
Method Article
CATCH-UP: ATAC-SEQ 및 ChIP-Seq 대량 데이터를 위한 고처리량 업스트림 파이프라인
요약
ATAC-seq 및 ChIP-seq는 유전자 조절에 대한 자세한 조사를 가능하게 합니다. 그러나 이러한 데이터 유형을 처리하는 것은 까다롭고 연구 그룹 간에 일관성이 없는 경우가 많습니다. 우리는 CATCH-UP을 제시합니다: 표준화되고 재현 가능한 데이터 처리 및 신규 및 게시된 ATAC/ChIP-seq 데이터 세트의 분석을 가능하게 하는 사용하기 쉬운 컴퓨팅 파이프라인입니다.
초록
차세대 염기서열분석(NGS)과 결합된 전이효소 접근 가능 염색질(ATAC) 및 염색질 면역침전(ChIP)에 대한 분석은 유전자 조절 연구에 혁명을 일으켰습니다. 이러한 기술에 의해 생성된 고차원 데이터 세트 분석의 표준화가 부족하면 재현성을 달성하기가 어려워져 게시 및 처리된 데이터에 불일치가 발생했습니다. 이 문제의 일부는 이러한 유형의 데이터 분석에 사용할 수 있는 다양한 생물정보학 도구에 기인합니다. 둘째, 원시 데이터를 완전히 처리되고 해석 가능한 출력으로 변환하기 위해 다양한 생물정보학 도구가 순차적으로 필요하며, 이러한 도구에는 다양한 수준의 계산 기술이 필요합니다. 또한 데이터 처리 중에 균일하게 사용되지 않는 품질 관리를 위한 많은 옵션이 있습니다. 자사는 전이효소 접근 가능한 염색질 염기서열분석(ATAC-seq) 및 염색질 면역침전성 염기서열분석(ChIP-seq) 업스트림 파이프라인(CATCH-UP)을 위한 완전한 분석법으로 이러한 문제를 해결하며, 원시 fastq 파일에서 시각화 가능한 bigwig 트랙 및 피크 콜에 이르기까지 대량 ChIP-seq 및 ATAC-seq 데이터 세트를 분석하기 위한 사용하기 쉬운 Python 기반 파이프라인입니다. 이 파이프라인은 설치 및 실행이 간단하여 최소한의 컴퓨팅 지식이 필요합니다. 파이프라인은 모듈식이며 다양한 컴퓨팅 인프라에서 확장 가능하고 병렬화할 수 있으므로 방법론을 쉽게 보고할 수 있으므로 신규 또는 게시된 데이터 세트를 재현 가능한 분석할 수 있습니다.
서문
세포가 올바른 생물학적 기능을 확립하고 유지하기 위해서는 유전자 발현을 엄격하게 조절해야 합니다. 비정상적인 유전자 발현이 많은 질병의 발병기전의 기저에 있다는 것은 잘 알려진 사실이며, 따라서 유전자 조절1의 메커니즘을 이해하는 데 많은 연구가 관심을 기울이고 있습니다. 유전자 발현은 프로모터(promoter) 및 인핸서(enhancer)와 같은 조절 요소에 의해 촉진됩니다. 염기서열 내에서 이러한 요소에는 전사 인자(TF) 결합 부위가 포함되어 있으며, 이 부위는 활성화될 때 TF 결합을 위한 플랫폼을 제공합니다. 이러한 부위에서 TF가 결합하면 뉴클레오솜이 변위되어 DNA 접근성이 증가하고 그에 따라 전사 기계에 대한 허용율이 증가합니다. 이러한 접근성 증가의 결과로, DNA의 이러한 영역은 전사 조절 2,3을 조사하는 연구자들에 의해 이용된 생화학적 특성인 DNase 및 Tn5와 같은 뉴클레아제 및 전이효소에 더 민감합니다.
DNase-seq 및 ATAC-seq를 통해 연구자는 게놈 전반에 걸쳐 열린 염색질 영역, TF 결합 부위 및 뉴클레오솜 위치를 매핑할 수 있습니다. 이 두 가지 기법 중 ATAC-seq는 간단한 2단계 프로토콜과 낮은 세포 수 요구 사항(DNase-seq의 경우 복제당 100만 개의 세포에 비해 50,000개의 세포)으로 인해 지난 10년 동안 인기가 높아졌습니다. ATAC-seq는 세포 집단의 일반적인 염색질 지형에 대한 개요를 제공하지만, 특정 단백질이 게놈에 결합하는 것은 대체로 불가지론적입니다 4,5. 특정 단백질이 게놈과 상호 작용하는 위치를 식별하기 위해 황금 표준 기술은 염색질 면역침전(ChIP)-seq입니다. ChIP-seq는 세포 내 단백질-DNA 상호작용을 화학적으로 고정한 다음, 관심 단백질에 특이적인 항체를 사용하여 관심 단백질(POI)에 결합된 DNA 단편을 선택하는 면역침전("풀다운")을 포함합니다. 이러한 DNA 단편은 TF와 같은 특정 단백질 또는 특정 히스톤 변형을 포함하는 부위의 게놈 결합 위치를 밝히기 위해 염기서열을 분석할 수 있습니다1. ATAC-seq 및 ChIP-seq 데이터 세트를 결합하면 세포 집단에 대한 조절 환경에 대한 자세한 그림을 도출할 수 있습니다.
분석에 필요한 기본 워크플로우는 다음과 같습니다: 원시 염기서열분석 판독은 참조 게놈에 정렬("매핑")하기 전에 품질 관리를 받아야 합니다. 그런 다음 성공적으로 매핑된 읽기를 필터링하여 저품질 읽기와 PCR 중복을 모두 제거할 수 있습니다. 이러한 매핑되고 필터링된 판독을 시각화하려면 게놈 전체에서 이러한 판독의 "범위"를 계산해야 합니다. 이렇게 하면 MLV(Multi-locus view) 또는 UCSC 게놈 브라우저와 같은 게놈 브라우저에 "트랙"으로 업로드할 수 있는 파일이 생성됩니다6,7. 이러한 커버리지 트랙의 피크 식별 또는 "피크 콜링"은 일반적으로 LanceOtron 또는 MACS2 8,9와 같은 도구를 사용하여 달성됩니다. 마지막으로, 피크 위치, 모양 및 크기 분석을 통해 샘플 또는 생물학적 조건 간에 비교를 수행할 수 있습니다. 이러한 데이터 세트의 분석 및 통합은 생물 정보학 도구의 다양한 조합을 구현할 수 있는 복잡한 다단계 프로세스입니다. 다른 버전의 도구는 서로 호환되지 않을 수 있으며 데이터 처리의 출력을 변경할 수 있습니다. 또한 nf-core10, panpipes11, genpipes12, PEPATAC13 또는 ChIP-AP14 파이프라인에서 볼 수 있듯이 데이터 처리의 다양한 부분을 구현하는 데 필요한 계산 능력과 사용자 숙련도도 매우 다양합니다.
전반적으로 이로 인해 분석과 분석 보고 모두에서 불일치가 발생했으며, 그 결과 생물정보학에 대한 지식이 부족한 사람에게 재현성, 접근성 및 편의성이 저하되었습니다. 당사는 ChIP-seq 및 ATAC/DNase-seq 데이터를 처리하기 위한 사용하기 쉽고 유연한 모듈식 파이프라인인 CATCH-UP(완전한 ATAC-seq 및 ChIP-seq 업스트림 파이프라인)을 통해 이러한 모든 문제를 해결합니다. CATCH-UP을 구현하려면 최소한의 생물정보학 경험이 필요합니다. 다양한 컴퓨팅 인프라에서 실행할 수 있으며 연구 그룹 내에서 그리고 연구 그룹 간에 재현 가능한 데이터 분석을 가능하게 합니다.
CATCH-UP은 ChIP-seq 및 ATAC-seq 데이터 분석을 표준화하기 위해 구축된 Python 기반 Snakemake 파이프라인입니다. 원시 염기서열 분석 데이터(fastq.gz 파일)를 입력으로 사용하고 각 단계에 대한 각각의 결과를 제공하는 피크(.bed) 파일 형식의 출력을 생성합니다. 사용자가 각 분석 단계의 매개변수를 편집할 수 있는 yaml 형식(config.yaml)의 구성 파일을 제공합니다. Snakemake 내에 구현된 관리 시스템을 사용하면 다양한 컴퓨팅 인프라(예: 서버, 클러스터, 클라우드 시스템 또는 개인용 컴퓨터)를 사용할 수 있으며 사용자가 많은 양의 데이터를 제공하는 경우 병렬로 사용할 수 있습니다.
아래에는 워크플로우의 각 단계에 대한 자세한 설명이 나와 있습니다(워크플로우 그림은 그림 1 참조). 이 설명은 프로토콜 섹션의 단계별 지침을 따르기 위해 필수적입니다.
fastq 이동: 파이프라인의 첫 번째 단계는 원시 fastq 파일을 명명된 분석 디렉터리에 복사하는 것입니다. 이렇게 하면 원시 데이터 파일이 손상되거나 수정되는 것을 방지하기 위해 원본 데이터는 그대로 유지됩니다.
연결: 원시 염기서열분석 데이터에 여러 레인이 포함된 경우 이 단계는 분석 전에 레인을 연결하는 데 필요합니다. 기본적으로 파이프라인은 모든 fastq 파일을 단일 샘플로 처리합니다. 이 연결 단계는 구성 파일에서 정의해야 합니다.
트리밍: 선택적 데이터 정리 단계. 이를 통해 trimmomatic15를 사용하여 저품질 읽기 또는 어댑터 시퀀스를 트리밍할 수 있습니다. 사용자는 어댑터 시퀀스의 사용자 정의 fasta 파일을 제공할 수 있습니다. 예제는 adapter 디렉토리에 제공됩니다. 추가 트리밍 매개변수는 구성 파일에서 정의할 수 있습니다. 기본적으로 워크플로는 이 규칙을 건너뜁니다.
얼라이너: 얼라인먼트의 경우 기본적으로 Bowtie216 이 적용됩니다. BWA-MEM217 과 같은 대체 정렬 도구도 지정할 수 있습니다. Bowtie2 정렬 도구는 상대적으로 짧은 판독값을 상대적으로 큰 게놈에 정렬하는 데 특히 능숙하기 때문에 기본적으로 선택되므로 ChIP-seq 및 ATAC-seq 데이터를 포유류 게놈에 정렬하는 데 적합합니다. 중간 파일을 피하기 위해 aligner는 bam 파일을 출력에 저장하기 위해 samtools 뷰로 파이프됩니다. 이 규칙의 경우 사용자는 판독값을 매핑할 기본 게놈 빌드(예: hg19/hg38(인간), mm10/mm39(마우스))를 지정해야 합니다.
필터링: 적절하게 매핑된 읽기는 유지되고 품질이 낮은 읽기는 필터링됩니다. 기본값: samtools 보기, 매개 변수 포함: -bShuF 4 -f 3 -q 30.
정렬: 정렬된 읽기는 가장 왼쪽 좌표 순서로 정렬됩니다. 기본값: samtools sort(snakemake wrapper), 매개변수 사용: -m 4G.
Mark duplicates: 모든 중복 읽기가 식별되고 플래그가 지정됩니다. 사용자는 구성 파일 매개변수를 변경하여 제거하도록 결정할 수 있습니다. 기본값: Picard MarkDuplicates (snakemake wrapper), 매개 변수: --REMOVE_DUPLICATES False 를 사용하여 중복을 표시하고 유지합니다.
bam 병합: 염기서열분석 데이터가 반복 실험 또는 샘플로 구성된 경우 사용자는 단일 bam으로 병합할 수 있습니다. 이 경우 사용자는 bam을 병합하거나 분석 전반에 걸쳐 bam 파일을 별도로 유지하도록 선택할 수 있습니다. 사용자가 bam을 병합하도록 선택하는 경우(samtools merge 사용) 병합된 bam에 대해 공통 접두사를 지정해야 합니다.
인덱스: 이 단계는 정렬된 좌표를 인덱싱합니다. 기본값: samtools 인덱스(snakemake wrapper), samtools에서 지정한 기본 매개변수 사용.
BamCoverage: 이 규칙은 정렬된 읽기에서 bigwig 커버리지 트랙을 만듭니다. deepTools의 bamCoverage 도구가 적용되고 coverage는 bin당 읽기 수로 계산되며, bin은 지정된 크기의 창을 나타냅니다. 이 파이프라인에서 bamCoverage는 기본적으로 -bs 1 -normalizeUsing RPKM -extendReads 매개변수와 함께 적용됩니다.
최대 호출: LanceOtron8 이 이 파이프라인의 기본 피크 호출자로 선택되었습니다. 대부분 통계 기반 피크 콜러와 달리 LanceOtron은 게놈 농축 측정 및 통계 테스트를 통합하고 업계 표준 피크 콜러인 MACS29를 능가하는 것으로 나타난 딥 러닝 기반 피크 콜러입니다. 거물들이 LanceOtron과 호환되려면 커버리지가 염기쌍별로 계산되고 RPKM이 정규화되어야 합니다. 이는 BamCoverage 단계의 기본 설정에 반영됩니다. MACS2는 대체 피크 호출자로 선택할 수 있습니다. 새로운 피크 호출자의 릴리스는 이 분석 파이프라인의 성능을 유지하고 최적화하기 위해 모니터링되고 해당되는 경우 통합됩니다.
TrackDb: MLV6 또는 UCSC Genome Browser18 플랫폼과 같은 도구에서 파일을 로드하고 시각화하기 위해 bigwig 파일의 키-값 쌍 연결을 만듭니다.
출력 데이터 외에도 파이프라인의 각 단계는 로그 파일을 출력하고 사용자가 분석 진행 상황을 추적할 수 있도록 적절한 품질 관리 검사가 제공됩니다. FastQC19는 원시 및 트리밍된(선택한 경우) 염기서열분석 데이터(1단계 - Fastq 이동 및 2단계 - 트리밍)에 적용됩니다. Samtools stats plus MultiQC20은 3 - Aligner, 6 - Mark duplicates 및 7 - Merge bam의 출력에서 bam 파일에 대한 품질 관리 보고서를 수집, 생성 및 시각화하는 데 사용됩니다. 위 단계에서 적용된 각 도구에 대한 자세한 내용은 표 1을 참조하십시오.
프로토콜
1. CATCH-UP 파이프라인 실행
- https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline 에서 UpStreamPipeline 저장소를 복제합니다.
선택한 작업 디렉터리로 이동하여 다음 코드를 복사하고 명령줄에서 실행합니다.
자식 클론 git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - cd UpStreamPipeline 명령을 사용하여 다운로드한 UpStreamPipeline 폴더 내부를 탐색합니다.
- 아나콘다 배포판을 설치합니다(필요한 경우).
- which conda 명령을 사용하여 anaconda가 이미 시스템에 설치되어 있는지 확인합니다. 명령에 conda 배포에 대한 경로가 표시되지 않으면 https://github.com/conda-forge/miniforge#mambaforge 에서 mambaforge를 다운로드하고 시스템에 적합한 배포 및 버전을 선택합니다. 예를 들어 Linux 사용자의 경우 wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh 사용합니다. 다양한 운영 체제에 대한 웹 페이지를 방문하십시오 : https://github.com/conda-forge/miniforge/.
- sh Mambaforge-Linux-x86_64.sh를 사용하여 설치 프로그램을 실행하고 conda init를 실행하여 시스템에서 conda를 초기화합니다.
- 업스트림 conda 환경을 설치하고 활성화합니다(업스트림 conda 환경의 요구 사항은 표 2에 나열되어 있음).
- mamba env create - file=envs/upstream.yml 명령을 사용하여 환경을 설치합니다.
- conda activate upstream 명령을 사용하여 환경을 활성화합니다.
- 업스트림 conda 환경이 성공적으로 설치되면 conda activate upstream 명령을 사용하여 환경을 활성화하고 cd genetics/CATCH-UP을 사용하여 CATCH-UP 폴더로 이동합니다.
- cd /config/analysis.yaml 명령을 사용하여 config 폴더 내에서 찾을 수 있는 구성 파일을 편집하고 텍스트 편집기를 사용하여 분석 사양에 따라 수정합니다. 줄별 지침에 따라 파일 자체 내의 각 매개 변수를 편집합니다. 이 파일은 분석 후에도 유지되며 재현성을 돕기 위해 실행 매개변수를 문서화하는 역할을 합니다.
- 텍스트 편집기에서 다음 세 개의 파일을 열고 편집합니다(예: Mac의 경우 TextEdit 또는 Windows의 경우 Notepad).
- 분석할 모든 fastq 파일 목록을 포함하도록 1_fastqfile_home_dir.txt 파일을 편집합니다.
참고: 읽기 번호 및 확장자(예: _R1/_R2 및 .fastq.gz)는 제외해야 합니다. 예를 들어, 프로젝트에 다음과 같은 fastq 파일 목록이 포함되어 있는 경우:
Sample1_conditionA_L001_R1 . 패스트큐 . GZ
Sample1_conditionA_L001_R2 . 패스트큐 . GZ
Sample1_conditionA_L002_R1 . 패스트큐 . GZ
Sample1_conditionA_L002_R2 . 패스트큐 . GZ
Sample1_conditionB_L001_R1 . 패스트큐 . GZ
Sample1_conditionB_L001_R2 . 패스트큐 . GZ
Sample1_conditionB_L002_R1 . 패스트큐 . GZ
Sample1_conditionB_L002_R2 . 패스트큐 . GZ
Sample2_conditionA_L001_R1 . 패스트큐 . GZ
Sample2_conditionA_L001_R2 . 패스트큐 . GZ
Sample2_conditionA_L002_R1 . 패스트큐 . GZ
Sample2_conditionA_L002_R2 . 패스트큐 . GZ
Sample2_conditionB_L001_R1 . 패스트큐 . GZ
Sample2_conditionB_L001_R2 . 패스트큐 . GZ
Sample2_conditionB_L002_R1 . 패스트큐 . GZ
Sample2_conditionB_L002_R2 . 패스트큐 . GZ
이 경우 1_fastqfile_home_dir.txt 사항은 다음과 같습니다.
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - 원시 데이터에 연결이 필요한 시퀀싱 레인이 포함되어 있는 경우 2_fastq le_concat.txt 파일을 편집하여 연결할 파일 이름의 접두사를 정의합니다. 연결할 시퀀싱 레인이 없는 경우 2_fastq le_concat.txt 편집하지 마십시오. 2_fastqfile_concat.txt의 모든 줄에 다음과 같이 하나의 샘플 접두사가 포함되어 있는지 확인합니다.
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - 서로 다른 샘플의 데이터를 병합해야 하는 경우 병합할 파일 이름의 접두사 3_merge_bams.txt 사용하여 파일을 편집합니다. 모든 줄에 다음과 같이 하나의 샘플 접두사가 포함되어 있는지 확인합니다.
샘플1
샘플2
그림 2 는 이러한 세 가지 파일을 요약하는 방법을 요약한 것입니다. 이 프로토콜은 단일 또는 쌍단 염기서열분석 데이터에 적용할 수 있습니다. 파이프라인은 달리 지정되지 않는 한 기본적으로 쌍단 분석으로 설정됩니다. 이는 구성 파일에서 수정할 수 있습니다(1.6단계 참조).
- 분석할 모든 fastq 파일 목록을 포함하도록 1_fastqfile_home_dir.txt 파일을 편집합니다.
- 필요한 모든 파일을 편집한 후 snakemake를 사용하여 snakemake --configfile=config/analysis_name.yaml all --cores 4와 같이 CATCH-UP을 실행합니다.
참고: 자세한 지침 및 설명서는 여기에서 사용할 수 있는 UpStreamPipeline GitHub 리포지토리 내의 CATCH-UP 폴더를 참조하십시오. 여기에는 데이터 파일 시퀀싱 경로 변경 및 결과 저장부터 각 단계의 매개 변수 편집에 이르기까지 구성 파일을 올바르게 수정하는 방법에 대한 자세한 설명서가 포함되어 있습니다.
결과
CATCH-UP 파이프라인은 각 단계에 대한 결과, 로그 및 품질 관리(QC) 출력을 생성합니다. 구성 파일 내에서 사용자는 필요한 스토리지 메모리를 줄이기 위해 출력 파일을 유지하거나 제거하도록 선택할 수 있습니다. 모든 출력은 다음과 같이 설명됩니다.
00. fastq_home_dir: 구성 파일, fastqfile_home_dir.txt 및 merge_bams.txt 참조 및 재현을 위해 이 폴더에 복사됩니다.
01. 읽기 : 워크 플로우 프로세스 중에 원본 원시 데이터의 변경을 방지하기 위해 FastQ 파일이이 폴더에 복사되며, 지정된 경우 레인을 연결할 수 있습니다.
02. 트리밍 : 지정된 경우 읽기 및 어댑터가 트리밍 된 FastQ 파일.
03. aligner: 선택한 게놈에 대한 정렬.
04. 여과 : 품질 관리 필터링.
05. Sorted: BAM 파일의 정렬.
06. 중복: 중복에 플래그를 지정합니다.
07. merge: config.yaml에 지정된 경우 BAM 파일을 병합합니다.
08. bam_coverages : 커버리지의 bigwig 파일.
09. peak_calling: LanceOtron 피크 호출 출력의 베드 파일.
10. track: 필요한 경우 Genome Browser에서 사용할 수 있는 서식이 지정된 텍스트 파일을 생성합니다.
01, 02, 03, 06 및 07 출력의 경우 QC 메트릭 및 HTML 파일이 제공됩니다. 또한 그림 3에서는 MLV 플랫폼을 통해 최종 출력을 시각화하여 CATCH-UP을 사용하여 처리된 데이터의 예를 제공합니다.

그림 1: CATCH-UP의 워크플로우. fastq 파일 목록이 주어지면 CATCH-UP은 모든 업스트림 단계를 통해 모든 샘플을 병렬로 처리합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
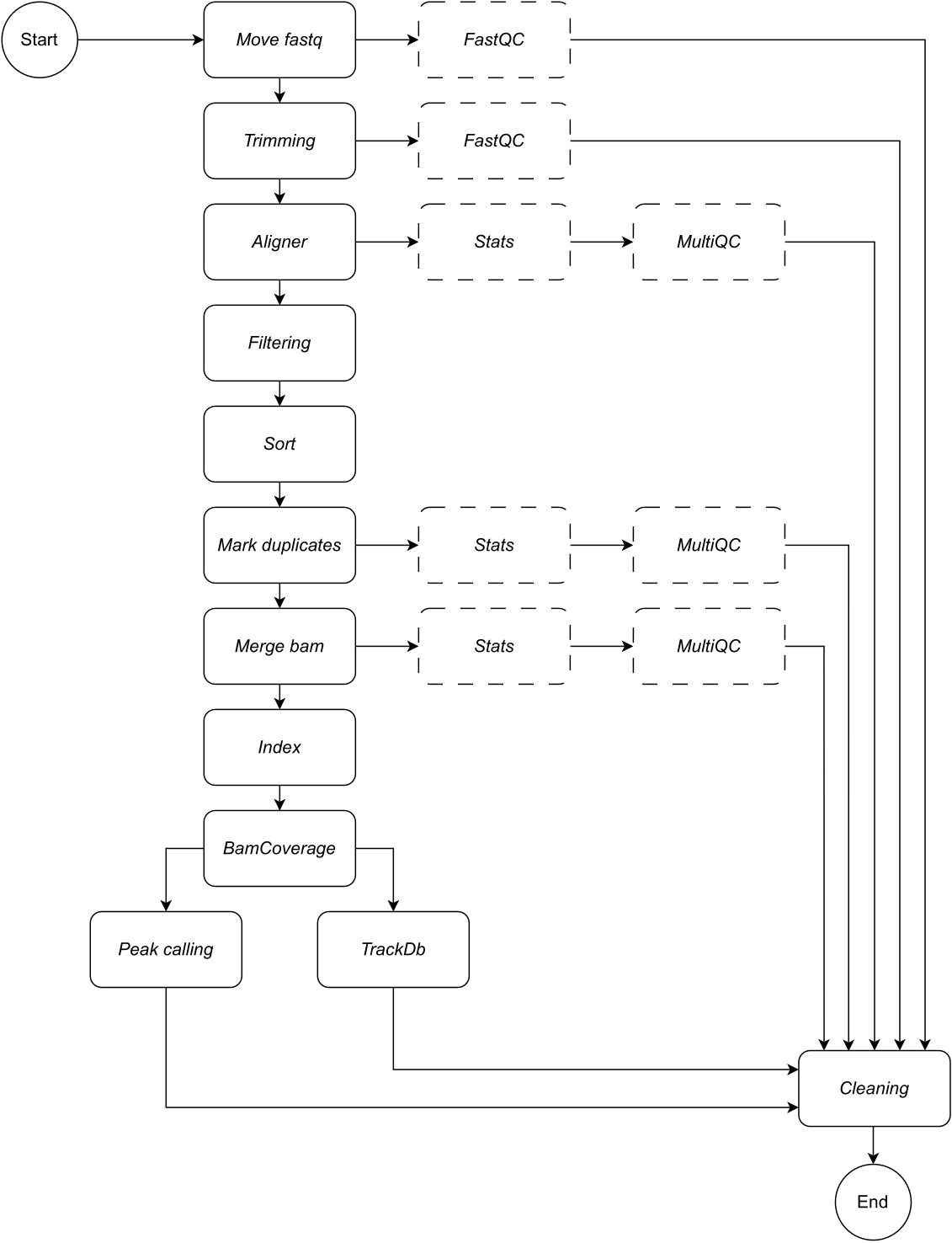{kind=link}

그림 2: CATCH-UP을 실행하기 위해 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt 및 3_merge_bams.txt를 올바르게 수정해야하는 방법을 설명하는 예시 표현. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 3: CATCH-UP 파이프라인의 출력 예. 원시 염기서열분석 데이터(fastq 파일)는 ENCODE21에서 다운로드되었습니다. DNase-seq 및 5가지 유형의 ChIP-seq(H3K4me1, H3K4me3, H3K27ac, CTCF 및 POLR2A)에 대한 fastq 파일을 처리하기 위해 CATCH-UP 파이프라인을 사용했습니다. Bigwig 출력 파일은 게놈 조절 요소의 시각화 및 식별을 위해 Multi Locus View에 업로드되었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
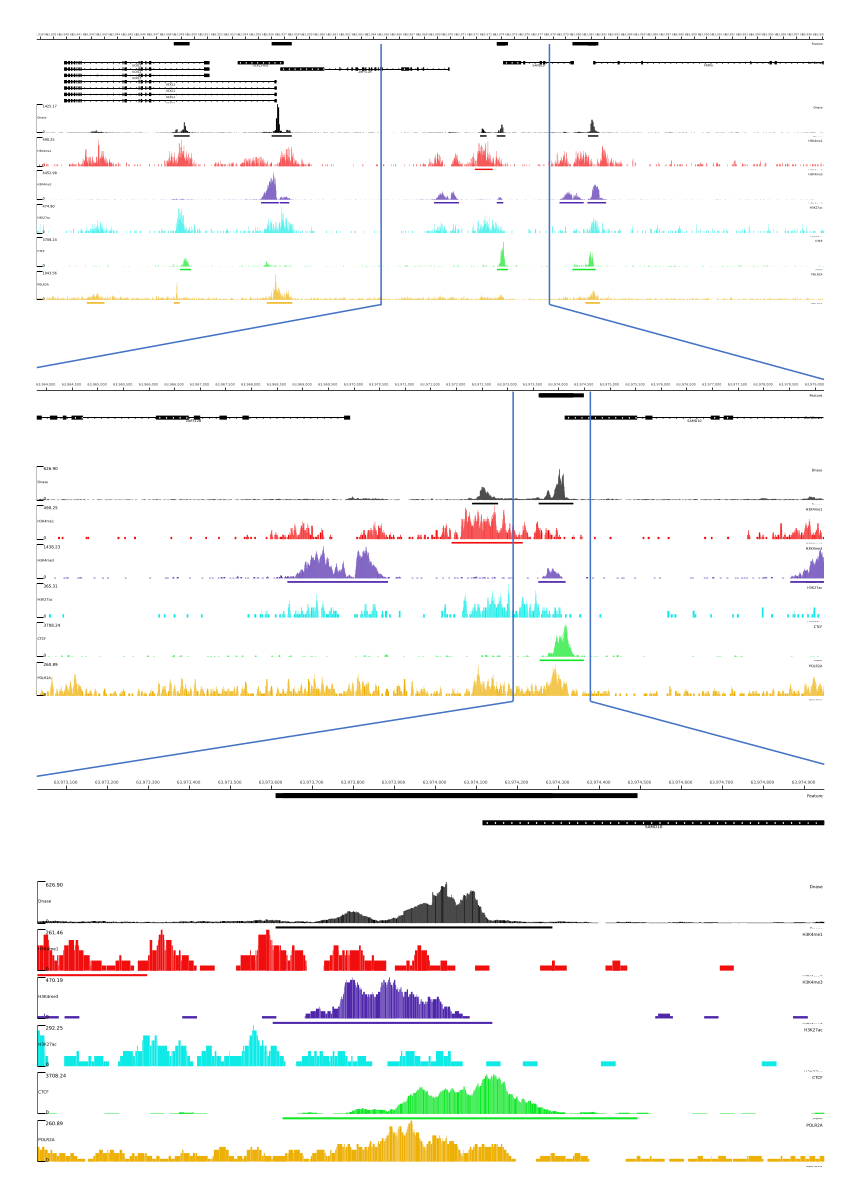{kind=link}
표 1: 설명서 리소스. 이 표에는 CATCH-UP 워크플로와 관련된 도구, 해당 설명서에 대한 링크 및 해당 참조가 표시됩니다. 이 표를 다운로드하려면 여기를 클릭하십시오.
표 2: 업스트림 conda 환경에 대한 채널 및 종속성 요구 사항 목록. 이 표를 다운로드하려면 여기를 클릭하십시오.
표 3: CATCH-UP을 테스트하는 데 사용되는 운영 체제. Ubuntu는 고성능 클러스터 및 로컬 컴퓨터에서 테스트되었습니다. 이 표를 다운로드하려면 여기를 클릭하십시오.
토론
게놈 데이터를 생성하기 위한 NGS 기술의 활용 및 활용이 증가함에 따라 이러한 데이터 분석을 위한 생물정보학 도구의 개발이 증가하고 있습니다. 데이터 분석의 각 단계에 적용할 수 있는 여러 도구와 각 도구 내에서 지정할 수 있는 다양한 매개변수가 있습니다 6,8,9,15,16,17,18,19,20,22,23,24 . 이로 인해 적용할 수 있는 분석 전략이 매우 다양하게 조합될 수 있으며, 각 전략은 결과에 차이를 생성할 수 있습니다. 여러 실험을 정확하게 비교하기 위해서는 생물정보학 분석의 표준화가 필수적입니다. 역사적으로 NGS 데이터는 습식 실험실 과학자에 의해 생성되고 데이터는 생물 정보학자에 의해 분석됩니다.
NGS 데이터 분석은 "업스트림" 파이프라인과 "다운스트림" 파이프라인으로 나눌 수 있으며, 여기서 업스트림에는 염기서열분석 기계의 원시 데이터 출력에서 연구자가 시각적으로 해석할 수 있는 형식으로 이동하는 데 필요한 단계가 포함됩니다. 다운스트림 분석에는 연구 질문 및 실험 설계에 맞춤화된 추가 단계가 포함됩니다. 따라서 업스트림 파이프라인은 일반화할 수 있으며 과학적 재현성 향상을 위해 표준화에 적합합니다. 반면에 다운스트림 파이프라인은 맞춤형이고 생물학적 질문에 의존하며 연구자의 통찰력이 필요하기 때문에 표준화에 적합하지 않습니다. 당사는 습식 실험실 과학자들이 생물정보학에 대한 사전 지식 없이도 자신의 데이터를 재현 가능하게 분석할 수 있도록 하는 사용자 친화적인 업스트림 파이프라인을 만들었습니다. 여기에서는 snakemake 프레임워크를 사용하여 구축되고 사용자 친화적이고 ChIP-seq 및 ATAC-seq 데이터 분석의 재현성 문제를 해결하도록 설계된 파이프라인인 CATCH-UP을 소개합니다. 이 파이프라인은 ChIP-seq 또는 ATAC-seq 데이터를 처리하도록 구축되었습니다. 사용자가 CATCH-UP을 다운로드하면 한 줄의 코드를 사용하여 명령줄에서 파이프라인을 실행하기 전에 먼저 분석 매개 변수와 샘플 이름을 정의해야 합니다. ChIP-seq 또는 ATAC-seq 분석을 위한 분석 매개변수를 사용자 정의하는 방법에 대한 간단한 단계별 지침은 구성 파일 자체 및 CATCH-UP GitHub 저장소의 단계별 가이드에 제공됩니다.
PEPATAC 및 ChIP-AP와 같은 ChIP-seq 또는 ATAC-seq 데이터에 대한 기존 분석 파이프라인이 있습니다. 이러한 파이프라인은 업스트림 및 다운스트림 분석을 단일 워크플로우에 통합하거나 그래픽 사용자 인터페이스(GUI)를 사용하는 등의 장점이 있지만, 이러한 도구는 중간 수준의 컴퓨터 교육을 받은 생물정보학자 및 과학자를 대상으로 합니다13,14. CATCH-UP은 생물정보학 교육을 받지 않은 습식 실험실 과학자가 자체 업스트림 분석을 수행할 수 있도록 하고, 실험실 전반에 걸쳐 간편한 보고와 정확한 재현성을 촉진하여 업스트림 분석의 표준화를 가능하게 하는 두 가지 문제를 해결하기 위해 설계되었습니다. CATCH-UP은 의도적으로 업스트림 분석으로 제한되지만 출력은 데이터 세트를 통계적으로 비교하거나 전사 인자 결합25,26을 추론하는 데 사용되는 도구와 같은 다운스트림 분석 도구와 호환됩니다.
복제 가능한 업스트림 분석을 수행하는 데 필요한 모든 중요한 단계는 견고성을 보장하기 위해 CATCH-UP 파이프라인 내에 사전 정의되어 있습니다. 이 파이프라인의 자세한 특성으로 인해 사용자는 파이프라인의 출력을 단계별로 따를 수 있으며, 이는 문제를 해결하고 분석 워크플로를 복제할 수 있도록 하는 데 유용합니다. NGS 기술의 빠르게 발전하는 특성을 감안할 때 이 파이프라인의 모듈식 특성은 도구 버전 업데이트 릴리스와 새로운 도구 구현을 모두 통합하도록 쉽게 조정할 수 있는 기능을 제공하므로 유용합니다. CATCH-UP은 Ubuntu, CentOS, macOS(Intel CPU) 및 Windows 운영 체제에 대해 성공적으로 테스트되었습니다(표 3). 파이프라인은 워크플로우를 병렬화하여 수십 개의 샘플이 포함된 대규모 실험을 처리할 수 있도록 구축되어 다양한 실험 설계에 적용할 수 있습니다. 전반적으로 ChIP-seq 및 ATAC-seq 데이터 분석에서 CATCH-UP을 구현하면 사용자 친화적이고 재현 가능하며 적응력이 뛰어난 분석 워크플로를 구현할 수 있습니다.
공개
J.R.H.는 Nucleome Therapeutics의 공동 창립자이자 이사이며 회사에 컨설팅을 제공합니다.
감사의 말
J.R.H.는 웰컴 트러스트(Wellcome Trust, 225220/Z/22/Z 및 106130/Z/14/Z)와 MRC(MC_UU_00029/3)의 보조금으로 지원되었습니다. M.B.는 Wellcome Trust 보조금(225220/Z/22/Z)의 지원을 받았습니다. ERG는 튀르키예 공화국 국가 교육부(Republic of Türkiye Ministry of National Education)의 대학원 교육(YLSY) 장학금을 위해 해외로 파견된 후보자의 국가 교육 선발 및 배치부의 지원을 받았습니다. 예를 들어, Wellcome Genomic Medicine and Statistics PhD Programme(108861/Z/15/Z)의 지원을 받았습니다. S.G.R.은 MRC(Medical Research Council) 보조금(MC_UU_00029/3)의 지원을 받았습니다.
자료
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
참고문헌
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유