
Method Article
CATCH-UP: una pipeline upstream ad alto rendimento per dati ATAC-Seq e ChIP-Seq di massa
In questo articolo
Riepilogo
ATAC-seq e ChIP-seq consentono di studiare in dettaglio la regolazione genica; Tuttavia, l'elaborazione di questi tipi di dati è impegnativa e spesso incoerente tra i gruppi di ricerca. Presentiamo CATCH-UP: una pipeline computazionale di facile utilizzo che consente l'elaborazione e l'analisi standardizzata e riproducibile dei dati di set di dati ATAC/ChIP-seq nuovi e pubblicati.
Abstract
I saggi per la cromatina accessibile alla trasposasi (ATAC) e l'immunoprecipitazione della cromatina (ChIP), abbinati al sequenziamento di nuova generazione (NGS), hanno rivoluzionato lo studio della regolazione genica. La mancanza di standardizzazione nell'analisi dei set di dati altamente dimensionali generati da queste tecniche ha reso difficile il raggiungimento della riproducibilità, portando a discrepanze nei dati pubblicati ed elaborati. Parte di questo problema è dovuto alla vasta gamma di strumenti bioinformatici disponibili per l'analisi di questi tipi di dati. In secondo luogo, sono necessari in sequenza diversi strumenti bioinformatici per convertire i dati grezzi in un output completamente elaborato e interpretabile, e questi strumenti richiedono diversi livelli di competenze computazionali. Inoltre, ci sono molte opzioni per il controllo della qualità che non vengono impiegate in modo uniforme durante l'elaborazione dei dati. Affrontiamo questi problemi con un saggio completo per il sequenziamento della cromatina accessibile alla trasposasi (ATAC-seq) e il sequenziamento dell'immunoprecipitazione della cromatina (ChIP-seq) a monte (CATCH-UP), una pipeline facile da usare basata su Python per l'analisi di set di dati ChIP-seq e ATAC-seq di massa dai file fastq grezzi alle tracce bigwig visualizzabili e alle chiamate di picco. Questa pipeline è semplice da installare ed eseguire e richiede una conoscenza computazionale minima. La pipeline è modulare, scalabile e parallelizzabile su varie infrastrutture di calcolo, consentendo una facile segnalazione della metodologia per consentire l'analisi riproducibile di set di dati nuovi o pubblicati.
Introduzione
L'espressione genica deve essere strettamente regolata affinché le cellule stabiliscano e mantengano la loro corretta funzione biologica. È noto che l'espressione genica aberrante è alla base della patogenesi di molte malattie e, pertanto, un grande interesse della ricerca risiede nella comprensione dei meccanismi di regolazione genica1. L'espressione genica è facilitata da elementi regolatori come promotori e potenziatori. All'interno della loro sequenza, questi elementi contengono siti di legame del fattore di trascrizione (TF) che, quando attivi, forniscono una piattaforma per il legame TF. Il legame dei TF in questi siti provoca uno spostamento dei nucleosomi, con conseguente aumento dell'accessibilità del DNA e un conseguente aumento della permessibilità al macchinario trascrizionale. Come risultato di questa maggiore accessibilità, queste regioni del DNA sono più sensibili alle nucleasi e alle trasposisi come la DNasi e il Tn5, una proprietà biochimica che è stata sfruttata dai ricercatori che studiano la regolazione trascrizionale 2,3.
DNase-seq e ATAC-seq consentono ai ricercatori di mappare le regioni della cromatina aperta, i siti di legame del TF e il posizionamento nucleosomico in tutto il genoma. Di queste due tecniche, ATAC-seq è cresciuta in popolarità nell'ultimo decennio grazie al semplice protocollo in due fasi e a un basso numero di cellule richiesto (50.000 cellule rispetto a 1 milione per replicazione per DNase-seq). Sebbene l'ATAC-seq fornisca una panoramica del panorama generale della cromatina in una popolazione di cellule, è in gran parte agnostico a quali proteine specifiche si legano al genoma 4,5. Al fine di identificare le posizioni in cui una specifica proteina interagisce con il genoma, la tecnica gold standard è l'immunoprecipitazione della cromatina (ChIP)-seq. ChIP-seq comporta la fissazione chimica delle interazioni proteina-DNA in una cellula, seguita da immunoprecipitazione ("pull-down") utilizzando un anticorpo specifico per la proteina di interesse per selezionare i frammenti di DNA legati dalla proteina di interesse (POI). Questi frammenti di DNA possono essere sequenziati per rivelare le posizioni di legame genomico di proteine specifiche come i TF o siti contenenti specifiche modificazioni istoniche1. Combinando i set di dati ATAC-seq e ChIP-seq, è possibile ricavare un quadro dettagliato del panorama normativo per una popolazione di cellule.
Il flusso di lavoro di base richiesto per l'analisi è il seguente: le letture di sequenziamento grezze devono essere controllate prima dell'allineamento a un genoma di riferimento ("mappatura"). Le letture mappate correttamente possono quindi essere filtrate per rimuovere sia le letture di bassa qualità che i duplicati PCR. Per visualizzare queste letture mappate e filtrate, è necessario calcolare la "copertura" di queste letture in tutto il genoma. Questo genera un file che può essere caricato su un browser del genoma come la vista multi-locus (MLV) o il browser del genoma UCSC come "traccia"6,7. L'identificazione dei picchi, o "chiamata dei picchi" di queste tracce di copertura, si ottiene in genere utilizzando strumenti come LanceOtron o MACS2 8,9. Infine, attraverso l'analisi della posizione, della forma e delle dimensioni dei picchi, è possibile effettuare confronti tra campioni o condizioni biologiche. L'analisi e l'integrazione di questi dataset è un processo complesso a più fasi in cui possono essere implementate diverse combinazioni di strumenti bioinformatici. Diverse versioni degli strumenti possono essere incompatibili tra loro e possono modificare l'output dell'elaborazione dei dati. C'è anche un'ampia varietà nella potenza di calcolo e nella competenza dell'utente necessarie per implementare diverse parti dell'elaborazione dei dati, come mostrato nelle pipeline nf-core10, panpipes11, genpipes12, PEPATAC13 o ChIP-AP14.
Nel complesso, ciò ha portato a incongruenze sia nell'analisi che nella segnalazione dell'analisi, il che, a sua volta, ha portato a scarsa riproducibilità, accessibilità e convenienza per chiunque abbia una conoscenza limitata della bioinformatica. Affrontiamo tutti questi problemi con CATCH-UP (complete ATAC-seq and ChIP-seq upstream pipeline), una pipeline facile da usare, flessibile e modulare per l'elaborazione di dati ChIP-seq e ATAC/DNase-seq. L'implementazione di CATCH-UP richiede un'esperienza minima in bioinformatica; Può essere eseguito su varie infrastrutture informatiche e consente un'analisi riproducibile dei dati all'interno e tra gruppi di ricerca.
CATCH-UP è una pipeline Snakemake basata su Python creata per standardizzare l'analisi dei dati ChIP-seq e ATAC-seq. Prende i dati di sequenziamento grezzi (file fastq.gz) come input e genera un output sotto forma di file di picco (.bed) che forniscono il rispettivo risultato per ogni fase. Forniamo un file di configurazione in formato yaml (config.yaml), in cui l'utente può modificare i parametri di ogni fase di analisi. Il sistema di gestione implementato all'interno di snakemake consente l'utilizzo di diverse infrastrutture informatiche (come server, cluster, sistemi cloud o personal computer) e in parallelo se l'utente fornisce una grande quantità di dati.
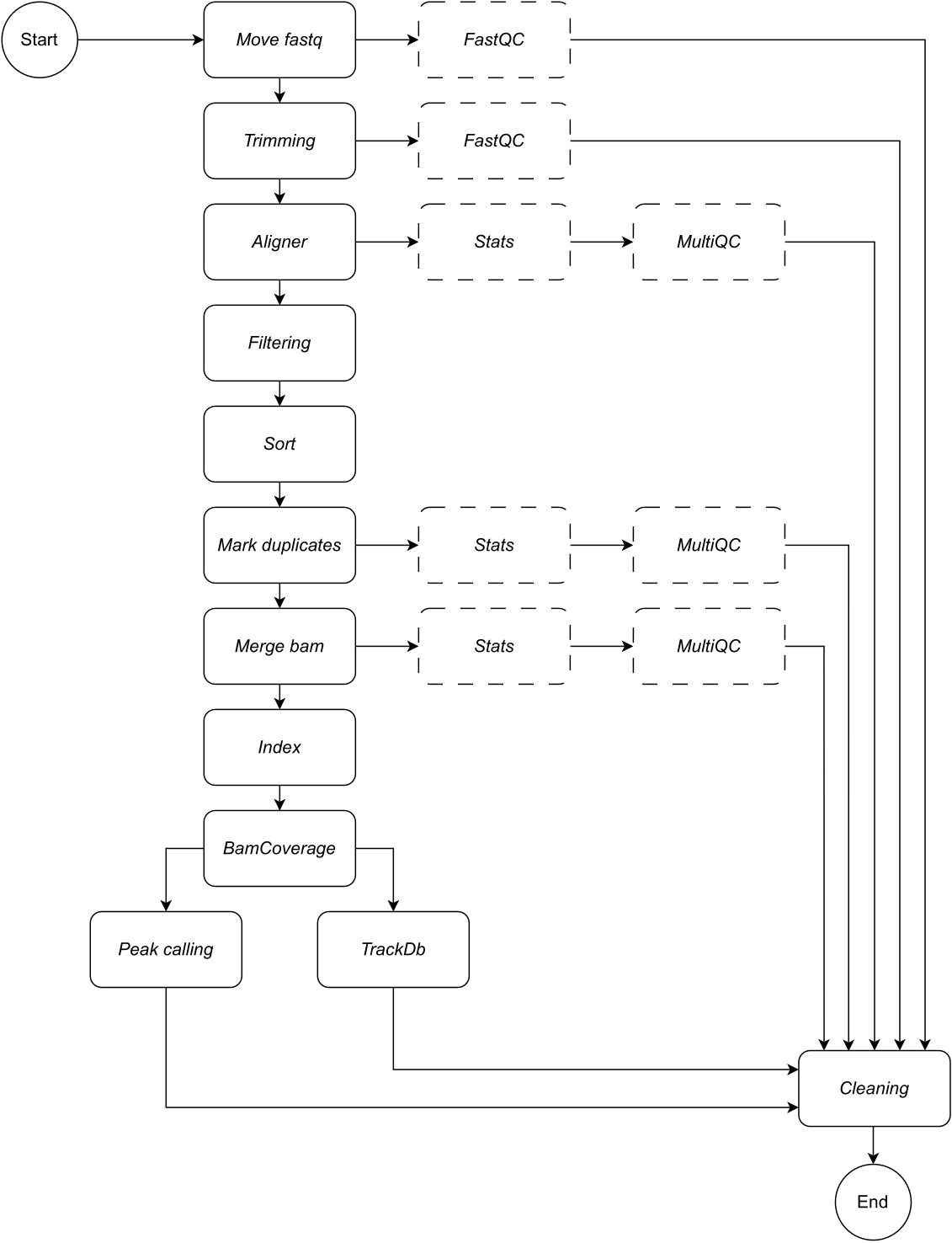
Di seguito, forniamo una descrizione dettagliata di ogni fase del flusso di lavoro (vedere la Figura 1 per l'illustrazione del flusso di lavoro). Questa spiegazione è essenziale per seguire passo dopo passo la sezione relativa al protocollo:
Move fastq: il primo passo della pipeline consiste nel copiare i file fastq grezzi nella directory di analisi denominata. In questo modo i dati originali rimangono intatti per evitare di danneggiare o modificare i file di dati non elaborati.
Concatenazione: se i dati di sequenziamento non elaborati contengono più corsie, questo passaggio è necessario per concatenare le corsie prima dell'analisi. Per impostazione predefinita, la pipeline gestisce tutti i file fastq come singoli campioni. Questo passaggio di concatenazione deve essere definito nel file di configurazione.
Rifilatura: fase opzionale di pulizia dei dati. Ciò consente di tagliare le letture di bassa qualità o le sequenze dell'adattatore utilizzando trimmomatic15. L'utente può fornire file fasta personalizzati di sequenze di adattatori; Un esempio è disponibile nella directory dell'adapter. Ulteriori parametri di ritaglio possono essere definiti nel file di configurazione. Per impostazione predefinita, il flusso di lavoro ignora questa regola.
Allineatore: per l'allineamento, Bowtie216 viene applicato di default; È possibile specificare anche strumenti di allineamento alternativi come BWA-MEM217 . Lo strumento di allineamento Bowtie2 è selezionato come predefinito in quanto è particolarmente abile nell'allineare letture relativamente brevi a genomi relativamente grandi ed è quindi adatto all'allineamento dei dati ChIP-seq e ATAC-seq ai genomi dei mammiferi. Per evitare file intermedi, l'allineatore viene convogliato nella vista samtools per salvare il file bam nell'output. Per questa regola, l'utente deve specificare la build del genoma preferita su cui mappare le letture, ad esempio hg19/hg38 (umano), mm10/mm39 (topo).
Filtraggio: le letture mappate correttamente vengono mantenute e le letture con bassa qualità vengono filtrate. Predefinito: vista samtools, con parametri: -bShuF 4 -f 3 -q 30.
Ordina: le letture allineate sono ordinate in base alla coordinata più a sinistra. Predefinito: samtools sort (snakemake wrapper), con parametro: -m 4G.
Contrassegna i duplicati: tutte le letture duplicate vengono identificate e contrassegnate. L'utente può decidere di rimuoverli modificando il parametro del file di configurazione. Predefinito: Picard MarkDuplicates (snakemake wrapper), con parametro: --REMOVE_DUPLICATES False per contrassegnare e conservare i duplicati.
Unisci bam: se i dati di sequenziamento sono composti da repliche o campioni, l'utente potrebbe voler unire in un singolo bam. In questo caso, l'utente può scegliere di unire i bams o di mantenere separati i file bam durante l'analisi. Se l'utente sceglie di unire i bams (utilizzando samtools merge), è necessario specificare un prefisso comune per i bam uniti.
Indice: questo passaggio indicizza le coordinate ordinate. Predefinito: indice samtools (involucro snakemake), utilizzando i parametri predefiniti specificati da samtools.
BamCoverage: questa regola crea una traccia di copertura più grande da letture allineate. Viene applicato lo strumento bamCoverage di deepTools e la copertura viene calcolata come il numero di letture per contenitore, in cui il contenitore rappresenta una finestra di una dimensione specificata. In questa pipeline, bamCoverage viene applicato con i parametri seguenti impostati come predefiniti: -bs 1 -normalizeUsing RPKM -extendReads.
Chiamata di picco: LanceOtron8 è stato selezionato come peakcaller predefinito per questa pipeline. A differenza dei tradizionali peak caller, che sono per lo più basati su test statistici, LanceOtron è un peak caller basato sul deep learning, che incorpora misurazioni di arricchimento genomico e test statistici e ha dimostrato di superare il peak caller standard del settore, MACS29. Affinché i pezzi grossi siano compatibili con LanceOtron, la copertura deve essere calcolata per coppia di basi e l'RPKM normalizzato; ciò si riflette nelle impostazioni predefinite per il passaggio BamCoverage. MACS2 può essere selezionato come chiamante di picco alternativo. Il rilascio di nuovi peak caller sarà monitorato e incorporato a seconda dei casi, al fine di mantenere e ottimizzare le prestazioni di questa pipeline di analisi.
TrackDb: crea un'associazione di coppie chiave-valore di file bigwig per caricarli e visualizzarli in strumenti come le piattaforme MLV6 o UCSC Genome Browser18 .
Oltre ai dati di output, ogni fase della pipeline genera un file di log e vengono forniti controlli di qualità appropriati in modo che l'utente possa tenere traccia dell'avanzamento dell'analisi. FastQC19 viene applicato ai dati di sequenziamento grezzi e tagliati (se selezionati) (passaggi 1 - Sposta fastq e 2 - Ritaglio). Le statistiche di Samtools e MultiQC20 vengono utilizzate per raccogliere, produrre e visualizzare i rapporti di controllo qualità sui file bam nell'output nei passaggi 3 - Allineatore, 6 - Contrassegna duplicati e 7 - Unisci bam. Per ulteriori informazioni su ciascuno degli strumenti applicati nei passaggi precedenti, vedere la Tabella 1.
Protocollo
1. Esecuzione della pipeline CATCH-UP
- Clonare il repository UpStreamPipeline da https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Passare alla directory di lavoro scelta, copiare il seguente codice ed eseguire l'operazione dalla riga di comando:
clone git git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Navigare all'interno della cartella UpStreamPipeline scaricata utilizzando il comando: cd UpStreamPipeline
- Installare la distribuzione anaconda (se necessario):
- Controlla se anaconda è già installato sul sistema utilizzando il comando which conda. Se il comando non mostra alcun percorso per nessuna distribuzione conda, scarica mambaforge da https://github.com/conda-forge/miniforge#mambaforge e seleziona la distribuzione e la versione corrette per il sistema. Ad esempio, per gli utenti Linux, utilizzare wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Visita questa pagina web per diversi sistemi operativi: https://github.com/conda-forge/miniforge/.
- Esegui l'installatore usando sh Mambaforge-Linux-x86_64.sh e inizializza conda nel sistema eseguendo conda init.
- Installare e attivare l'ambiente conda upstream (i requisiti dell'ambiente conda upstream sono elencati nella Tabella 2):
- Installa l'ambiente utilizzando il comando mamba env create - file=envs/upstream.yml.
- Attivare l'ambiente utilizzando il comando conda activate upstream.
- Una volta installato correttamente l'ambiente conda upstream, attivare l'ambiente utilizzando il comando conda activate upstream e passare alla cartella CATCH-UP utilizzando cd genetics/CATCH-UP.
- Modificare il file di configurazione, che si trova all'interno della cartella config utilizzando il comando cd /config/analysis.yaml, e modificarlo di conseguenza con la specifica di analisi utilizzando un editor di testo. Segui le istruzioni riga per riga per modificare ogni parametro all'interno del file stesso. Questo file verrà conservato dopo l'analisi e fungerà da documentare i parametri di esecuzione per facilitare la riproducibilità.
- Apri e modifica i seguenti tre file in un editor di testo (ad esempio, TextEdit per Mac o Blocco note per Windows):
- Modifica 1_fastqfile_home_dir.txt file in modo che contenga un elenco di tutti i file fastq da analizzare.
NOTA: I numeri di lettura e gli interni (ad es. _R1/_R2 e .fastq.gz) devono essere esclusi. Ad esempio, se un progetto contiene questo elenco di file fastq:
Sample1_conditionA_L001_R1 . Fastq . gz
Sample1_conditionA_L001_R2 . Fastq . gz
Sample1_conditionA_L002_R1 . Fastq . gz
Sample1_conditionA_L002_R2 . Fastq . gz
Sample1_conditionB_L001_R1 . Fastq . gz
Sample1_conditionB_L001_R2 . Fastq . gz
Sample1_conditionB_L002_R1 . Fastq . gz
Sample1_conditionB_L002_R2 . Fastq . gz
Sample2_conditionA_L001_R1 . Fastq . gz
Sample2_conditionA_L001_R2 . Fastq . gz
Sample2_conditionA_L002_R1 . Fastq . gz
Sample2_conditionA_L002_R2 . Fastq . gz
Sample2_conditionB_L001_R1 . Fastq . gz
Sample2_conditionB_L001_R2 . Fastq . gz
Sample2_conditionB_L002_R1 . Fastq . gz
Sample2_conditionB_L002_R2 . Fastq . gz
In questo caso, il 1_fastqfile_home_dir.txt è il seguente:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Se i dati grezzi contengono corsie di sequenziazione che richiedono la concatenazione, modificare 2_fastqfile file_concat.txt per definire il prefisso dei nomi dei file da concatenare. Se non sono presenti corsie di sequenziamento da concatenare, non modificare 2_fastqfile_concat.txt. Assicurarsi che ogni riga di 2_fastqfile_concat.txt contenga un prefisso di esempio come segue:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Se è necessario unire i dati di campioni diversi, modificare 3_merge_bams.txt file con il prefisso dei nomi dei file da unire. Assicurarsi che ogni riga contenga un prefisso di esempio come indicato di seguito:
Campione1
Campione 2
Nella Figura 2 viene illustrato un riepilogo di come riepilogare questi tre file. Il protocollo può essere applicato a dati di sequenziamento a estremità singola o accoppiata. Per impostazione predefinita, la pipeline utilizza l'analisi paired-end, se non diversamente specificato. Questo può essere modificato nel file di configurazione (vedere il passaggio 1.6).
- Modifica 1_fastqfile_home_dir.txt file in modo che contenga un elenco di tutti i file fastq da analizzare.
- Una volta che tutti i file richiesti sono stati modificati, usa snakemake per eseguire CATCH-UP come segue: snakemake --configfile=config/analysis_name.yaml all --cores 4.
NOTA: per istruzioni e documentazione più dettagliate, consulta la cartella CATCH-UP all'interno del repository GitHub di UpStreamPipeline, disponibile qui. Ciò include una documentazione dettagliata sulla corretta modifica del file di configurazione, dalla modifica dei percorsi per la sequenziazione dei file di dati e l'archiviazione dei risultati alla modifica dei parametri per ogni passaggio.
Risultati
La pipeline CATCH-UP produce un risultato, un log e un output di controllo qualità (QC) per ogni fase. All'interno del file di configurazione, l'utente può scegliere di mantenere o rimuovere i file di output per ridurre la memoria di archiviazione richiesta. Tutti i risultati sono spiegati come segue:
00. fastq_home_dir: Il file di configurazione, il le_home_dir.txt FastQFi e merge_bams.txt vengono copiati in questa cartella per riferimento e riproducibilità.
01. Legge: i file FastQ vengono copiati in questa cartella per evitare alterazioni dei dati grezzi originali durante il processo del flusso di lavoro, le corsie possono essere concatenate se specificato.
02. Ritaglio: file FastQ con lettura e adattatori tagliati se specificato.
03. Allineatore: allineamento rispetto al genoma selezionato.
04. Filtraggio: filtraggio del controllo qualità.
05. Ordinato: ordinamento dei file BAM.
06. Duplicati: Segnalazione di duplicati.
07. Unisci: unione di file BAM se questo è stato specificato in config.yaml.
08. bam_coverages: file bigwig della copertura.
09. peak_calling: un file letto dell'uscita di chiamata di picco di LanceOtron.
10. traccia: produce un file di testo formattato pronto per essere utilizzato su Genome Browser, se necessario.
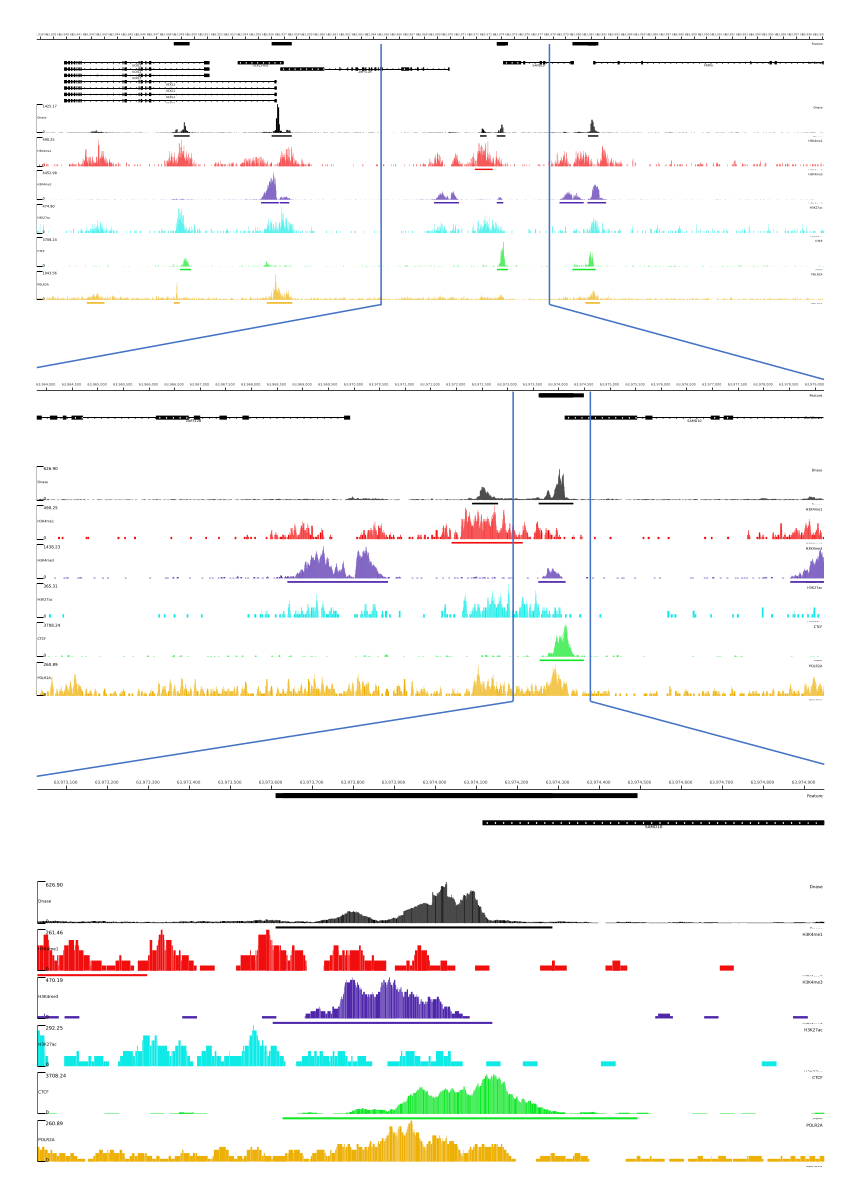
Per gli output 01, 02, 03, 06 e 07, vengono fornite le metriche QC e i file HTML. Inoltre, in Figura 3, forniamo un esempio di dati elaborati utilizzando CATCH-UP, visualizzando l'output finale attraverso la piattaforma MLV.

Figura 1: Flusso di lavoro di CATCH-UP. Dato un elenco di file fastq, CATCH-UP elabora in parallelo tutti i campioni attraverso tutti i passaggi a monte. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Rappresentazione illustrativa che spiega come 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt e 3_merge_bams.txt devono essere modificati correttamenteper eseguire il CATCH-UP. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Esempio di output dalla pipeline CATCH-UP. I dati grezzi di sequenziamento (file fastq) sono stati scaricati da ENCODE21. La pipeline CATCH-UP è stata utilizzata per elaborare i file fastq per DNase-seq e 5 tipi di ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF e POLR2A). I file di output di Bigwig sono stati caricati su Multi Locus View per la visualizzazione e l'identificazione degli elementi regolatori genomici. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tabella 1: Risorse per la documentazione. Questa tabella mostra gli strumenti coinvolti nel flusso di lavoro CATCH-UP, il link per la loro documentazione e i rispettivi riferimenti. Clicca qui per scaricare questa tabella.
Tabella 2: Elenco dei requisiti di canale e dipendenza per l'ambiente conda upstream. Clicca qui per scaricare questa tabella.
Tabella 3: Sistemi operativi utilizzati per testare CATCH-UP. Ubuntu è stato testato su un cluster ad alte prestazioni e su una macchina locale. Clicca qui per scaricare questa tabella.
Discussione
L'aumento dell'adozione e dell'utilizzo delle tecniche NGS per generare dati genomici è stato accompagnato da un aumento dello sviluppo di strumenti bioinformatici per l'analisi di questi dati. Esistono più strumenti che possono essere applicati per ogni fase dell'analisi dei dati, nonché molti parametri diversi che possono essere specificati all'interno di ogni strumento 6,8,9,15,16,17,18,19,20,22,23,24 . Ciò crea una combinazione molto diversificata di strategie di analisi che potrebbero essere applicate, ognuna delle quali potrebbe produrre variazioni nel risultato. Al fine di effettuare un confronto accurato tra gli esperimenti, la standardizzazione dell'analisi bioinformatica è essenziale. Storicamente, i dati NGS sono generati da scienziati di laboratorio umido e i dati vengono analizzati da bioinformatici.
L'analisi dei dati NGS può essere suddivisa in pipeline "a monte" e "a valle", dove a monte include i passaggi necessari per passare dall'output dei dati grezzi da una macchina di sequenziamento a un formato che sia visivamente interpretabile da un ricercatore. L'analisi a valle include fasi aggiuntive su misura per la domanda di ricerca e il disegno sperimentale. Le pipeline a monte sono quindi generalizzabili e suscettibili di standardizzazione per una migliore riproducibilità scientifica. Le condutture a valle, d'altra parte, sono su misura, dipendono dalla questione biologica e richiedono l'intuizione del ricercatore, rendendole meno appropriate per la standardizzazione. Abbiamo creato una pipeline upstream di facile utilizzo che consente agli scienziati di laboratorio umido di analizzare in modo riproducibile i propri dati senza bisogno di alcuna conoscenza preliminare di bioinformatica. Qui presentiamo CATCH-UP, una pipeline costruita utilizzando il framework snakemake e progettata per essere sia facile da usare che per combattere il problema della riproducibilità nell'analisi dei dati ChIP-seq e ATAC-seq. Questa pipeline è stata creata per gestire dati ChIP-seq o ATAC-seq. Una volta che l'utente ha scaricato CATCH-UP, i parametri di analisi e la denominazione del campione devono essere definiti prima di eseguire la pipeline sulla riga di comando utilizzando una singola riga di codice. Semplici istruzioni dettagliate su come personalizzare i parametri di analisi per l'analisi ChIP-seq o ATAC-seq sono fornite all'interno del file di configurazione stesso e nella nostra guida passo passo nel repository GitHub CATCH-UP.
Esistono pipeline di analisi esistenti per i dati ChIP-seq o ATAC-seq, come PEPATAC e ChIP-AP. Sebbene queste pipeline presentino vantaggi come l'incorporazione di analisi a monte e a valle in un unico flusso di lavoro o l'uso di un'interfaccia utente grafica (GUI), questi strumenti sono destinati a bioinformatici e scienziati con un livello moderato di formazione computazionale13,14. CATCH-UP è stato progettato per risolvere due problemi: consentire agli scienziati di laboratorio senza formazione bioinformatica di eseguire le proprie analisi a monte e consentire la standardizzazione delle analisi a monte facilitando la creazione di report e l'esatta riproducibilità tra i laboratori. CATCH-UP è intenzionalmente limitato all'analisi a monte, ma i risultati sono compatibili con gli strumenti di analisi a valle, come quelli utilizzati per confrontare statisticamente i set di dati o dedurre il legame del fattore di trascrizione25,26.
Tutti i passaggi critici necessari per eseguire un'analisi upstream replicabile sono predefiniti all'interno della pipeline CATCH-UP per garantire la robustezza. La natura dettagliata di questa pipeline consente all'utente di seguire passo dopo passo l'output della pipeline, il che è utile sia per la risoluzione dei problemi che per consentire la replica del flusso di lavoro analitico. Data la natura in rapida evoluzione delle tecniche NGS, la natura modulare di questa pipeline è vantaggiosa in quanto fornisce la capacità di essere facilmente adattata per incorporare sia il rilascio di aggiornamenti della versione dello strumento che l'implementazione di nuovi strumenti. CATCH-UP è stato testato con successo per i seguenti sistemi operativi: Ubuntu, CentOS, macOS (CPU Intel) e Windows (Tabella 3). La pipeline è stata costruita per gestire esperimenti di grandi dimensioni contenenti decine di campioni parallelizzando il flusso di lavoro, rendendola adattabile a diversi disegni sperimentali. Nel complesso, l'implementazione di CATCH-UP nell'analisi dei dati ChIP-seq e ATAC-seq consente un flusso di lavoro di analisi facile da usare, riproducibile e altamente adattabile.
Divulgazioni
J.R.H. è co-fondatore e direttore di Nucleome Therapeutics e fornisce consulenza all'azienda.
Riconoscimenti
J.R.H. è stato sostenuto da sovvenzioni del Wellcome Trust (225220/Z/22/Z e 106130/Z/14/Z) e dell'MRC (MC_UU_00029/3). M.B. è stato sostenuto dalla sovvenzione Wellcome Trust (225220/Z/22/Z). E.R.G è stato sostenuto dal Ministero dell'Istruzione Nazionale per la selezione e il collocamento dei candidati inviati all'estero per la borsa di studio Post-Laurea (YLSY), Ministero dell'Educazione Nazionale della Repubblica di Turchia. E.G. è stato supportato dal programma di dottorato Wellcome Genomic Medicine and Statistics (108861/Z/15/Z). S.G.R. è stato sostenuto dalla sovvenzione del Medical Research Council (MRC) (MC_UU_00029/3).
Materiali
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Riferimenti
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati