
Method Article
CATCH-UP: Toplu ATAC-Seq ve ChIP-Seq verileri için yüksek verimli bir yukarı akış boru hattı
Bu Makalede
Özet
ATAC-seq ve ChIP-seq, gen regülasyonunun ayrıntılı olarak araştırılmasına izin verir; Ancak, bu veri türlerinin işlenmesi zordur ve araştırma grupları arasında genellikle tutarsızdır. CATCH-UP'ı sunuyoruz: yeni ve yayınlanmış ATAC/ChIP-seq veri kümelerinin standartlaştırılmış ve tekrarlanabilir veri işlemesine ve analizine olanak tanıyan, kullanımı kolay bir hesaplama hattı.
Özet
Transpozaz ile erişilebilir kromatin (ATAC) ve kromatin immünopresipitasyon (ChIP) testi, yeni nesil dizileme (NGS) ile birleştiğinde, gen regülasyonu çalışmasında devrim yaratmıştır. Bu teknikler tarafından oluşturulan yüksek boyutlu veri kümelerinin analizinde standardizasyon eksikliği, tekrarlanabilirliğin elde edilmesini zorlaştırmış ve yayınlanmış, işlenmiş verilerde tutarsızlıklara yol açmıştır. Bu sorunun bir kısmı, bu tür verilerin analizi için mevcut olan çeşitli biyoinformatik araçlardan kaynaklanmaktadır. İkinci olarak, ham verileri tamamen işlenmiş ve yorumlanabilir bir çıktıya dönüştürmek için sırayla bir dizi farklı biyoinformatik araç gereklidir ve bu araçlar farklı düzeylerde hesaplama becerileri gerektirir. Ayrıca, veri işleme sırasında tek tip olarak kullanılmayan kalite kontrol için birçok seçenek vardır. Bu sorunları, transpozaz erişilebilir kromatin dizilimi (ATAC-seq) ve kromatin immünopresipitasyon dizilimi (ChIP-seq) yukarı akış boru hattı (CATCH-UP) için eksiksiz bir tahlil ile ele alıyoruz, ham fastq dosyalarından görselleştirilebilir bigwig parçalarına ve tepe çağrılarına kadar toplu ChIP-seq ve ATAC-seq veri kümelerinin analizi için kullanımı kolay, Python tabanlı bir boru hattı. Bu işlem hattının kurulumu ve çalıştırılması kolaydır ve minimum hesaplama bilgisi gerektirir. İşlem hattı modüler, ölçeklenebilir ve çeşitli bilgi işlem altyapılarında paralelleştirilebilir olup, yeni veya yayınlanmış veri kümelerinin tekrarlanabilir analizini sağlamak için metodolojinin kolay raporlanmasına olanak tanır.
Giriş
Hücrelerin doğru biyolojik işlevlerini oluşturmaları ve sürdürmeleri için gen ekspresyonu sıkı bir şekilde düzenlenmelidir. Anormal gen ekspresyonunun birçok hastalığın patogenezinin altında yattığı iyi bilinmektedir ve bu nedenle, gen düzenleme mekanizmalarının anlaşılmasında çok fazla araştırma ilgisi yatmaktadır1. Gen ekspresyonu, destekleyiciler ve güçlendiriciler gibi düzenleyici unsurlar tarafından kolaylaştırılır. Dizileri içinde, bu elemanlar, aktif olduklarında TF bağlanması için bir platform sağlayan transkripsiyon faktörü (TF) bağlama bölgeleri içerir. TF'lerin bu bölgelerdeki bağlanması, nükleozomların yer değiştirmesine neden olur, bu da DNA erişilebilirliğinde bir artışa ve ardından transkripsiyonel mekanizmaya izin verilebilirlikte bir artışa neden olur. Bu artan erişilebilirliğin bir sonucu olarak, DNA'nın bu bölgeleri, transkripsiyonel düzenlemeyiaraştıran araştırmacılar tarafından yararlanılan biyokimyasal bir özellik olan DNaz ve Tn5 gibi nükleazlara ve transpozazlara karşı daha hassastır 2,3.
DNase-seq ve ATAC-seq, araştırmacıların genom boyunca açık kromatin, TF bağlanma bölgeleri ve nükleozomal konumlandırma bölgelerini haritalamasına olanak tanır. Bu iki teknikten ATAC-seq, basit iki aşamalı protokol ve düşük hücre sayısı gereksinimi (DNase-seq için replikat başına 1 milyona kıyasla 50.000 hücre) nedeniyle son on yılda popülerlik kazanmıştır. ATAC-seq, bir hücre popülasyonundaki genel kromatin manzarasına genel bir bakış sağlarken, hangi spesifik proteinlerin genomabağlandığı büyük ölçüde agnostiktir 4,5. Spesifik bir proteinin genom ile etkileşime girdiği yerleri belirlemek için altın standart teknik Kromatin İmmünopresipitasyon (ChIP)-seq.'dir. ChIP-seq, bir hücrede protein-DNA etkileşimlerinin kimyasal olarak sabitlenmesini ve ardından ilgilenilen protein (POI) tarafından bağlanan DNA fragmanlarını seçmek için ilgilenilen proteine özgü bir antikor kullanılarak immünopresipitasyonu ("aşağı çekme") içerir. Bu DNA fragmanları, TF'ler gibi spesifik proteinlerin genomik bağlanma konumlarını veya spesifik histon modifikasyonları içeren bölgeleri ortaya çıkarmak için dizilenebilir1. ATAC-seq ve ChIP-seq veri kümelerini birleştirerek, bir hücre popülasyonu için düzenleyici ortamın ayrıntılı bir resmi elde edilebilir.
Analiz için gerekli olan temel iş akışı şu şekildedir: ham dizileme okumaları, bir referans genoma hizalanmadan ("haritalama") önce kalite kontrolünden geçirilmelidir. Başarıyla eşlenen okumalar daha sonra hem düşük kaliteli okumaları hem de PCR kopyalarını kaldırmak için filtrelenebilir. Bu eşlenmiş ve filtrelenmiş okumaları görselleştirmek için, bu okumaların genom boyunca "kapsamını" hesaplamak gerekir. Bu, çok lokuslu görünüm (MLV) veya UCSC genom tarayıcısı gibi bir genom tarayıcısına bir "parça" olarak yüklenebilen bir dosya oluşturur6,7. Bu kapsama yollarının tepe tanımlaması veya "tepe çağrısı" tipik olarak LanceOtron veya MACS2 8,9 gibi araçlar kullanılarak elde edilir. Son olarak, tepe konumu, şekil ve boyut analizi yoluyla numuneler veya biyolojik koşullar arasında karşılaştırmalar yapılabilir. Bu veri kümelerinin analizi ve entegrasyonu, farklı biyoinformatik araç kombinasyonlarının uygulanabildiği karmaşık, çok adımlı bir süreçtir. Araçların farklı sürümleri birbiriyle uyumsuz olabilir ve veri işlemenin çıktısını değiştirebilir. NF-core10, panpipes11, genpipes12, PEPATAC13 veya ChIP-AP14 boru hatlarında gösterildiği gibi veri işlemenin farklı bölümlerini uygulamak için gereken hesaplama gücü ve kullanıcı yeterliliğinde de çok çeşitli vardır.
Genel olarak, bu, hem analizde hem de analizin raporlanmasında tutarsızlıklara yol açmış ve bu da biyoinformatik hakkında sınırlı bilgiye sahip herkes için zayıf tekrarlanabilirlik, erişilebilirlik ve rahatlığa yol açmıştır. Tüm bu sorunları, ChIP-seq ve ATAC/DNase-seq verilerini işlemek için kullanımı kolay, esnek ve modüler bir boru hattı olan CATCH-UP (tam ATAC-seq ve ChIP-seq yukarı akış boru hattı) ile ele alıyoruz. CATCH-UP'ın uygulanması minimum biyoinformatik deneyimi gerektirir; Çeşitli bilgi işlem altyapılarında çalıştırılabilir ve araştırma grupları içinde ve arasında tekrarlanabilir veri analizi sağlar.
CATCH-UP, ChIP-seq ve ATAC-seq verilerinin analizini standartlaştırmak için oluşturulmuş Python tabanlı bir Snakemake boru hattıdır. Ham sıralama verilerini (fastq.gz dosyaları) girdi olarak alır ve her adım için ilgili sonucu sağlayan tepe (.bed) dosyaları biçiminde bir çıktı oluşturur. Kullanıcının her analiz adımının parametrelerini düzenleyebileceği yaml biçiminde (config.yaml) bir yapılandırma dosyası sağlıyoruz. Snakemake içinde uygulanan yönetim sistemi, farklı bilgi işlem altyapılarının (sunucular, kümeler, bulut sistemleri veya kişisel bilgisayarlar gibi) ve kullanıcının büyük miktarda veri sağlaması durumunda paralel olarak kullanılmasını sağlar.
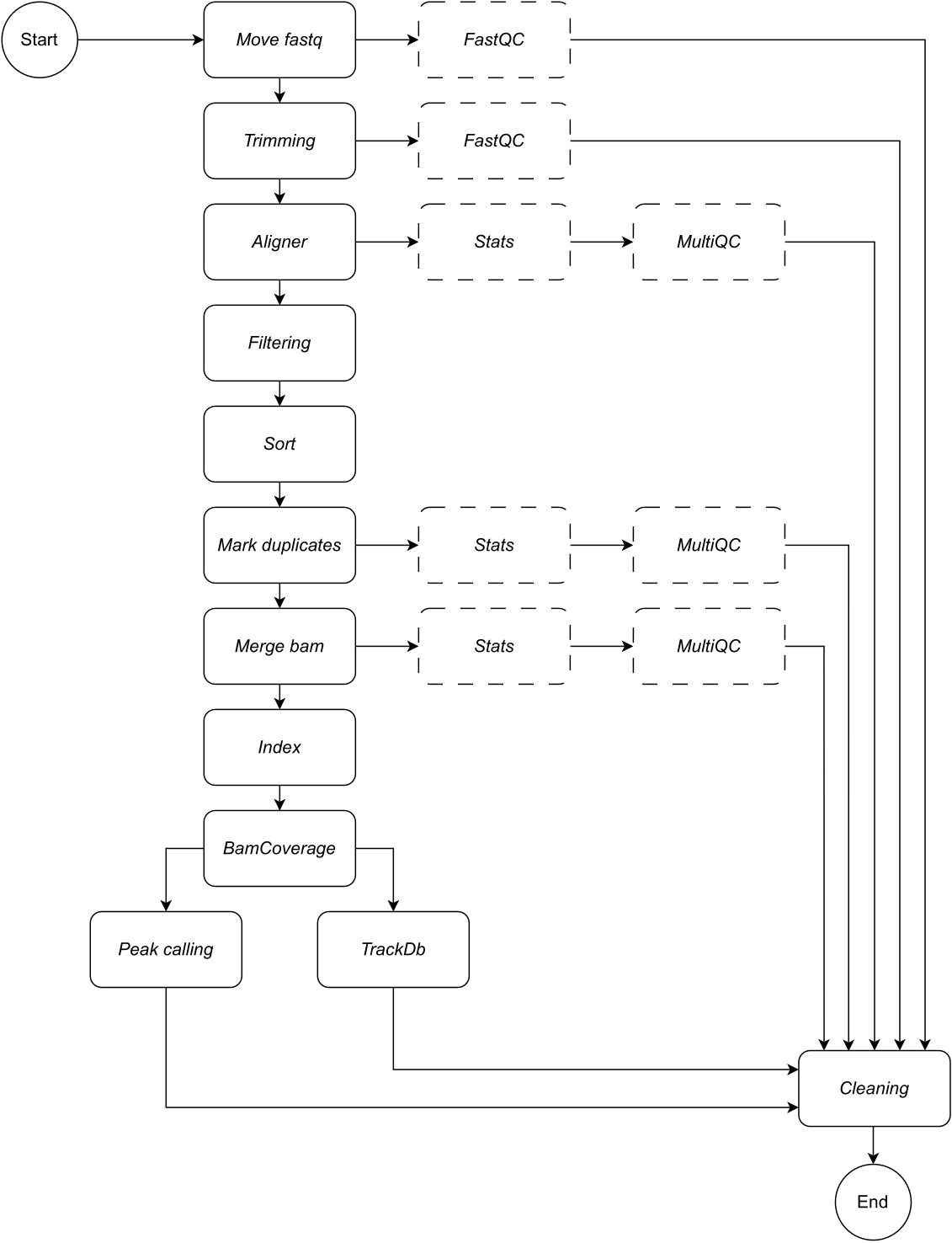
Aşağıda, iş akışının her adımının ayrıntılı bir açıklamasını sunuyoruz (iş akışı çizimi için Şekil 1'e bakın). Bu açıklama, protokol bölümündeki adım adım takip etmek için gereklidir:
Fastq'yu Taşı: İşlem hattının ilk adımı, ham FastQ dosyalarını adlandırılmış analiz dizinine kopyalamaktır. Bu, ham veri dosyalarının bozulmasını veya değiştirilmesini önlemek için orijinal verilere dokunulmaz.
Birleştirme: ham sıralama verileri birden fazla şerit içeriyorsa, analizden önce şeritleri birleştirmek için bu adım gereklidir. Varsayılan olarak, işlem hattı tüm fastq dosyalarını tek örnekler olarak işler. Bu birleştirme adımı, yapılandırma dosyasında tanımlanmalıdır.
Kırpma: isteğe bağlı veri temizleme adımı. Bu, trimmomatic15 kullanılarak düşük kaliteli okumaların veya adaptör dizilerinin kırpılmasına olanak tanır. Kullanıcı, adaptör dizilerinin özel fasta dosyalarını sağlayabilir; Bağdaştırıcı dizininde bir örnek verilmiştir. Konfigürasyon dosyasında ek kırpma parametreleri tanımlanabilir. Varsayılan olarak, iş akışı bu kuralı atlar.
Hizalayıcı: hizalama için Papyon216 varsayılan olarak uygulanır; BWA-MEM217 gibi alternatif hizalama araçları da belirtilebilir. Bowtie2 hizalama aracı, nispeten kısa okumaları nispeten büyük genomlara hizalamada özellikle usta olduğu ve bu nedenle ChIP-seq ve ATAC-seq verilerinin memeli genomlarına hizalanması için çok uygun olduğu için varsayılan olarak seçilir. Herhangi bir ara dosyadan kaçınmak için, hizalayıcı, bam dosyasını çıktıya kaydetmek için samtools görünümüne aktarılır. Bu kural için kullanıcı, okumaların eşleneceği tercih edilen genom yapısını belirtmelidir, örneğin, hg19/hg38 (insan), mm10/mm39 (fare).
Filtreleme: Düzgün eşlenmiş okumalar korunur ve düşük kaliteli okumalar filtrelenir. Varsayılan: samtools görünümü, parametrelerle: -bShuF 4 -f 3 -q 30.
Sırala: hizalanmış okumalar en soldaki koordinat sırasına göre sıralanır. Varsayılan: samtools sıralama (snakemake wrapper), parametre ile: -m 4G.
Yinelenenleri işaretle: tüm yinelenen okumalar tanımlanır ve işaretlenir. Kullanıcı, yapılandırma dosyası parametresini değiştirerek bunları kaldırmaya karar verebilir. Varsayılan: Picard MarkDuplicates (yılan sarmalayıcı), parametresiyle: --REMOVE_DUPLICATES False kopyaları işaretlemek ve korumak için.
Bam'ı birleştirme: Sıralama verileri çoğaltmalardan veya örneklerden oluşuyorsa, kullanıcı tek bir bam'da birleştirmek isteyebilir. Bu durumda, kullanıcı bamları birleştirmeyi veya bam dosyalarını analiz boyunca ayrı tutmayı seçebilir. Kullanıcı bams'ı birleştirmeyi seçerse (samtools merge kullanarak), birleştirilen bams için ortak bir önek belirtilmelidir.
İndeks: Bu adım, sıralanan koordinatları indeksler. Varsayılan: samtools tarafından belirtilen varsayılan parametreleri kullanan samtools dizini (snakemake wrapper).
BamCoverage: Bu kural, hizalanmış okumalardan büyük bir kapsama alanı parçası oluşturur. deepTools'tan bamCoverage aracı uygulanır ve kapsam, bölmenin belirli bir boyuttaki bir pencereyi temsil ettiği bölme başına okuma sayısı olarak hesaplanır. Bu işlem hattında, bamCoverage varsayılan olarak ayarlanan aşağıdaki parametrelerle uygulanır: -bs 1 -normalizeUsing RPKM -extendReads.
Tepe çağrısı: LanceOtron8 , bu işlem hattı için varsayılan tepe arayan olarak seçildi. Çoğunlukla istatistiksel olarak test tabanlı olan geleneksel tepe arayanların aksine, LanceOtron, genomik zenginleştirme ölçümlerini ve istatistiksel testleri içeren ve endüstri standardı tepe arayan MACS29'dan daha iyi performans gösterdiği gösterilen derin öğrenme tabanlı bir tepe arayandır. Büyük perukların LanceOtron ile uyumlu olması için, kapsama alanı baz çifti başına hesaplanmalı ve RPKM normalleştirilmelidir; bu, BamCoverage adımının varsayılan ayarlarına yansıtılır. MACS2, alternatif bir tepe arayan olarak seçilebilir. Bu analiz hattının performansını korumak ve optimize etmek için yeni tepe arayanların serbest bırakılması izlenecek ve uygun olduğu şekilde dahil edilecektir.
TrackDb: Bu, büyük peruk dosyalarını MLV 6 veya UCSCGenome Browser18 platformları gibi araçlarda yüklemek ve görselleştirmek için bir anahtar-değer çifti ilişkilendirmesi oluşturur.
Çıktı verilerine ek olarak, boru hattının her adımı bir günlük dosyası çıkarır ve kullanıcının analiz ilerlemesini takip edebilmesi için uygun kalite kontrol kontrolleri sağlanır. FastQC19, ham ve kırpılmış (seçilirse) sıralama verilerine uygulanır (adım 1 - Hızlı hareket et ve 2- Kırpma). Samtools istatistikleri ve MultiQC20, adım 3 - Hizalayıcı, 6 - Kopyaları işaretle ve 7 - Bam'ı birleştir adımlarında çıktıdaki bam dosyalarında kalite kontrol raporlarını toplamak, üretmek ve görselleştirmek için kullanılır. Yukarıdaki adımlarda uygulanan araçların her biri hakkında daha fazla bilgi için Tablo 1'e bakın.
Protokol
1. CATCH-UP boru hattının çalıştırılması
- UpStreamPipeline deposunu https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline'dan kopyalayın:
Seçilen çalışma dizinine gidin, aşağıdaki kodu kopyalayın ve komut satırında çalıştırın:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Şu komut kullanılarak indirilen UpStreamPipeline klasörünün içinde gezinin: cd UpStreamPipeline
- Anakonda dağıtımını kurun (gerekirse):
- Komutu kullanarak anaconda'nın sistemde zaten kurulu olup olmadığını kontrol edin conda. Komut herhangi bir conda dağıtımına giden herhangi bir yol göstermiyorsa, https://github.com/conda-forge/miniforge#mambaforge'dan mambaforge'u indirin ve sistem için doğru dağıtımı ve sürümü seçin. Örneğin, linux kullanıcıları için wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh kullanın. Farklı İşletim Sistemleri için bu web sayfasını ziyaret edin: https://github.com/conda-forge/miniforge/.
- sh kullanarak yükleyiciyi çalıştırın Mambaforge-Linux-x86_64.sh ve conda init komutunu çalıştırarak sistemde conda'yı başlatın.
- Yukarı akış conda ortamını kurun ve etkinleştirin (yukarı akış conda ortamının gereksinimleri Tablo 2'de listelenmiştir):
- mamba env create - file=envs/upstream.yml komutunu kullanarak ortamı yükleyin.
- conda activate upstream komutunu kullanarak ortamı etkinleştirin.
- Yukarı akış conda ortamı başarıyla yüklendikten sonra, conda activate upstream komutunu kullanarak ortamı etkinleştirin ve cd genetics/CATCH-UP kullanarak CATCH-UP klasörüne gidin.
- cd /config/analysis.yaml komutunu kullanarak config klasöründe bulunabilen yapılandırma dosyasını düzenleyin ve bir metin düzenleyicisi kullanarak analiz belirtimine uygun şekilde değiştirin. Dosyanın içindeki her parametreyi düzenlemek için satır satır yönergeleri izleyin. Bu dosya, analizden sonra saklanacak ve tekrarlanabilirliğe yardımcı olmak için çalışma parametrelerini belgelemek için hareket edecektir.
- Aşağıdaki üç dosyayı bir metin düzenleyicide açın ve düzenleyin (örneğin, Mac için TextEdit veya Windows için Not Defteri):
- Analiz edilecek tüm fastq dosyalarının bir listesini içerecek şekilde 1_fastqfile_home_dir.txt dosyasını düzenleyin.
NOT: Okuma numaraları ve uzantıları (örn. _R1/_R2 ve .fastq.gz) hariç tutulmalıdır. Örneğin, bir proje bu fastq dosyaları listesini içeriyorsa:
Sample1_conditionA_L001_R1 . hızlı . GZ (İngilizce)
Sample1_conditionA_L001_R2 . hızlı . GZ (İngilizce)
Sample1_conditionA_L002_R1 . hızlı . GZ (İngilizce)
Sample1_conditionA_L002_R2 . hızlı . GZ (İngilizce)
Sample1_conditionB_L001_R1 . hızlı . GZ (İngilizce)
Sample1_conditionB_L001_R2 . hızlı . GZ (İngilizce)
Sample1_conditionB_L002_R1 . hızlı . GZ (İngilizce)
Sample1_conditionB_L002_R2 . hızlı . GZ (İngilizce)
Sample2_conditionA_L001_R1 . hızlı . GZ (İngilizce)
Sample2_conditionA_L001_R2 . hızlı . GZ (İngilizce)
Sample2_conditionA_L002_R1 . hızlı . GZ (İngilizce)
Sample2_conditionA_L002_R2 . hızlı . GZ (İngilizce)
Sample2_conditionB_L001_R1 . hızlı . GZ (İngilizce)
Sample2_conditionB_L001_R2 . hızlı . GZ (İngilizce)
Sample2_conditionB_L002_R1 . hızlı . GZ (İngilizce)
Sample2_conditionB_L002_R2 . hızlı . GZ (İngilizce)
Bu durumda, 1_fastqfile_home_dir.txt aşağıdaki gibidir:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Ham veriler, birleştirme gerektiren sıralama şeritleri içeriyorsa, birleştirilecek dosya adlarının önekini tanımlamak için 2_fastq file_concat.txt dosyasını düzenleyin. Birleştirilecek sıralama şeritleri yoksa, fi le_concat.txt 2_fastq düzenlemeyin. Her 2_fastqfile_concat.txt satırının aşağıdaki gibi bir örnek öneki içerdiğinden emin olun:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Farklı örneklerin verilerinin birleştirilmesi gerekiyorsa, 3_merge_bams.txt dosyayı birleştirilecek dosya adlarının önekiyle düzenleyin. Her satırın aşağıdaki gibi bir örnek öneki içerdiğinden emin olun:
Örnek1
Örnek2
Şekil 2'de bu üç dosyanın nasıl özetleneceğinin bir özeti gösterilmektedir. Protokol, tek veya çift uçlu sıralama verilerine uygulanabilir. İşlem hattı, aksi belirtilmedikçe varsayılan olarak eşleştirilmiş uç analizine geçer; Bu, yapılandırma dosyasında değiştirilebilir (bkz. adım 1.6).
- Analiz edilecek tüm fastq dosyalarının bir listesini içerecek şekilde 1_fastqfile_home_dir.txt dosyasını düzenleyin.
- Gerekli tüm dosyalar düzenlendikten sonra, CATCH-UP'ı aşağıdaki gibi çalıştırmak için snakemake'i kullanın: snakemake --configfile=config/analysis_name.yaml all --cores 4.
NOT: Daha ayrıntılı yönergeler ve belgeler için, burada bulunan UpStreamPipeline GitHub deposundaki CATCH-UP klasörüne bakın. Bu, veri dosyalarını sıralamak ve sonuçları depolamak için yolları değiştirmekten her adım için parametreleri düzenlemeye kadar yapılandırma dosyasını doğru şekilde değiştirmeye ilişkin ayrıntılı belgeleri içerir.
Sonuçlar
CATCH-UP işlem hattı, her adım için bir sonuç, günlük ve kalite kontrol (QC) çıktısı üretir. Yapılandırma dosyası içinde kullanıcı, gereken depolama belleğini azaltmak için çıktı dosyalarını tutmayı veya kaldırmayı seçebilir. Çıktıların tamamı şu şekilde açıklanmıştır:
00. fastq_home_dir: yapılandırma dosyası, fastqfile_home_dir.txt ve merge_bams.txt , referans ve tekrarlanabilirlik için bu klasöre kopyalanır.
01. Okumalar: FastQ dosyaları, iş akışı işlemi sırasında orijinal ham verilerin değiştirilmesini önlemek için bu klasöre kopyalanır, belirtilirse şeritler birleştirilebilir.
02. Kırpma: Belirtilirse kırpılmış okuma ve adaptörlere sahip FastQ dosyaları.
03. Hizalayıcı: Seçilen genoma karşı hizalama.
04. Dosyalama: Kalite kontrol filtrelemesi.
05. Sıralanmış: BAM dosyalarının sıralanması.
06. Kopyalar: Kopyaları işaretleme.
07. Birleştirme: config.yaml'de belirtilmişse BAM dosyalarını birleştirme.
08. bam_coverages: Haberin büyük peruk dosyası.
09. peak_calling: LanceOtron tepe çağrı çıktısının bir yatak dosyası.
10. parça: Gerekirse Genome Browser'da kullanılmaya hazır biçimlendirilmiş bir metin dosyası üretir.
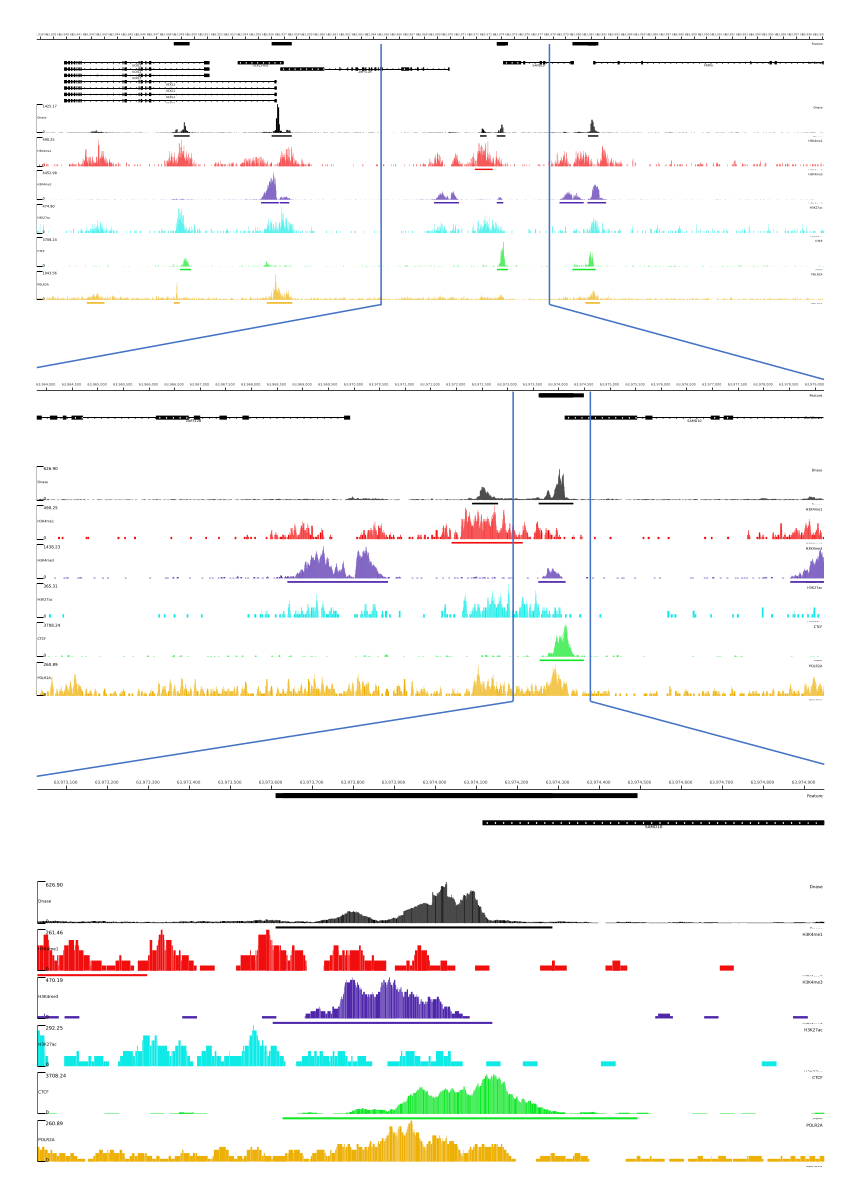
01, 02, 03, 06 ve 07 çıktıları için QC metrikleri ve HTML dosyaları sağlanır. Ek olarak, Şekil 3'te, MLV platformu aracılığıyla nihai çıktıyı görselleştiren CATCH-UP kullanarak işlenmiş verilerin bir örneğini sunuyoruz.

Şekil 1: CATCH-UP iş akışı. Fastq dosyalarının bir listesi verildiğinde, CATCH-UP tüm örnekleri tüm yukarı akış adımlarında paralel olarak işler. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}

Şekil 2: CATCH-UP'ı çalıştırmak için 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt ve 3_merge_bams.txt nasıl doğru şekilde değiştirilmesi gerektiğiniaçıklayan açıklayıcı gösterim. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}

Şekil 3: CATCH-UP işlem hattından örnek çıktı. Ham sıralama verileri (fastq dosyaları) ENCODE21'den indirildi. DNase-seq ve 5 tür ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF ve POLR2A) için fastq dosyalarını işlemek için CATCH-UP işlem hattı kullanıldı. Bigwig çıktı dosyaları, genomik düzenleyici unsurların görselleştirilmesi ve tanımlanması için Multi Locus View'a yüklendi. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
{kind=link}
Tablo 1: Belge kaynakları. Bu tablo, CATCH-UP iş akışında yer alan araçları, belgelerinin bağlantısını ve ilgili referansları gösterir. Bu Tabloyu indirmek için lütfen buraya tıklayın.
Tablo 2: Yukarı akış conda ortamı için kanal ve bağımlılık gereksinimlerinin listesi. Bu Tabloyu indirmek için lütfen buraya tıklayın.
Tablo 3: CATCH-UP'ı test etmek için kullanılan İşletim Sistemleri. Ubuntu, yüksek performanslı bir küme ve yerel bir makine üzerinde test edilmiştir. Bu Tabloyu indirmek için lütfen buraya tıklayın.
Tartışmalar
Genomik veri üretmek için NGS tekniklerinin artan alımı ve kullanımı, bu verilerin analizi için biyoinformatik araçların geliştirilmesindeki artışla eşleştirilmiştir. Veri analizinin her adımı için uygulanabilecek birden fazla aracın yanı sıra her bir araçta belirtilebilecek birçok farklı parametrevardır 6,8,9,15,16,17,18,19,20,22,23,24 . Bu, uygulanabilecek ve her biri sonuçta farklılıklar üretebilecek çok çeşitli analiz stratejilerinin bir kombinasyonunu oluşturur. Deneyler arasında doğru bir şekilde karşılaştırma yapmak için biyoinformatik analizin standardizasyonu esastır. Tarihsel olarak, NGS verileri ıslak laboratuvar bilim adamları tarafından üretilir ve veriler biyoinformatikçiler tarafından analiz edilir.
NGS veri analizi, "yukarı akış" ve "aşağı akış" boru hatlarına ayrılabilir, burada yukarı akış, bir sıralama makinesinden gelen ham veri çıktısından bir araştırmacı tarafından görsel olarak yorumlanabilir bir formata geçmek için gerekli adımları içerir. Aşağı akış analizi, araştırma sorusuna ve deneysel tasarıma özel ek adımlar içerir. Bu nedenle, yukarı akış boru hatları genelleştirilebilir ve gelişmiş bilimsel tekrarlanabilirlik için standardizasyona uygundur. Öte yandan, aşağı akış boru hatları ısmarlamadır, biyolojik soruya bağlıdır ve araştırmacının içgörüsünü gerektirir, bu da onları standardizasyon için daha az uygun hale getirir. Islak laboratuvar bilim adamlarının herhangi bir ön biyoinformatik bilgisine ihtiyaç duymadan kendi verilerini tekrarlanabilir bir şekilde analiz etmelerine olanak tanıyan kullanıcı dostu bir yukarı akış boru hattı oluşturduk. Burada, snakemake çerçevesi kullanılarak oluşturulmuş ve hem kullanıcı dostu olacak hem de ChIP-seq ve ATAC-seq veri analizinde tekrarlanabilirlik sorunuyla mücadele etmek için tasarlanmış bir boru hattı olan CATCH-UP'ı sunuyoruz. Bu işlem hattı, ChIP-seq veya ATAC-seq verilerini işlemek için oluşturulmuştur. Kullanıcı CATCH-UP'ı indirdikten sonra, işlem hattını tek bir kod satırı kullanarak komut satırında çalıştırmadan önce analiz parametreleri ve örnek adlandırma tanımlanmalıdır. ChIP-seq veya ATAC-seq analizi için analiz parametrelerinin nasıl özelleştirileceğine ilişkin basit adım adım talimatlar, yapılandırma dosyasının kendisinde ve CATCH-UP GitHub deposundaki adım adım kılavuzumuzda sağlanır.
ChIP-seq veya ATAC-seq verileri için PEPATAC ve ChIP-AP gibi mevcut analiz işlem hatları vardır. Bu boru hatları, hem yukarı hem de aşağı analizlerin tek bir iş akışına dahil edilmesi veya bir grafik kullanıcı arayüzünün (GUI) kullanılması gibi avantajlara sahip olsa da, bu araçlar orta düzeyde hesaplama eğitimine sahip biyoinformatikçileri ve bilim adamlarını hedef almaktadır13,14. CATCH-UP iki sorunu çözmek için tasarlanmıştır: biyoinformatik eğitimi olmayan ıslak laboratuvar bilim adamlarının kendi yukarı akış analizlerini gerçekleştirmelerini sağlamak ve laboratuvarlar arasında kolay raporlamayı ve tam tekrarlanabilirliği kolaylaştırarak yukarı akış analizinin standardizasyonunu sağlamak. CATCH-UP kasıtlı olarak yukarı akış analiziyle sınırlıdır, ancak çıktılar, veri kümelerini istatistiksel olarak karşılaştırmak veya transkripsiyon faktörü bağlamasınıçıkarmak için kullanılanlar gibi aşağı akış analiz araçlarıyla uyumludur 25,26.
Tekrarlanabilir bir yukarı akış analizi gerçekleştirmek için gerekli tüm kritik adımlar, sağlamlığı sağlamak için CATCH-UP boru hattı içinde önceden tanımlanmıştır. Bu işlem hattının ayrıntılı yapısı, kullanıcının işlem hattının çıkışını adım adım izlemesine olanak tanır ve bu da hem sorun giderme hem de analitik iş akışının çoğaltılmasını sağlamak için yararlıdır. NGS tekniklerinin hızla gelişen doğası göz önüne alındığında, bu boru hattının modüler yapısı, hem araç sürümü güncellemelerinin yayınlanmasını hem de yeni araçların uygulanmasını içerecek şekilde kolayca uyarlanabilme yeteneği sağladığı için faydalıdır. CATCH-UP aşağıdaki işletim sistemleri için başarıyla test edilmiştir: Ubuntu, CentOS, macOS (Intel CPU) ve Windows (Tablo 3). Boru hattı, iş akışını paralelleştirerek onlarca örnek içeren büyük deneyleri işlemek için inşa edilmiştir ve bu da onu farklı deneysel tasarımlara uyarlanabilir hale getirir. Genel olarak, ChIP-seq ve ATAC-seq verilerinin analizinde CATCH-UP'ın uygulanması, kullanıcı dostu, tekrarlanabilir ve son derece uyarlanabilir bir analiz iş akışı sağlar.
Açıklamalar
J.R.H., Nucleome Therapeutics'in kurucu ortağı ve direktörüdür ve şirkete danışmanlık yapmaktadır.
Teşekkürler
J.R.H., Wellcome Trust (225220/Z/22/Z ve 106130/Z/14/Z) ve MRC (MC_UU_00029/3) tarafından sağlanan hibelerle desteklenmiştir. M.B., Wellcome Trust hibesi (225220/Z/22/Z) tarafından desteklenmiştir. E.R.G, Millî Eğitim Bakanlığı Lisansüstü Eğitim Amacıyla Yurt Dışına Gönderilen Adayların Seçme ve Yerleştirme (YLSY) bursu ve T.C. Millî Eğitim Bakanlığı tarafından desteklenmiştir. E.G., Wellcome Genomik Tıp ve İstatistik Doktora Programı (108861/Z/15/Z) tarafından desteklenmiştir. S.G.R., Tıbbi Araştırma Konseyi (MRC) hibesi (MC_UU_00029/3) tarafından desteklenmiştir.
Malzemeler
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Referanslar
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiThis article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır