
Method Article
CATCH-UP: A High-Throughput Upstream-Pipeline for Bulk ATAC-Seq and ChIP-Seq Data
In This Article
Summary
ATAC-seq and ChIP-seq allow detailed investigation of gene regulation; however, processing these data types is challenging and often inconsistent between research groups. We present CATCH-UP: an easy-to-use computational pipeline that allows standardized and reproducible data processing and analysis of new and published ATAC/ChIP-seq datasets.
Abstract
Assay for transposase-accessible chromatin (ATAC) and chromatin immunoprecipitation (ChIP), coupled with next-generation sequencing (NGS), have revolutionized the study of gene regulation. A lack of standardization in the analysis of the highly dimensional datasets generated by these techniques has made reproducibility difficult to achieve, leading to discrepancies in the published, processed data. Part of this problem is due to the diverse range of bioinformatic tools available for the analysis of these types of data. Secondly, a number of different bioinformatic tools are required sequentially to convert raw data into a fully processed and interpretable output, and these tools require varying levels of computational skills. Furthermore, there are many options for quality control that are not uniformly employed during data processing. We address these issues with a complete assay for transposase-accessible chromatin sequencing (ATAC-seq) and chromatin immunoprecipitation sequencing (ChIP-seq) upstream pipeline (CATCH-UP), an easy-to-use, Python-based pipeline for the analysis of bulk ChIP-seq and ATAC-seq datasets from raw fastq files to visualizable bigwig tracks and peaks calls. This pipeline is simple to install and run, requiring minimal computational knowledge. The pipeline is modular, scalable, and parallelizable on various computing infrastructures, allowing for easy reporting of methodology to enable reproducible analysis of novel or published datasets.
Introduction
Gene expression must be tightly regulated for cells to establish and maintain their correct biological function. It is well known that aberrant gene expression underlies the pathogenesis of many diseases, and therefore, a great deal of research interest lies in understanding the mechanisms of gene regulation1. Gene expression is facilitated by regulatory elements such as promoters and enhancers. Within their sequence, these elements contain transcription factor (TF) binding sites, which, when active, provide a platform for TF binding. The binding of TFs at these sites results in a displacement of nucleosomes, resulting in an increase in DNA accessibility and a subsequent increase in permissibility to the transcriptional machinery. As a result of this increased accessibility, these regions of DNA are more sensitive to nucleases and transposases such as DNase and Tn5, a biochemical property that has been exploited by researchers investigating transcriptional regulation2,3.
DNase-seq and ATAC-seq allow researchers to map regions of open chromatin, TF binding sites, and nucleosomal positioning across the genome. Of these two techniques, ATAC-seq has grown in popularity over the past decade due to the simple two-step protocol and a low cell number requirement (50,000 cells compared to 1 million per replicate for DNase-seq). Whilst ATAC-seq provides an overview of the general chromatin landscape in a population of cells, it is largely agnostic to which specific proteins are binding to the genome4,5. In order to identify the locations where a specific protein is interacting with the genome, the gold standard technique is Chromatin Immunoprecipitation (ChIP)-seq. ChIP-seq involves chemically fixing protein-DNA interactions in a cell, followed by immunoprecipitation ("pull-down") using an antibody specific to the protein of interest to select for DNA fragments bound by the protein of interest (POI). These DNA fragments can be sequenced to reveal the genomic binding locations of specific proteins such as TFs, or sites containing specific histone modifications1. By combining ATAC-seq and ChIP-seq datasets, a detailed picture of the regulatory landscape can be derived for a population of cells.
The basic workflow required for the analysis is as follows: raw sequencing reads must be quality controlled before alignment to a reference genome ("mapping"). The successfully mapped reads may then be filtered to remove both low-quality reads and PCR duplicates. In order to visualize these mapped and filtered reads, it is necessary to calculate the "coverage" of these reads across the genome. This generates a file that can be uploaded to a genome browser such as multi-locus view (MLV) or the UCSC genome browser as a "track"6,7. Peak identification, or "peak calling" of these coverage tracks is typically achieved using tools such as LanceOtron or MACS28,9. Finally, through the analysis of peak location, shape, and size comparisons can be made between samples or biological conditions. The analysis and integration of these datasets is a complex multi-step process in which different combinations of bioinformatic tools can be implemented. Different versions of the tools may be incompatible with one another and may change the output of the data processing. There is also a wide variety in the computational power and user proficiency required to implement different parts of data processing as shown in nf-core10, panpipes11, genpipes12, PEPATAC13, or ChIP-AP14 pipelines.
Overall, this has led to inconsistencies in both the analysis and the reporting of the analysis, which has, in turn, led to poor reproducibility, accessibility, and convenience for anybody with limited knowledge of bioinformatics. We address all these problems with CATCH-UP (complete ATAC-seq and ChIP-seq upstream pipeline), an easy-to-use, flexible, and modular pipeline for processing ChIP-seq and ATAC/DNase- seq data. The implementation of CATCH-UP requires minimal bioinformatics experience; it can be run on various computing infrastructures and enables reproducible data analysis within and across research groups.
CATCH-UP is a Python-based Snakemake pipeline built to standardize the analysis of ChIP-seq and ATAC-seq data. It takes raw sequencing data (fastq.gz files) as input and generates an output in the form of peak (.bed) files providing the respective outcome for each step. We provide a configuration file in yaml format (config.yaml), in which the user can edit the parameters of each analysis step. The management system implemented within snakemake enables the use of different computing infrastructures (such as servers, clusters, cloud systems, or personal computers) and in parallel if the user provides a large amount of data.
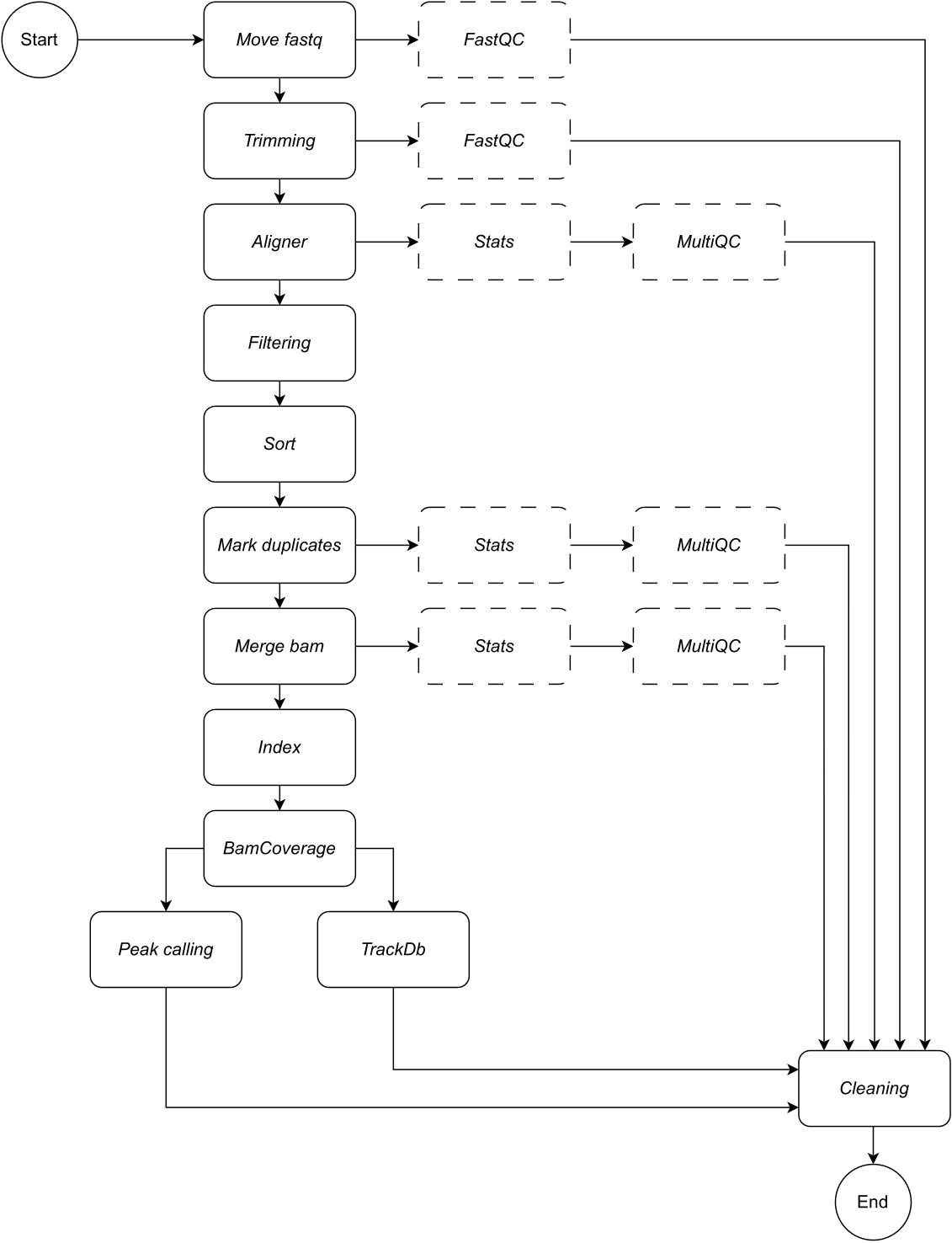
Below, we provide a detailed description of each step of the workflow (see Figure 1 for the workflow illustration). This explanation is essential in order to follow the step-by-step in the protocol section:
Move fastq: the first step of the pipeline is to copy the raw fastq files into the named analysis directory. This leaves the original data untouched to avoid corrupting or modifying the raw data files.
Concatenating: if raw sequencing data contains multiple lanes, this step is required to concatenate the lanes prior to analysis. By default, the pipeline handles all fastq files as single samples. This concatenation step must be defined in the configuration file.
Trimming: optional data cleaning step. This allows the trimming of low-quality reads or adapter sequences by using trimmomatic15. The user can provide custom fasta files of adapter sequences; an example is provided in the adapter directory. Additional trimming parameters can be defined in the configuration file. By default, the workflow skips this rule.
Aligner: for alignment, Bowtie216 is applied by default; alternative alignment tools such as bwa-mem217 can also be specified. The Bowtie2 alignment tool is selected as default as it is particularly adept at aligning relatively short reads to relatively large genomes and is therefore, well suited to the alignment of ChIP-seq and ATAC-seq data to mammalian genomes. To avoid any intermediate files, the aligner is piped into samtools view to save the bam file in output. For this rule, the user must specify the preferred genome build on which to map the reads e.g., hg19/hg38 (human), mm10/mm39 (mouse).
Filtering: properly mapped reads are retained, and reads with low quality are filtered out. Default: samtools view, with parameters: -bShuF 4 -f 3 -q 30.
Sort: aligned reads are sorted in order of the leftmost coordinate. Default: samtools sort (snakemake wrapper), with parameter: -m 4G.
Mark duplicates: all duplicate reads are identified and flagged. The user can decide to remove them by changing the configuration file parameter. Default: Picard MarkDuplicates (snakemake wrapper), with parameter: --REMOVE_DUPLICATES False to flag and retain duplicates.
Merge bam: If the sequencing data is composed of replicates or samples, the user may want to merge into a single bam. In this case, the user can choose to merge the bams or keep bam files separate throughout the analysis. If the user chooses to merge bams (employing samtools merge), a common prefix must be specified for the merged bams.
Index: this step indexes the sorted coordinates. Default: samtools index (snakemake wrapper), using default parameters specified by samtools.
BamCoverage: this rule creates a bigwig coverage track from aligned reads. The bamCoverage tool from deepTools is applied, and coverage is calculated as the number of reads per bin, in which the bin represents a window of a specified size. In this pipeline, bamCoverage is applied with the following parameters set as default: -bs 1 -normalizeUsing RPKM -extendReads.
Peak calling: LanceOtron8 was selected as the default peakcaller for this pipeline. Unlike traditional peak callers, which are mostly statistically test-based, LanceOtron is a deep learning-based peak caller, which incorporates genomic enrichment measurements and statistical testing and has been shown to outperform the industry standard peak caller, MACS29. For bigwigs to be compatible with LanceOtron, the coverage must be calculated per base-pair, and RPKM normalized; this is reflected in the default settings for the BamCoverage step. MACS2 can be selected as an alternative peak caller. The release of new peak callers will be monitored and incorporated as applicable in order to maintain and optimize the performance of this analysis pipeline.
TrackDb: this creates a key-value pair association of bigwig files in order to load and visualize them in tools such as MLV6 or UCSC Genome Browser18 platforms.
In addition to the output data, each step of the pipeline outputs a log file, and appropriate quality control checks are provided so that the user can track the analysis progress. FastQC19 is applied to raw and trimmed (if selected) sequencing data (steps 1 - Move fastq and 2- Trimming). Samtools stats plus MultiQC20 are used to collect, produce, and visualize quality control reports on bam files in output in steps 3 - Aligner, 6 - Mark duplicates, and 7 - Merge bam. For further information on each of the tools applied in the above steps, see Table 1.
Protocol
1. Running CATCH-UP pipeline
- Clone the UpStreamPipeline repository from https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Navigate to the chosen working directory, copy the following code and run on the command line:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Navigate inside the UpStreamPipeline folder downloaded using the command: cd UpStreamPipeline
- Install the anaconda distribution (if necessary):
- Check if anaconda is already installed on the system using the command which conda. If the command does not show any path to any conda distribution, download mambaforge from https://github.com/conda-forge/miniforge#mambaforge and select the right distribution and version for the system. For example, for linux users, use wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Visit this web page for different Operating Systems: https://github.com/conda-forge/miniforge/.
- Run the installer using sh Mambaforge-Linux-x86_64.sh, and initialize conda in the system by running conda init.
- Install and activate the upstream conda environment (requirements of the upstream conda environment are listed in Table 2):
- Install the environment using the command mamba env create - file=envs/upstream.yml.
- Activate the environment using the command conda activate upstream.
- Once the upstream conda environment is successfully installed, activate the environment using the command conda activate upstream and navigate to the CATCH-UP folder using cd genetics/CATCH-UP.
- Edit the configuration file, which can be found within the config folder using the command cd /config/analysis.yaml, and modify it accordingly with the analysis specification using a text editor. Follow the line-by-line instructions to edit each parameter within the file itself. This file will be retained after the analysis and act to document the run parameters to aid reproducibility.
- Open and edit the following three files in a text editor (e.g., TextEdit for Mac or Notepad for Windows):
- Edit 1_fastqfile_home_dir.txt file to contain a list of all fastq files to be analyzed.
NOTE: Read numbers and extensions (e.g., _R1/_R2 and .fastq.gz) must be excluded. For example, if a project contains this list of fastq files:
Sample1_conditionA_L001_R1 . fastq . gz
Sample1_conditionA_L001_R2 . fastq . gz
Sample1_conditionA_L002_R1 . fastq . gz
Sample1_conditionA_L002_R2 . fastq . gz
Sample1_conditionB_L001_R1 . fastq . gz
Sample1_conditionB_L001_R2 . fastq . gz
Sample1_conditionB_L002_R1 . fastq . gz
Sample1_conditionB_L002_R2 . fastq . gz
Sample2_conditionA_L001_R1 . fastq . gz
Sample2_conditionA_L001_R2 . fastq . gz
Sample2_conditionA_L002_R1 . fastq . gz
Sample2_conditionA_L002_R2 . fastq . gz
Sample2_conditionB_L001_R1 . fastq . gz
Sample2_conditionB_L001_R2 . fastq . gz
Sample2_conditionB_L002_R1 . fastq . gz
Sample2_conditionB_L002_R2 . fastq . gz
in this case, the 1_fastqfile_home_dir.txt is as follows:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - If raw data contains sequencing lanes that require concatenation, edit 2_fastqfile_concat.txt file to define the prefix of the file names to be concatenated. If there are no sequencing lanes to concatenate, then do not edit 2_fastqfile_concat.txt. Ensure that every line of 2_fastqfile_concat.txt contains one sample prefix as follows:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - If merging the data of different samples is required, then edit 3_merge_bams.txt file with the prefix of the file names to be merged. Ensure that every line contains one sample prefix as follows:
Sample1
Sample2
Figure 2 shows a summary of how to summarize these three files. The protocol can be applied to either single or paired-end sequencing data. The pipeline defaults to paired-end analysis unless otherwise specified; this can be modified in the configuration file (see step 1.6).
- Edit 1_fastqfile_home_dir.txt file to contain a list of all fastq files to be analyzed.
- Once all of the required files have been edited, use snakemake to run CATCH-UP as follows: snakemake --configfile=config/analysis_name.yaml all --cores 4.
NOTE: For more detailed instructions and documentation, see the CATCH-UP folder within the UpStreamPipeline GitHub repository, available here. This includes detailed documentation on correctly modifying the configuration file, from changing paths for sequencing data files and storing results to editing parameters for each step.
Results
The CATCH-UP pipeline produces a result, log, and quality control (QC) output for each step. Within the configuration file, the user can choose to either keep or remove output files to reduce the storage memory required. All of the outputs are explained as follows:
00. fastq_home_dir: config file, fastqfile_home_dir.txt, and merge_bams.txt are copied into this folder for reference and reproducibility.
01. reads: fastq files are copied into this folder to avoid alterations of the original raw data during the workflow process, lanes can be concatenated if specified.
02. trimming: fastq files with read and adapters trimmed if specified.
03. aligner: alignment against the selected genome.
04. filtering: quality control filtering.
05. sorted: sorting of bam files.
06. duplicates: flagging duplicates.
07. merge: merging bam files if this was specified in config.yaml.
08. bam_coverages: bigwig file of the coverage.
09. peak_calling: a bed file of LanceOtron peak calling output.
10. track: produces a formatted text file ready to be used on Genome Browser if needed.
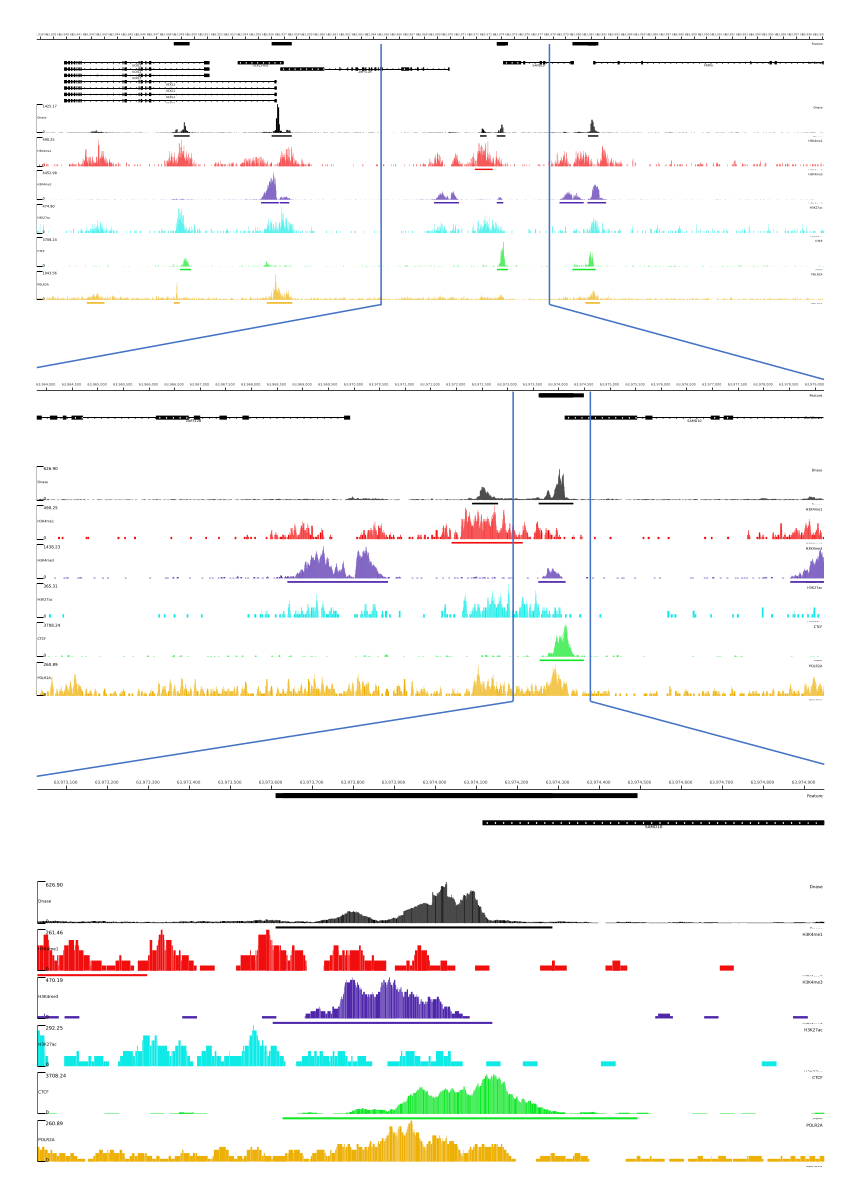
For 01, 02, 03, 06, and 07 outputs, QC metrics and HTML files are provided. In addition, in Figure 3, we provide an example of processed data using CATCH-UP, visualizing the final output through the MLV platform.

Figure 1: Workflow of CATCH-UP. Given a list of fastq files, CATCH-UP processes in parallel all samples through all upstream steps. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Illustrative representation explaining how 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt, and 3_merge_bams.txt must be correctly modified in order to run CATCH-UP. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Example output from CATCH-UP pipeline. Raw sequencing data (fastq files) were downloaded from ENCODE21. CATCH-UP pipeline was used to process the fastq files for DNase-seq and 5 types of ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF, and POLR2A). Bigwig output files were uploaded to Multi Locus View for visualization and identification of genomic regulatory elements. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Documentation resources. This table shows the tools involved in the CATCH-UP workflow, the link for their documentation, and the respective references. Please click here to download this Table.
Table 2: List of channel and dependency requirements for upstream conda environment. Please click here to download this Table.
Table 3: Operating Systems used to test CATCH-UP. Ubuntu was tested on a high-performance cluster and a local machine. Please click here to download this Table.
Discussion
The increased uptake and utilization of NGS techniques to generate genomic data have been matched by an increase in the development of bioinformatics tools for the analysis of these data. There are multiple tools that could be applied for each step of the data analysis, as well as many different parameters that can be specified within each tool6,8,9,15,16,17,18,19,20,22,23,24. This makes for a vastly diverse combination of analysis strategies that could be applied, each of which could produce variations in the outcome. In order to accurately compare across experiments, standardization of bioinformatic analysis is essential. Historically, NGS data is generated by wet lab scientists, and the data is analyzed by bioinformaticians.
NGS data analysis can be divided into "upstream" and "downstream" pipelines, where upstream includes the necessary steps to go from raw data output from a sequencing machine to a format that is visually interpretable by a researcher. Downstream analysis includes additional steps that are bespoke to the research question and experimental design. Upstream pipelines are therefore, generalizable and amenable to standardization for improved scientific reproducibility. Downstream pipelines, on the other hand, are bespoke, dependent on the biological question, and require insight from the investigator, making them less appropriate for standardization. We have created a user-friendly upstream pipeline that allows wet-lab scientists to reproducibly analyze their own data without needing any prior knowledge of bioinformatics. Here, we present CATCH-UP, a pipeline built using the snakemake framework and designed to be both user-friendly and to combat the issue of reproducibility in ChIP-seq and ATAC-seq data analysis. This pipeline has been built to handle either ChIP-seq or ATAC-seq data. Once the user has downloaded CATCH-UP, the analysis parameters and sample naming must first be defined before running the pipeline on the command line using a single line of code. Simple step-by-step instructions on how to customize the analysis parameters for either ChIP-seq or ATAC-seq analysis are provided within the configuration file itself and in our step-by-step guide in the CATCH-UP GitHub repository.
There are existing analysis pipelines for ChIP-seq or ATAC-seq data, such as PEPATAC and ChIP-AP. Whilst these pipelines have advantages such as the incorporation of both upstream and downstream analyses in a single workflow or the use of a graphical user interface (GUI), these tools are targeted at bioinformaticians and scientists with a moderate level of computational training13,14. CATCH-UP has been designed to solve two problems: enable wet lab scientists with no bioinformatic training to perform their own upstream analysis and enable standardization of upstream analysis by facilitating easy reporting and exact reproducibility across labs. CATCH-UP is intentionally limited to upstream analysis, but the outputs are compatible with downstream analysis tools such as those used to statistically compare datasets or infer transcription factor binding25,26.
All critical steps necessary to perform a replicable upstream analysis are predefined within the CATCH-UP pipeline to ensure robustness. The verbose nature of this pipeline allows the user to follow the pipeline's output step-by-step, which is useful for both troubleshooting and enabling the analytical workflow to be replicated. Given the rapidly evolving nature of NGS techniques, the modular nature of this pipeline is beneficial as it provides the capability to be easily adapted to incorporate both the release of tool version updates and the implementation of new tools. CATCH-UP has been successfully tested for the following operating systems: Ubuntu, CentOS, macOS (Intel CPU), and Windows (Table 3). The pipeline has been built to handle large experiments containing tens of samples by parallelizing the workflow, making it adaptable to different experimental designs. Overall, implementing CATCH-UP in the analysis of ChIP-seq and ATAC-seq data enables a user-friendly, reproducible, and highly adaptable analysis workflow.
Disclosures
J.R.H. is a co-founder and director of Nucleome Therapeutics and provides consultancy to the company.
Acknowledgements
J.R.H. was supported by grants from the Wellcome Trust (225220/Z/22/Z and 106130/Z/14/Z) and the MRC (MC_UU_00029/3). M.B. was supported by the Wellcome Trust grant (225220/Z/22/Z). E.R.G was supported by The Ministry of National Education Selection and Placement of Candidates Sent Abroad for Postgraduate Education (YLSY) scholarship, Republic of Türkiye Ministry of National Education. E.G. was supported by the Wellcome Genomic Medicine and Statistics PhD Programme (108861/Z/15/Z). S.G.R. was supported by the Medical Research Council (MRC) grant (MC_UU_00029/3).
Materials
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
References
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623(2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521(2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037(2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101(2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537(2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008(2021).
- Picard Toolkit. , http://broadinstitute.github.io/picard/ (2019).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved