マッピング」)。正常にマッピングされたリードは、低品質リードとPCR重複の両方を削除するためにフィルタリングできます。これらのマッピングおよびフィルタリングされたリードを視覚化するには、ゲノム全体のこれらのリードの「カバレッジ」を計算する必要があります。これにより、マルチローカスビュー(MLV)やUCSCゲノムブラウザなどのゲノムブラウザに「トラック」6,7としてアップロードできるファイルが生成されます。これらのカバレッジトラックのピーク同定、または「ピークコール」は、通常、LanceOtron や MACS2 8,9 などのツールを使用して実現されます。最後に、ピークの位置、形状、サイズの分析を通じて、サンプル間または生物学的条件間で比較を行うことができます。これらのデータセットの分析と統合は、バイオインフォマティクスツールのさまざまな組み合わせを実装できる複雑な多段階のプロセスです。ツールの異なるバージョンは互いに互換性がなく、データ処理の出力を変更する可能性があります。また、nf-core10、panpipes11、genpipes12、PEPATAC13、または ChIP-AP14 パイプラインに示されているように、データ処理のさまざまな部分を実装するために必要な計算能力とユーザーの習熟度にも多岐にわたる。
全体として、これは分析と分析の報告の両方に一貫性を欠くことにつながり、その結果、バイオインフォマティクスの知識が限られている人にとっては、再現性、アクセシビリティ、利便性が低下しています。私たちは、ChIP-seqおよびATAC/DNase-seqデータを処理するための使いやすく柔軟性のあるモジュール式パイプラインであるCATCH-UP(Complete ATAC-seq and ChIP-seq upstream pipeline)で、これらの問題をすべて解決します。CATCH-UPの実装には、最小限のバイオインフォマティクスの経験が必要です。さまざまなコンピューティングインフラストラクチャで実行でき、研究グループ内および研究グループ間で再現性のあるデータ分析が可能になります。
CATCH-UPは、ChIP-seqおよびATAC-seqデータの解析を標準化するために構築されたPythonベースのSnakemakeパイプラインです。生のシーケンシングデータ(fastq.gzファイル)を入力として受け取り、各ステップのそれぞれの結果を提供するピーク(.bed)ファイルの形式で出力を生成します。設定ファイルはyaml形式(config.yaml)で提供しており、ユーザーは各分析ステップのパラメータを編集することができます。snakemakeに実装された管理システムは、ユーザーが大量のデータを提供する場合に、さまざまなコンピューティングインフラストラクチャ(サーバー、クラスター、クラウドシステム、パーソナルコンピューターなど)を並行して使用できます。
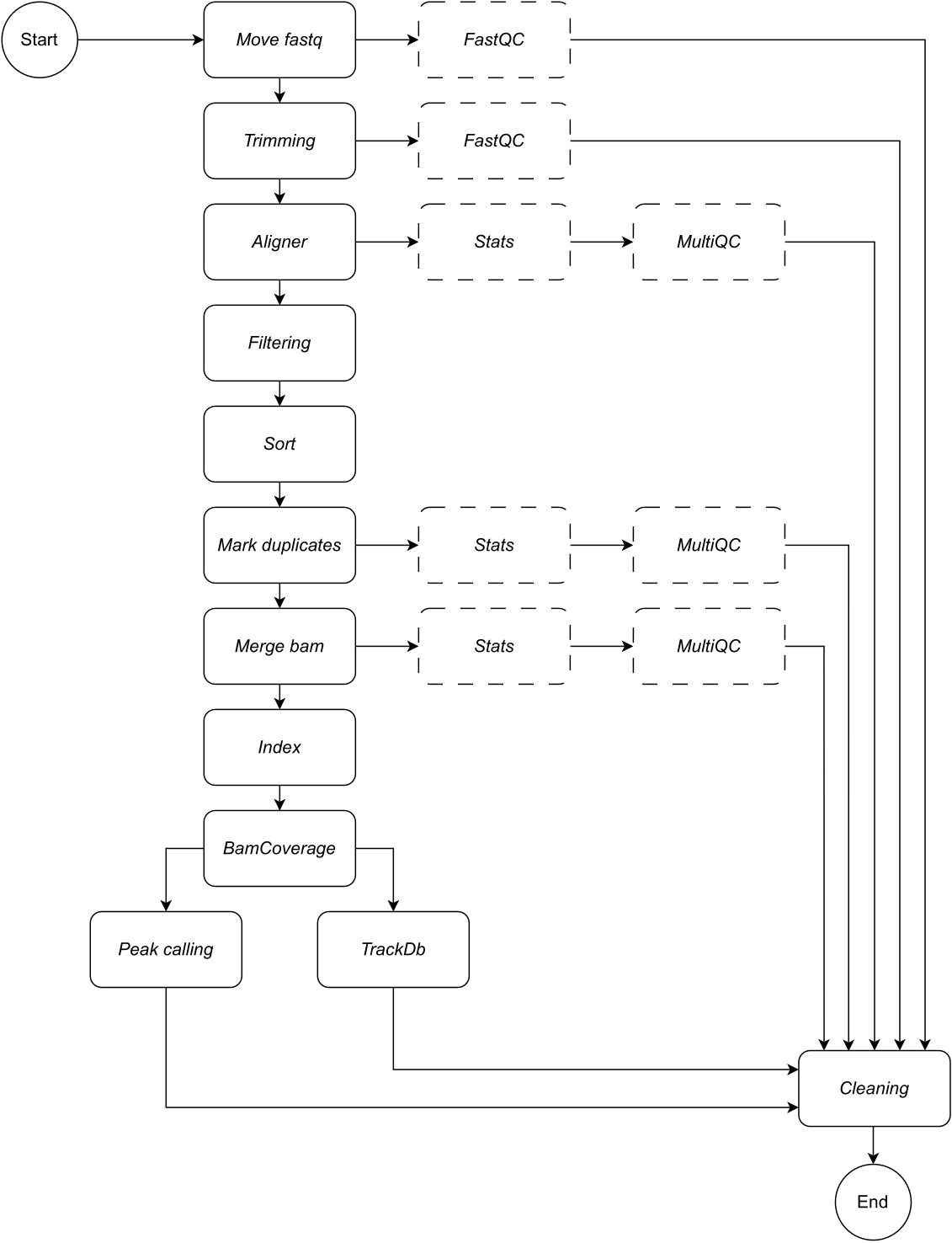
以下に、ワークフローの各ステップについて詳しく説明します (ワークフローの図については、 図 1 を参照してください)。この説明は、プロトコルセクションのステップバイステップに従うために不可欠です。
fastq の移動: パイプラインの最初のステップは、生の fastq ファイルを名前付きの分析ディレクトリにコピーすることです。これにより、生データファイルの破損や変更を避けるために、元のデータはそのまま残ります。
連結:生のシーケンシングデータに複数のレーンが含まれている場合、解析前にレーンを連結するためにこの手順が必要です。デフォルトでは、パイプラインはすべての fastq ファイルを 1 つのサンプルとして扱います。この連結ステップは、構成ファイルで定義する必要があります。
トリミング:オプションのデータクリーニングステップ。これにより、trimmomatic15 を使用して低品質の読み取りまたはアダプター シーケンスをトリミングできます。ユーザーは、アダプター シーケンスのカスタム fasta ファイルを提供できます。例は、アダプター・ディレクトリーにあります。追加のトリミング パラメーターは、構成ファイルで定義できます。デフォルトでは、ワークフローはこのルールをスキップします。
アライナー:アライメントの場合、Bowtie216 がデフォルトで適用されます。BWA-MEM217 などの代替アライメントツールも指定できます。Bowtie2 アライメントツールは、比較的短いリードを比較的大きなゲノムにアラインメントするのに特に長けており、ChIP-seq および ATAC-seq データを哺乳類のゲノムにアラインメントするのに適しているため、デフォルトとして選択されています。中間ファイルを避けるために、アライナーは samtools ビューにパイプされ、bam ファイルが出力に保存されます。このルールでは、ユーザーは、読み取りをマッピングする優先ゲノムビルドを指定する必要があります。例:hg19/hg38(ヒト)、mm10/mm39(マウス)。
フィルタリング: 適切にマッピングされた読み取りは保持され、低品質の読み取りはフィルタリングされます。デフォルト: samtools ビュー、パラメータ: -bShuF 4 -f 3 -q 30。
ソート: 整列された読み取りは、左端の座標の順にソートされます。デフォルト: samtools sort (snakemake wrapper), パラメータ: -m 4G.
重複をマークする: すべての重複読み取りが識別され、フラグが付けられます。ユーザーは、構成ファイルのパラメーターを変更することで、これらのファイルを削除することを決定できます。デフォルト: Picard MarkDuplicates (snakemake ラッパー)、パラメータ付き: --REMOVE_DUPLICATES False は重複にフラグを立てて保持します。
マージ bam: シーケンシングデータがレプリケートまたはサンプルで構成されている場合、ユーザーは 1 つの bam にマージしたい場合があります。この場合、ユーザーは bams をマージするか、分析全体で bam ファイルを分離するかを選択できます。ユーザーが bams をマージすることを選択した場合 (samtools merge を使用)、マージされた bams に共通のプレフィックスを指定する必要があります。
インデックス:この手順では、ソートされた座標にインデックスを付けます。デフォルト: samtools index (snakemake wrapper)、samtools によって指定されたデフォルトのパラメータを使用。
BamCoverage: このルールは、アライメントされた読み取りからビッグウィッグカバレッジトラックを作成します。deepTools の bamCoverage ツールが適用され、カバレッジはビンごとの読み取り数として計算され、ビンは指定されたサイズのウィンドウを表します。このパイプラインでは、bamCoverage は、既定値として設定されたパラメーター -bs 1 -normalizeUsing RPKM -extendReads で適用されます。
ピーク コーリング: LanceOtron8 は、このパイプラインの既定のピーク コーラーとして選択されました。従来のピークコーラーは、ほとんどが統計テストベースであるのに対し、LanceOtronはディープラーニングベースのピークコーラーであり、ゲノムエンリッチメント測定と統計テストを組み込んでおり、業界標準のピークコーラーであるMACS29を凌駕することが示されています。bigwigs が LanceOtron と互換性を持つためには、カバレッジを塩基対ごとに計算し、RPKM を正規化する必要があります。これは、BamCoverage ステップのデフォルト設定に反映されます。MACS2 は、代替のピークコーラーとして選択できます。新しいピークコーラーのリリースは、この分析パイプラインのパフォーマンスを維持および最適化するために、必要に応じて監視および組み込まれます。
TrackDb: これにより、MLV6 や UCSC Genome Browser18 プラットフォームなどのツールで bigwig ファイルをロードして視覚化するために、bigwig ファイルのキーと値のペアの関連付けが作成されます。
出力データに加えて、パイプラインの各ステップはログファイルを出力し、ユーザーが分析の進行状況を追跡できるように適切な品質管理チェックが提供されます。FastQC19は、生およびトリミングされた(選択されている場合)シーケンシングデータに適用されます(ステップ1 - Fastqの移動および2 - トリミング)。Samtoolsの統計とMultiQC20は、ステップ3 - Aligner、6 - Mark duplicates、および7 - Merge bamの出力で、bamファイルの品質管理レポートを収集、作成、視覚化するために使用されます。上記の手順で適用した各ツールの詳細については、表 1 を参照してください。
プロトコル
1. CATCH-UPパイプラインの実行
- https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline からUpStreamPipelineリポジトリをクローンします。
選択した作業ディレクトリに移動し、次のコードをコピーして、コマンドラインで実行します。
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git (英語) - ダウンロードしたUpStreamPipelineフォルダ内を、cd UpStreamPipelineコマンドを使用してナビゲートします。
- anaconda ディストリビューションをインストールします (必要な場合)。
- anacondaがシステムにすでにインストールされているかどうかを確認するには、コマンドwhich condaを使用します。コマンドでcondaディストリビューションへのパスが表示されない場合は、https://github.com/conda-forge/miniforge#mambaforge からmambaforgeをダウンロードし、システムに適したディストリビューションとバージョンを選択してください。たとえば、Linux ユーザーの場合は、wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh を使用します。さまざまなオペレーティングシステムについては、次のWebページにアクセスしてください: https://github.com/conda-forge/miniforge/。
- sh Mambaforge-Linux-x86_64.sh を使用してインストーラーを実行し、 conda init を実行してシステム内の conda を初期化します。
- アップストリームconda環境をインストールしてアクティブ化します(アップストリームconda環境の要件を 表2に示します)。
- コマンド mamba env create - file=envs/upstream.yml を使用して環境をインストールします。
- conda activate upstream コマンドを使用して環境をアクティブ化します。
- アップストリームのconda環境が正常にインストールされたら、 conda activate upstream コマンドを使用して環境をアクティブ化し、cd genetics/CATCH-UPを使用してCATCH-UPフォルダに移動します。
- config フォルダ内にある設定ファイルを cd /config/analysis.yaml コマンドを使用して編集し、テキストエディタを使用して解析仕様に合わせて変更します。行ごとの指示に従って、ファイル自体の各パラメーターを編集します。このファイルは解析後も保持され、再現性を高めるために実行パラメータを文書化する役割を果たします。
- テキストエディタで次の3つのファイルを開いて編集します(Macの場合はテキストエディット、Windowsの場合はメモ帳など)。
- 1_fastqfile_home_dir.txtファイルを編集して 、 解析するすべてのfastqファイルのリストを含めます。
注:読み取り番号と拡張子( _R1/_R2 や .fastq.gzなど)は除外する必要があります。たとえば、プロジェクトに次の fastq ファイルのリストが含まれているとします。
Sample1_conditionA_L001_R1。ファストク .ジーゼット
Sample1_conditionA_L001_R2。ファストク .ジーゼット
Sample1_conditionA_L002_R1 .ファストク .ジーゼット
Sample1_conditionA_L002_R2。ファストク .ジーゼット
Sample1_conditionB_L001_R1。ファストク .ジーゼット
Sample1_conditionB_L001_R2 .ファストク .ジーゼット
Sample1_conditionB_L002_R1。ファストク .ジーゼット
Sample1_conditionB_L002_R2。ファストク .ジーゼット
Sample2_conditionA_L001_R1。ファストク .ジーゼット
Sample2_conditionA_L001_R2。ファストク .ジーゼット
Sample2_conditionA_L002_R1。ファストク .ジーゼット
Sample2_conditionA_L002_R2 .ファストク .ジーゼット
Sample2_conditionB_L001_R1。ファストク .ジーゼット
Sample2_conditionB_L001_R2。ファストク .ジーゼット
Sample2_conditionB_L002_R1。ファストク .ジーゼット
Sample2_conditionB_L002_R2。ファストク .ジーゼット
この場合、 1_fastqfile_home_dir.txtは次のとおりです。
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - 生データに連結が必要なシーケンシングレーンが含まれている場合は、2_fastq file_concat.txtファイルを編集して、連結するファイル名のプレフィックスを定義します。連結するシーケンス レーンがない場合は、2_fastqfile_concat.txtを編集しないでください。2_fastqfile_concat.txtの各行に、次のように 1 つのサンプル プレフィックスが含まれていることを確認します。
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - 異なるサンプルのデータをマージする必要がある場合は、マージ するファイル名の プレフィックスを使用して3_merge_bams.txtファイルを編集します。次のように、すべての行に 1 つのサンプル プレフィックスが含まれていることを確認します。
サンプル1
サンプル2
図 2 は、これら 3 つのファイルをまとめる方法の概要を示しています。このプロトコルは、シングルエンドまたはペアエンドのシーケンシングデータに適用できます。パイプラインは、特に指定がない限り、既定でペアエンド分析になります。これは、設定ファイルで変更できます(ステップ1.6を参照)。
- 1_fastqfile_home_dir.txtファイルを編集して 、 解析するすべてのfastqファイルのリストを含めます。
- 必要なファイルをすべて編集したら、snakemake を使用して次のように CATCH-UP を実行します: snakemake --configfile=config/analysis_name.yaml all --cores 4.
注: 詳細な手順とドキュメントについては、 こちらから入手できる UpStreamPipeline GitHub リポジトリ内の CATCH-UP フォルダーを参照してください。これには、データファイルのシーケンスパスの変更や結果の保存から各ステップのパラメータの編集まで、設定ファイルを正しく変更するための詳細なドキュメントが含まれています。
結果
CATCH-UP パイプラインは、各ステップの結果、ログ、および品質管理 (QC) の出力を生成します。設定ファイル内では、ユーザーは出力ファイルを保持するか削除するかを選択して、必要なストレージメモリを減らすことができます。すべての出力は次のように説明されています。
00. fastq_home_dir:設定ファイル、 fastqfile_home_dir.txt、 およびmerge_bams.txt は、参照と再現性のためにこのフォルダにコピーされます。
01. 読み取り:ワークフロープロセス中に元の生データが変更されないように、FASTQファイルはこのフォルダにコピーされ、レーンが指定されている場合は連結できます。
02. トリミング:FastQファイルの読み取りとアダプタが指定されている場合はトリミングされます。
03. Aligner:選択したゲノムに対するアライメント。
04. Fiのltering:品質管理のろ過。
05. ソート:BAMファイルのソート。
06. 重複:重複にフラグを立てます。
07. マージ: これがconfig.yamlで指定されている場合、BAMファイルをマージします。
08. bam_coverages:カバレッジのbigwigファイル。
09. peak_calling:LanceOtronピークコール出力のベッドファイル。
10. track: 必要に応じてGenome Browserで使用できるフォーマット済みのテキストファイルを生成します。
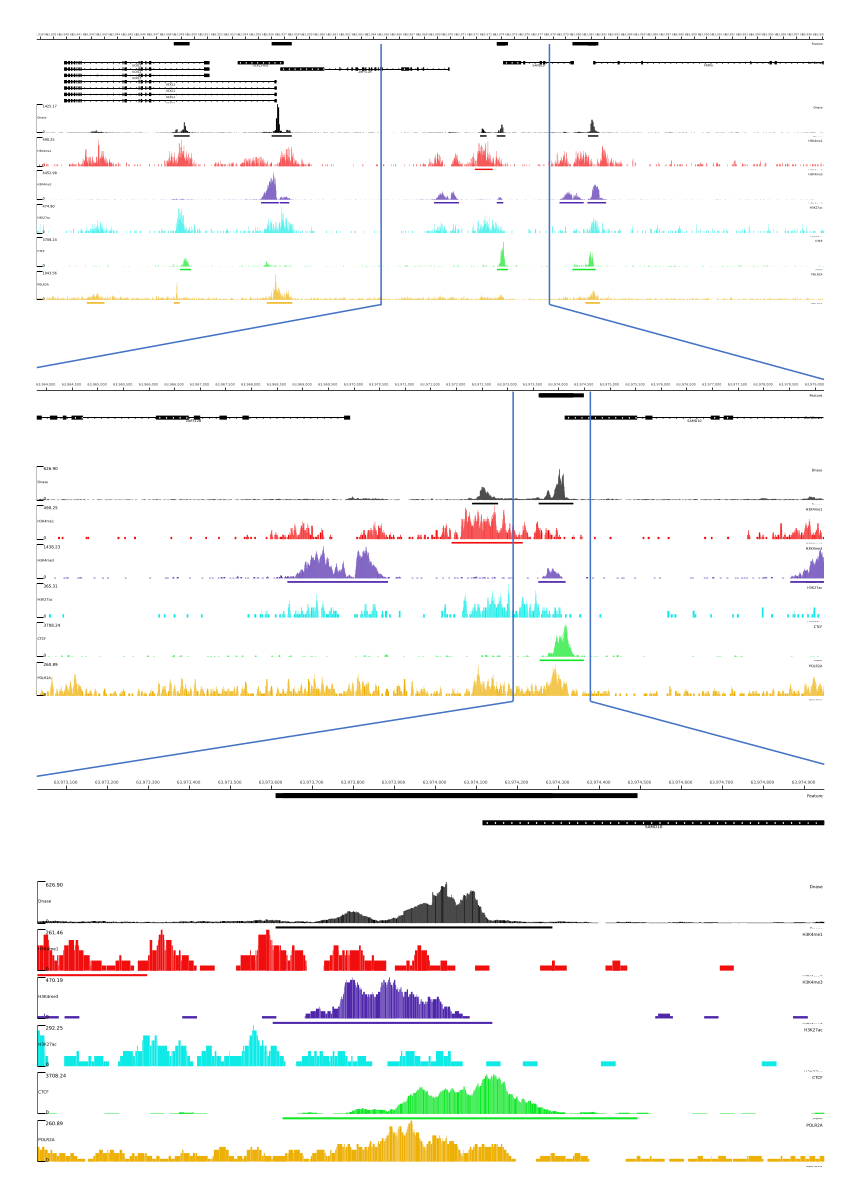
01、02、03、06、および 07 の出力には、QC メトリックと HTML ファイルが用意されています。また、 図3では、CATCH-UPを用いてMLVプラットフォームを通じて最終出力を可視化した処理データの例を示しています。

図1:CATCH-UPのワークフロー。 fastq ファイルのリストが与えられると、CATCH-UP はすべてのアップストリームステップを通じてすべてのサンプルを並行して処理します。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:CATCH-UPを実行するために 、1_fastqfile_home_dir.txt、 2_fastqfile_concat.txt、 および3_merge_bams.txtをどのように正しく変更する必要があるかを説明する説明的な表現。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図 3: CATCH-UP パイプラインからの出力例。 生のシーケンシングデータ(fastqファイル)はENCODE21からダウンロードしました。CATCH-UPパイプラインは、DNase-seqおよび5種類のChIP-seq(H3K4me1、H3K4me3、H3K27ac、CTCF、およびPOLR2A)のfastqファイルの処理に使用されました。Bigwigの出力ファイルをMulti Locus Viewにアップロードし、ゲノム調節要素の可視化と同定を行いました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
表 1: ドキュメント リソース。 この表は、CATCH-UP ワークフローに関連するツール、それらのドキュメントへのリンク、およびそれぞれの参照を示しています。 この表をダウンロードするには、ここをクリックしてください。
表 2: アップストリーム conda 環境のチャネルと依存関係の要件の一覧。この表をダウンロードするには、ここをクリックしてください。
表 3: CATCH-UP のテストに使用したオペレーティング システム。 Ubuntuは、高性能クラスターとローカルマシンでテストされました。 この表をダウンロードするには、ここをクリックしてください。
ディスカッション
ゲノムデータを生成するためのNGS技術の採用と利用の増加は、これらのデータを分析するためのバイオインフォマティクスツールの開発の増加と一致しています。データ分析の各ステップに適用できる複数のツールがあり、各ツール内で指定できる多くの異なるパラメーターがあります 6,8,9,15,16,17,18,19,20,22,23,24 .これにより、適用可能な分析戦略の組み合わせが非常に多様になり、それぞれが結果にばらつきをもたらす可能性があります。実験間で正確に比較するためには、バイオインフォマティクス解析の標準化が不可欠です。歴史的に、NGSデータはウェットラボの科学者によって生成され、データはバイオインフォマティシャンによって分析されます。
NGSデータ解析は、「アップストリーム」パイプラインと「ダウンストリーム」パイプラインに分けることができ、アップストリームには、シーケンシングマシンから出力された生データから研究者が視覚的に解釈できる形式に移行するために必要なステップが含まれます。ダウンストリーム解析には、研究課題と実験デザインに合わせた追加のステップが含まれます。したがって、アップストリームパイプラインは一般化可能で、科学の再現性を向上させるための標準化に適しています。一方、下流のパイプラインは、生物学的な問題に依存したオーダーメイドであり、研究者からの洞察を必要とするため、標準化にはあまり適していません。私たちは、ウェットラボの科学者がバイオインフォマティクスの予備知識を必要とせずに自分のデータを再現性よく分析できるように、ユーザーフレンドリーなアップストリームパイプラインを作成しました。ここでは、snakemakeフレームワークを使用して構築され、ChIP-seqおよびATAC-seqデータ解析における再現性の問題に対処するために設計された、ユーザーフレンドリーであるように設計されたパイプラインであるCATCH-UPを紹介します。このパイプラインは、ChIP-seqまたはATAC-seqデータを処理するために構築されています。ユーザーがCATCH-UPをダウンロードしたら、1行のコードを使用してコマンドラインでパイプラインを実行する前に、まず分析パラメータとサンプルの命名を定義する必要があります。ChIP-seqまたはATAC-seq解析の解析パラメータをカスタマイズする方法に関する簡単なステップバイステップの説明は、設定ファイル自体とCATCH-UP GitHubリポジトリのステップバイステップガイドに記載されています。
ChIP-seqまたはATAC-seqデータ用の既存の解析パイプラインは、PEPATACやChIP-APなどにあります。これらのパイプラインには、アップストリームとダウンストリームの両方の分析を単一のワークフローに組み込むことや、グラフィカルユーザーインターフェース(GUI)の使用などの利点がありますが、これらのツールは、中程度の計算トレーニングを受けたバイオインフォマティシャンや科学者を対象としています13,14。CATCH-UPは、バイオインフォマティクスのトレーニングを受けていないウェットラボの科学者が独自のアップストリーム分析を実行できるようにすることと、ラボ間での簡単なレポート作成と正確な再現性を促進することでアップストリーム分析の標準化を可能にするという2つの問題を解決するように設計されています。CATCH-UPは意図的にアップストリーム解析に限定されていますが、出力は、データセットを統計的に比較したり、転写因子の結合を推測するために使用されるようなダウンストリーム解析ツールと互換性があります25,26。
複製可能なアップストリーム解析を実行するために必要なすべての重要なステップは、堅牢性を確保するためにCATCH-UPパイプライン内で事前定義されています。このパイプラインの冗長な性質により、ユーザーはパイプラインの出力を段階的に追跡でき、トラブルシューティングと分析ワークフローのレプリケートの両方に役立ちます。NGS技術の急速に進化する性質を考えると、このパイプラインのモジュール性は、ツールバージョン更新のリリースと新しいツールの実装の両方を簡単に組み込むために適応できる機能を提供するため、有益です。CATCH-UP は、Ubuntu、CentOS、macOS (Intel CPU)、および Windows (表 3) のオペレーティング システムで正常にテストされています。パイプラインは、ワークフローを並列化することで数十のサンプルを含む大規模な実験を処理するように構築されており、さまざまな実験デザインに適応できます。全体として、ChIP-seqおよびATAC-seqデータの解析にCATCH-UPを実装することで、ユーザーフレンドリーで再現性が高く、適応性の高い解析ワークフローが可能になります。
開示事項
J.R.H.は、Nucleome Therapeuticsの共同設立者兼取締役であり、同社にコンサルティングを提供しています。
謝辞
J.R.H.は、ウェルカム・トラスト(225220/Z/22/Zおよび106130/Z/14/Z)およびMRC(MC_UU_00029/3)からの助成金によって支援されました。M.B.は、ウェルカム・トラストの助成金(225220/Z/22/Z)の支援を受けました。E.R.Gは、トルコ共和国国民教育省の国民教育省の大学院教育(YLSY)奨学金の国民教育省の選考と配置の支援を受けました。E.G.は、Wellcome Genomic Medicine and Statistics PhD Programme(108861/Z/15/Z)の支援を受けました。S.G.R.は、Medical Research Council(MRC)の助成金(MC_UU_00029/3)の支援を受けました。
資料
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
参考文献
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623(2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521(2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037(2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101(2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537(2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008(2021).
- Picard Toolkit. , http://broadinstitute.github.io/picard/ (2019).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved