
Method Article
RATTRAPAGE : un pipeline en amont à haut débit pour les données ATAC-Seq et ChIP-Seq en masse
Dans cet article
Résumé
L’ATAC-seq et le ChIP-seq permettent d’étudier en détail la régulation des gènes ; Cependant, le traitement de ces types de données est difficile et souvent incohérent entre les groupes de recherche. Nous présentons CATCH-UP : un pipeline de calcul facile à utiliser qui permet le traitement et l’analyse de données standardisées et reproductibles d’ensembles de données ATAC/ChIP-seq nouveaux et publiés.
Résumé
Le dosage de la chromatine accessible à la transposase (ATAC) et de l’immunoprécipitation de la chromatine (ChIP), associé au séquençage de nouvelle génération (NGS), a révolutionné l’étude de la régulation des gènes. Un manque de standardisation dans l’analyse des ensembles de données hautement dimensionnels générés par ces techniques a rendu la reproductibilité difficile à atteindre, ce qui a entraîné des divergences dans les données publiées et traitées. Une partie de ce problème est due à la diversité des outils bioinformatiques disponibles pour l’analyse de ces types de données. Deuxièmement, un certain nombre d’outils bioinformatiques différents sont nécessaires de manière séquentielle pour convertir les données brutes en une sortie entièrement traitée et interprétable, et ces outils nécessitent différents niveaux de compétences informatiques. En outre, il existe de nombreuses options de contrôle de la qualité qui ne sont pas utilisées de manière uniforme lors du traitement des données. Nous abordons ces problèmes avec un test complet pour le séquençage de la chromatine accessible par transposase (ATAC-seq) et le pipeline amont de séquençage par immunoprécipitation de la chromatine (ChIP-seq) (CATCH-UP), un pipeline facile à utiliser, basé sur Python, pour l’analyse d’ensembles de données ChIP-seq et ATAC-seq en vrac, des fichiers fastq bruts aux traces et appels de pics bigwig visualisables. Ce pipeline est simple à installer et à exécuter, et nécessite des connaissances informatiques minimales. Le pipeline est modulaire, évolutif et parallélisable sur diverses infrastructures informatiques, ce qui permet de rendre facilement compte de la méthodologie pour permettre une analyse reproductible d’ensembles de données nouveaux ou publiés.
Introduction
L’expression des gènes doit être étroitement régulée pour que les cellules puissent établir et maintenir leur fonction biologique correcte. Il est bien connu que l’expression aberrante des gènes est à l’origine de la pathogenèse de nombreuses maladies et, par conséquent, une grande partie de l’intérêt de la recherche réside dans la compréhension des mécanismes de régulation des gènes1. L’expression des gènes est facilitée par des éléments régulateurs tels que les promoteurs et les amplificateurs. Au sein de leur séquence, ces éléments contiennent des sites de liaison de facteur de transcription (TF) qui, lorsqu’ils sont actifs, fournissent une plate-forme pour la liaison TF. La liaison des TF à ces sites entraîne un déplacement des nucléosomes, ce qui entraîne une augmentation de l’accessibilité de l’ADN et une augmentation ultérieure de la licéité de la machinerie transcriptionnelle. En raison de cette accessibilité accrue, ces régions de l’ADN sont plus sensibles aux nucléases et aux transposases telles que la DNase et la Tn5, une propriété biochimique qui a été exploitée par les chercheurs qui étudient la régulation transcriptionnelle 2,3.
DNase-seq et ATAC-seq permettent aux chercheurs de cartographier les régions de chromatine ouverte, les sites de liaison TF et le positionnement nucléosomique à travers le génome. De ces deux techniques, ATAC-seq a gagné en popularité au cours de la dernière décennie en raison du protocole simple en deux étapes et d’un faible nombre de cellules requis (50 000 cellules contre 1 million par réplique pour DNase-seq). Alors que l’ATAC-seq fournit une vue d’ensemble du paysage général de la chromatine dans une population de cellules, il est largement agnostique quant aux protéines spécifiques qui se lient au génome 4,5. Afin d’identifier les endroits où une protéine spécifique interagit avec le génome, la technique de référence est l’immunoprécipitation de la chromatine (ChIP)-seq. Le ChIP-seq consiste à fixer chimiquement les interactions protéine-ADN dans une cellule, suivies d’une immunoprécipitation (« pull-down ») à l’aide d’un anticorps spécifique de la protéine d’intérêt pour sélectionner les fragments d’ADN liés par la protéine d’intérêt (POI). Ces fragments d’ADN peuvent être séquencés pour révéler les emplacements de liaison génomique de protéines spécifiques telles que les TF, ou des sites contenant des modifications d’histones spécifiques1. En combinant les ensembles de données ATAC-seq et ChIP-seq, il est possible d’obtenir une image détaillée du paysage réglementaire pour une population de cellules.
Le flux de travail de base requis pour l’analyse est le suivant : la qualité des lectures de séquençage brutes doit être contrôlée avant d’être alignée sur un génome de référence (« cartographie »). Les lectures mappées avec succès peuvent ensuite être filtrées pour supprimer à la fois les lectures de mauvaise qualité et les doublons PCR. Afin de visualiser ces lectures cartographiées et filtrées, il est nécessaire de calculer la « couverture » de ces lectures sur l’ensemble du génome. Cela génère un fichier qui peut être téléchargé sur un navigateur de génome tel que la vue multi-locus (MLV) ou le navigateur de génome UCSC en tant que « piste »6,7. L’identification des pics, ou « appel de pointe », de ces pistes de couverture est généralement réalisée à l’aide d’outils tels que LanceOtron ou MACS2 8,9. Enfin, grâce à l’analyse de l’emplacement, de la forme et de la taille des pics, des comparaisons peuvent être faites entre les échantillons ou les conditions biologiques. L’analyse et l’intégration de ces ensembles de données est un processus complexe en plusieurs étapes dans lequel différentes combinaisons d’outils bioinformatiques peuvent être mises en œuvre. Les différentes versions des outils peuvent être incompatibles les unes avec les autres et peuvent modifier le résultat du traitement des données. Il existe également une grande variété de puissance de calcul et de compétences de l’utilisateur requises pour mettre en œuvre différentes parties du traitement des données, comme le montrent les pipelines nf-core10, panpipes11, genpipes12, PEPATAC13 ou ChIP-AP14.
Dans l’ensemble, cela a entraîné des incohérences dans l’analyse et la présentation de l’analyse, ce qui a entraîné une reproductibilité, une accessibilité et une commodité médiocres pour toute personne ayant une connaissance limitée de la bio-informatique. Nous répondons à tous ces problèmes avec CATCH-UP (pipeline amont complet ATAC-seq et ChIP-seq), un pipeline facile à utiliser, flexible et modulaire pour le traitement des données ChIP-seq et ATAC/DNase-seq. La mise en œuvre de CATCH-UP nécessite une expérience minimale en bioinformatique ; Il peut être exécuté sur diverses infrastructures informatiques et permet une analyse reproductible des données au sein et entre les groupes de recherche.
CATCH-UP est un pipeline Snakemake basé sur Python conçu pour standardiser l’analyse des données ChIP-seq et ATAC-seq. Il prend des données de séquençage brutes (fichiers fastq.gz) en entrée et génère une sortie sous la forme de fichiers de pic (.bed) fournissant le résultat respectif pour chaque étape. Nous fournissons un fichier de configuration au format yaml (config.yaml), dans lequel l’utilisateur peut modifier les paramètres de chaque étape d’analyse. Le système de gestion mis en œuvre au sein de snakemake permet d’utiliser différentes infrastructures de calcul (telles que des serveurs, des clusters, des systèmes cloud ou des ordinateurs personnels) et en parallèle si l’utilisateur fournit une grande quantité de données.
Vous trouverez ci-dessous une description détaillée de chaque étape du flux de travail (voir la figure 1 pour l’illustration du flux de travail). Cette explication est essentielle afin de suivre le pas à pas dans la section protocole :
Déplacer fastq : la première étape du pipeline consiste à copier les fichiers fastq bruts dans le répertoire d’analyse nommé. Cela laisse les données d’origine intactes pour éviter d’endommager ou de modifier les fichiers de données brutes.
Concaténation : si les données de séquençage brutes contiennent plusieurs voies, cette étape est nécessaire pour concaténer les voies avant l’analyse. Par défaut, le pipeline gère tous les fichiers fastq comme des échantillons uniques. Cette étape de concaténation doit être définie dans le fichier de configuration.
Découpage : étape facultative de nettoyage des données. Cela permet de réduire les lectures de mauvaise qualité ou les séquences d’adaptateur à l’aide de trimmomatic15. L’utilisateur peut fournir des fichiers fasta personnalisés de séquences d’adaptateurs ; Un exemple est fourni dans le répertoire de l’adaptateur. Des paramètres de découpage supplémentaires peuvent être définis dans le fichier de configuration. Par défaut, le flux de travail ignore cette règle.
Aligneur : pour l’alignement, Bowtie216 est appliqué par défaut ; D’autres outils d’alignement tels que BWA-MEM217 peuvent également être spécifiés. L’outil d’alignement Bowtie2 est sélectionné par défaut car il est particulièrement apte à aligner des lectures relativement courtes sur des génomes relativement grands et est donc bien adapté à l’alignement des données ChIP-seq et ATAC-seq sur les génomes de mammifères. Pour éviter tout fichier intermédiaire, l’aligneur est redirigé vers la vue samtools pour enregistrer le fichier bam en sortie. Pour cette règle, l’utilisateur doit spécifier la construction du génome préférée sur laquelle cartographier les lectures, par exemple, hg19/hg38 (humain), mm10/mm39 (souris).
Filtrage : les lectures correctement mappées sont conservées, et les lectures de faible qualité sont filtrées. Par défaut : vue samtools, avec les paramètres : -bShuF 4 -f 3 -q 30.
Trier : les lectures alignées sont triées dans l’ordre de la coordonnée la plus à gauche. Par défaut : samtools sort (wrapper snakemake), avec le paramètre : -m 4G.
Marquer les doublons : toutes les lectures en double sont identifiées et signalées. L’utilisateur peut décider de les supprimer en modifiant le paramètre du fichier de configuration. Par défaut : Picard MarkDuplicates (wrapper snakemake), avec le paramètre : --REMOVE_DUPLICATES False pour marquer et conserver les doublons.
Fusionner bam : si les données de séquençage sont composées de répétitions ou d’échantillons, l’utilisateur peut souhaiter fusionner en un seul bam. Dans ce cas, l’utilisateur peut choisir de fusionner les bams ou de garder les fichiers bam séparés tout au long de l’analyse. Si l’utilisateur choisit de fusionner les bams (en utilisant samtools merge), un préfixe commun doit être spécifié pour les bams fusionnés.
Index : cette étape indexe les coordonnées triées. Par défaut : index samtools (wrapper snakemake), en utilisant les paramètres par défaut spécifiés par samtools.
BamCoverage : cette règle crée une piste de couverture bigwig à partir de lectures alignées. L’outil bamCoverage de deepTools est appliqué et la couverture est calculée comme le nombre de lectures par bac, dans lequel le bac représente une fenêtre d’une taille spécifiée. Dans ce pipeline, bamCoverage est appliqué avec les paramètres suivants définis par défaut : -bs 1 -normalizeUsing RPKM -extendReads.
Appel de pointe : LanceOtron8 a été sélectionné comme appel de pointe par défaut pour ce pipeline. Contrairement aux appelants de pointe traditionnels, qui sont principalement basés sur des tests statistiques, LanceOtron est un appelant de pic basé sur l’apprentissage profond, qui intègre des mesures d’enrichissement génomique et des tests statistiques et s’est avéré plus performant que l’appelant de pic standard de l’industrie, MACS29. Pour que les gros bonnets soient compatibles avec LanceOtron, la couverture doit être calculée par paire de bases, et RPKM normalisé ; cela se reflète dans les paramètres par défaut de l’étape BamCoverage. MACS2 peut être sélectionné comme appelant de pic alternatif. La libération de nouveaux appelants de pointe sera surveillée et intégrée, le cas échéant, afin de maintenir et d’optimiser le rendement de ce pipeline d’analyse.
TrackDb : cela crée une association de paires clé-valeur de fichiers bigwig afin de les charger et de les visualiser dans des outils tels que les plateformes MLV6 ou UCSC Genome Browser18 .
En plus des données de sortie, chaque étape du pipeline génère un fichier journal et des contrôles de qualité appropriés sont fournis afin que l’utilisateur puisse suivre la progression de l’analyse. FastQC19 s’applique aux données de séquençage brutes et rognées (si sélectionnées) (étapes 1 - Déplacer fastq et 2- Découpage). Samtools stats plus MultiQC20 sont utilisés pour collecter, produire et visualiser des rapports de contrôle qualité sur les fichiers bam en sortie dans les étapes 3 - Aligneur, 6 - Marquer les doublons et 7 - Fusionner bam. Pour plus d’informations sur chacun des outils utilisés dans les étapes ci-dessus, consultez le tableau 1.
Protocole
1. Exécution du pipeline CATCH-UP
- Clonez le référentiel UpStreamPipeline à partir de https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline :
Accédez au répertoire de travail choisi, copiez le code suivant et exécutez-le sur la ligne de commande :
git clone git@github.com :Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Naviguez dans le dossier UpStreamPipeline téléchargé à l’aide de la commande : cd UpStreamPipeline
- Installez la distribution anaconda (si nécessaire) :
- Vérifiez si anaconda est déjà installé sur le système à l’aide de la commande which conda. Si la commande n’affiche aucun chemin d’accès à une distribution conda, téléchargez mambaforge à partir de https://github.com/conda-forge/miniforge#mambaforge et sélectionnez la distribution et la version appropriées pour le système. Par exemple, pour les utilisateurs de Linux, utilisez wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Visitez cette page Web pour découvrir les différents systèmes d’exploitation : https://github.com/conda-forge/miniforge/.
- Exécutez le programme d’installation à l’aide de sh Mambaforge-Linux-x86_64.sh et initialisez conda dans le système en exécutant conda init.
- Installez et activez l’environnement conda en amont (les exigences de l’environnement conda en amont sont répertoriées dans le tableau 2) :
- Installez l’environnement à l’aide de la commande mamba env create - file=envs/upstream.yml.
- Activez l’environnement à l’aide de la commande conda activate en amont.
- Une fois l’environnement conda en amont installé, activez-le à l’aide de la commande conda activate upstream et accédez au dossier CATCH-UP à l’aide de cd genetics/CATCH-UP.
- Modifiez le fichier de configuration, qui se trouve dans le dossier config à l’aide de la commande cd /config/analysis.yaml, et modifiez-le en conséquence avec la spécification d’analyse à l’aide d’un éditeur de texte. Suivez les instructions ligne par ligne pour modifier chaque paramètre dans le fichier lui-même. Ce fichier sera conservé après l’analyse et servira à documenter les paramètres de passage pour faciliter la reproductibilité.
- Ouvrez et modifiez les trois fichiers suivants dans un éditeur de texte (par exemple, TextEdit pour Mac ou Bloc-notes pour Windows) :
- Modifiez 1_fastqfile_home_dir.txt fichier pour qu’il contienne une liste de tous les fichiers fastq à analyser.
REMARQUE : Les numéros de lecture et les extensions (par exemple, _R1/_R2 et .fastq.gz) doivent être exclus. Par exemple, si un projet contient cette liste de fichiers fastq :
Sample1_conditionA_L001_R1 . fastq . GZ
Sample1_conditionA_L001_R2 . fastq . GZ
Sample1_conditionA_L002_R1 . fastq . GZ
Sample1_conditionA_L002_R2 . fastq . GZ
Sample1_conditionB_L001_R1 . fastq . GZ
Sample1_conditionB_L001_R2 . fastq . GZ
Sample1_conditionB_L002_R1 . fastq . GZ
Sample1_conditionB_L002_R2 . fastq . GZ
Sample2_conditionA_L001_R1 . fastq . GZ
Sample2_conditionA_L001_R2 . fastq . GZ
Sample2_conditionA_L002_R1 . fastq . GZ
Sample2_conditionA_L002_R2 . fastq . GZ
Sample2_conditionB_L001_R1 . fastq . GZ
Sample2_conditionB_L001_R2 . fastq . GZ
Sample2_conditionB_L002_R1 . fastq . GZ
Sample2_conditionB_L002_R2 . fastq . GZ
Dans ce cas, la 1_fastqfile_home_dir.txt est la suivante :
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Si les données brutes contiennent des voies de séquençage qui nécessitent une concaténation, éditez 2_fastqfichier fi le_concat.txt pour définir le préfixe des noms de fichiers à concaténér. S’il n’y a pas de voies de séquençage à concaténer, ne modifiez pas 2_fastqfi le_concat.txt. Assurez-vous que chaque ligne de 2_fastqfile_concat.txt contient un exemple de préfixe comme suit :
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Si la fusion des données de différents échantillons est nécessaire, éditez 3_merge_bams.txt fichier avec le préfixe des noms de fichiers à fusionner. Assurez-vous que chaque ligne contient un exemple de préfixe comme suit :
Échantillon1
Échantillon2
La figure 2 présente un résumé de la façon de résumer ces trois fichiers. Le protocole peut être appliqué à des données de séquençage à une extrémité ou à une extrémité. Sauf indication contraire, le pipeline utilise par défaut l’analyse par paires d’extrémités. Cela peut être modifié dans le fichier de configuration (voir étape 1.6).
- Modifiez 1_fastqfile_home_dir.txt fichier pour qu’il contienne une liste de tous les fichiers fastq à analyser.
- Une fois que tous les fichiers requis ont été modifiés, utilisez snakemake pour exécuter CATCH-UP comme suit : snakemake --configfile=config/analysis_name.yaml all --cores 4.
REMARQUE : Pour obtenir des instructions et de la documentation plus détaillées, consultez le dossier CATCH-UP dans le référentiel GitHub UpStreamPipeline, disponible ici. Cela inclut une documentation détaillée sur la modification correcte du fichier de configuration, de la modification des chemins d’accès pour le séquençage des fichiers de données et le stockage des résultats à l’édition des paramètres pour chaque étape.
Résultats
Le pipeline CATCH-UP produit un résultat, un journal et une sortie de contrôle qualité (CQ) pour chaque étape. Dans le fichier de configuration, l’utilisateur peut choisir de conserver ou de supprimer les fichiers de sortie pour réduire la mémoire de stockage requise. Tous les résultats sont expliqués comme suit :
00. fastq_home_dir : le fichier de configuration, fastqfile_home_dir.txt et merge_bams.txt sont copiés dans ce dossier à des fins de référence et de reproductibilité.
01. Lectures : Les fichiers fastQ sont copiés dans ce dossier pour éviter les altérations des données brutes d’origine pendant le processus de flux de travail, les voies peuvent être concaténées si spécifiées.
02. Découpage : fichiers FastQ avec lecture et adaptateurs coupés si spécifié.
03. aligneur : alignement contre le génome sélectionné.
04. Filtering : filtrage de contrôle de la qualité.
05. Sorted : Tri des fichiers BAM.
06. Doublons : Marquage des doublons.
07. merge : fusion des fichiers BAM si cela a été spécifié dans config.yaml.
08. bam_coverages : Fichier Bigwig de la couverture.
09. peak_calling : un fichier de lit de la sortie d’appel de crête de LanceOtron.
10. track : produit un fichier texte formaté prêt à être utilisé sur Genome Browser si nécessaire.
Pour les sorties 01, 02, 03, 06 et 07, des mesures de contrôle qualité et des fichiers HTML sont fournis. De plus, dans la figure 3, nous fournissons un exemple de données traitées à l’aide de CATCH-UP, en visualisant le résultat final via la plate-forme MLV.

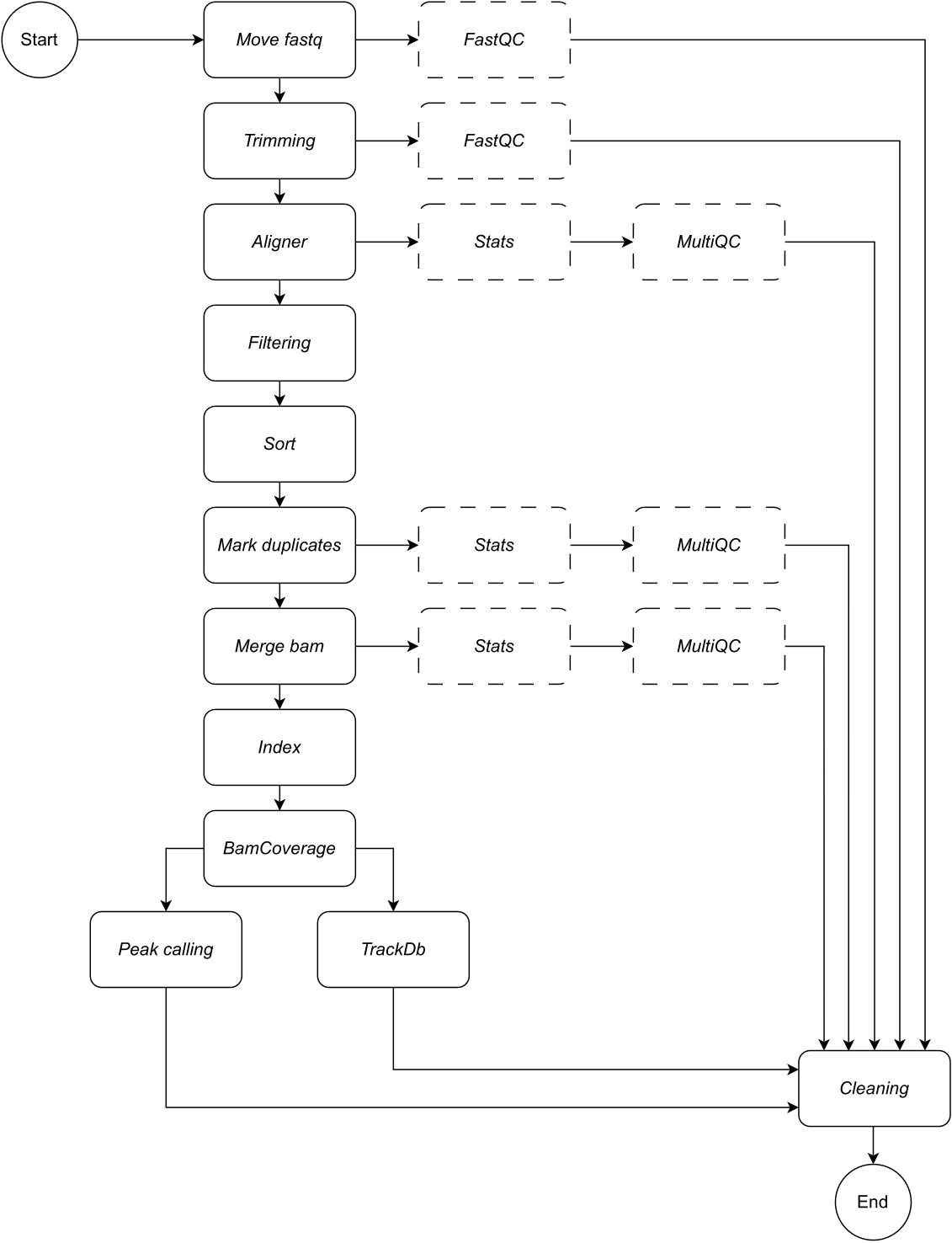
Figure 1 : Flux de travail de CATCH-UP. À partir d’une liste de fichiers fastq, CATCH-UP traite en parallèle tous les échantillons à travers toutes les étapes en amont. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Représentation illustrative expliquant comment les 1_fastqfile_home_dir.txt, les 2_fastqfile_concat.txt et les 3_merge_bams.txt doivent être correctement modifiéspour exécuter CATCH-UP. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

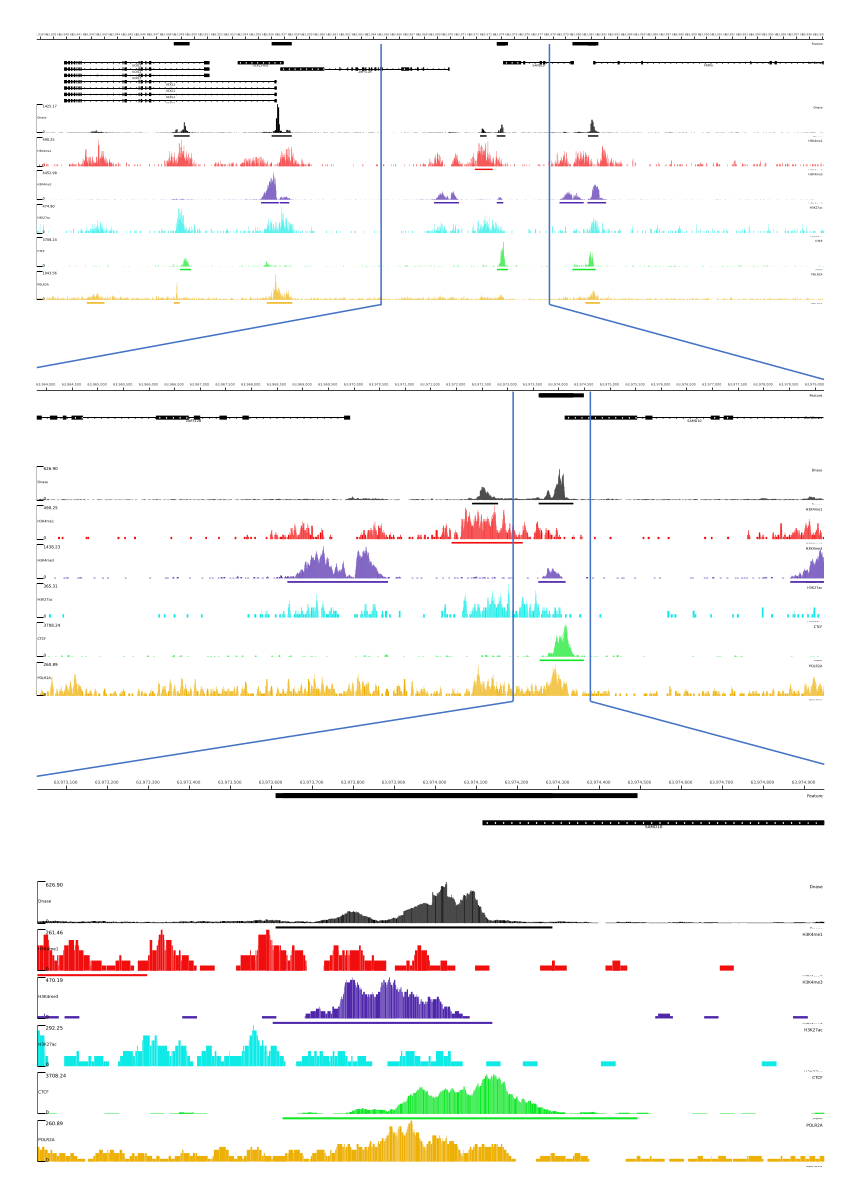
Figure 3 : Exemple de sortie du pipeline CATCH-UP. Les données de séquençage brutes (fichiers fastq) ont été téléchargées à partir d’ENCODE21. Le pipeline CATCH-UP a été utilisé pour traiter les fichiers fastq pour DNase-seq et 5 types de ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF et POLR2A). Les fichiers de sortie de Bigwig ont été téléchargés sur Multi Locus View pour la visualisation et l’identification des éléments régulateurs génomiques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau 1 : Ressources documentaires. Ce tableau présente les outils impliqués dans le flux de travail CATCH-UP, le lien vers leur documentation et les références respectives. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 2 : Liste des exigences en matière de canaux et de dépendances pour l’environnement conda en amont. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 3 : Systèmes d’exploitation utilisés pour tester CATCH-UP. Ubuntu a été testé sur un cluster hautes performances et une machine locale. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
L’adoption et l’utilisation accrues des techniques NGS pour générer des données génomiques ont été accompagnées d’une augmentation du développement d’outils bioinformatiques pour l’analyse de ces données. Il existe plusieurs outils qui peuvent être appliqués pour chaque étape de l’analyse des données, ainsi que de nombreux paramètres différents qui peuvent être spécifiés dans chaque outil 6,8,9,15,16,17,18,19,20,22,23,24 . Cela donne une combinaison très diversifiée de stratégies d’analyse qui pourraient être appliquées, chacune d’entre elles pouvant produire des variations dans le résultat. Afin de comparer avec précision les expériences, la normalisation de l’analyse bioinformatique est essentielle. Historiquement, les données NGS sont générées par des scientifiques de laboratoire humide, et les données sont analysées par des bioinformaticiens.
L’analyse des données NGS peut être divisée en pipelines « en amont » et « en aval », où l’amont comprend les étapes nécessaires pour passer de la sortie de données brutes d’une machine de séquençage à un format visuellement interprétable par un chercheur. L’analyse en aval comprend des étapes supplémentaires adaptées à la question de recherche et à la conception expérimentale. Les pipelines en amont sont donc généralisables et susceptibles d’être normalisés pour une meilleure reproductibilité scientifique. Les pipelines en aval, en revanche, sont sur mesure, dépendent de la question biologique et nécessitent l’éclairage de l’investigateur, ce qui les rend moins appropriés pour la normalisation. Nous avons créé un pipeline en amont convivial qui permet aux scientifiques de laboratoire humide d’analyser de manière reproductible leurs propres données sans avoir besoin de connaissances préalables en bioinformatique. Nous présentons ici CATCH-UP, un pipeline construit à l’aide du framework snakemake et conçu pour être à la fois convivial et pour lutter contre la question de la reproductibilité dans l’analyse de données ChIP-seq et ATAC-seq. Ce pipeline a été conçu pour gérer les données ChIP-seq ou ATAC-seq. Une fois que l’utilisateur a téléchargé CATCH-UP, les paramètres d’analyse et le nommage de l’échantillon doivent d’abord être définis avant d’exécuter le pipeline sur la ligne de commande à l’aide d’une seule ligne de code. Des instructions simples, étape par étape, sur la façon de personnaliser les paramètres d’analyse pour l’analyse ChIP-seq ou ATAC-seq sont fournies dans le fichier de configuration lui-même et dans notre guide étape par étape dans le référentiel GitHub CATCH-UP.
Il existe des pipelines d’analyse pour les données ChIP-seq ou ATAC-seq, tels que PEPATAC et ChIP-AP. Bien que ces pipelines présentent des avantages tels que l’incorporation d’analyses en amont et en aval dans un seul flux de travail ou l’utilisation d’une interface utilisateur graphique (GUI), ces outils s’adressent aux bioinformaticiens et aux scientifiques ayant un niveau modéré de formation informatique13,14. CATCH-UP a été conçu pour résoudre deux problèmes : permettre aux scientifiques de laboratoire humide sans formation en bioinformatique d’effectuer leur propre analyse en amont et permettre la standardisation de l’analyse en amont en facilitant la création de rapports et la reproductibilité exacte entre les laboratoires. CATCH-UP est intentionnellement limité à l’analyse en amont, mais les résultats sont compatibles avec les outils d’analyse en aval tels que ceux utilisés pour comparer statistiquement des ensembles de données ou déduire la liaison du facteur de transcription25,26.
Toutes les étapes critiques nécessaires à la réalisation d’une analyse amont reproductible sont prédéfinies dans le pipeline CATCH-UP pour garantir la robustesse. La nature détaillée de ce pipeline permet à l’utilisateur de suivre la sortie du pipeline étape par étape, ce qui est utile à la fois pour le dépannage et la réplication du flux de travail analytique. Compte tenu de la nature en évolution rapide des techniques NGS, la nature modulaire de ce pipeline est avantageuse car elle permet de l’adapter facilement pour intégrer à la fois la publication de mises à jour de versions d’outils et la mise en œuvre de nouveaux outils. CATCH-UP a été testé avec succès pour les systèmes d’exploitation suivants : Ubuntu, CentOS, macOS (processeur Intel) et Windows (Tableau 3). Le pipeline a été conçu pour gérer de grandes expériences contenant des dizaines d’échantillons en parallélisant le flux de travail, ce qui le rend adaptable à différents modèles expérimentaux. Dans l’ensemble, la mise en œuvre de CATCH-UP dans l’analyse des données ChIP-seq et ATAC-seq permet un flux de travail d’analyse convivial, reproductible et hautement adaptable.
Déclarations de divulgation
J.R.H. est cofondateur et directeur de Nucleome Therapeutics et fournit des conseils à l’entreprise.
Remerciements
J.R.H. a été soutenu par des subventions du Wellcome Trust (225220/Z/22/Z et 106130/Z/14/Z) et du MRC (MC_UU_00029/3). M.B. a été soutenu par la subvention du Wellcome Trust (225220/Z/22/Z). E.R.G a été soutenu par le ministère de l’Éducation nationale pour la sélection et le placement des candidats envoyés à l’étranger pour l’enseignement supérieur (YLSY), ministère de l’Éducation nationale de la République de Türkiye. E.G. a été soutenu par le programme de doctorat en médecine génomique et statistiques de Wellcome (108861/Z/15/Z). S.G.R. a été soutenu par la subvention du Conseil de recherches médicales (MRC) (MC_UU_00029/3).
matériels
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Références
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.