
Method Article
CATCH-UP: Um pipeline upstream de alta taxa de transferência para dados ATAC-Seq e ChIP-Seq em massa
Neste Artigo
Resumo
ATAC-seq e ChIP-seq permitem uma investigação detalhada da regulação gênica; no entanto, o processamento desses tipos de dados é desafiador e muitas vezes inconsistente entre os grupos de pesquisa. Apresentamos o CATCH-UP: um pipeline computacional fácil de usar que permite o processamento e análise de dados padronizados e reprodutíveis de conjuntos de dados ATAC/ChIP-seq novos e publicados.
Resumo
O ensaio para cromatina acessível por transposase (ATAC) e imunoprecipitação da cromatina (ChIP), juntamente com o sequenciamento de próxima geração (NGS), revolucionaram o estudo da regulação gênica. A falta de padronização na análise dos conjuntos de dados altamente dimensionais gerados por essas técnicas dificultou a reprodutibilidade, levando a discrepâncias nos dados publicados e processados. Parte desse problema se deve à diversidade de ferramentas de bioinformática disponíveis para a análise desses tipos de dados. Em segundo lugar, várias ferramentas bioinformáticas diferentes são necessárias sequencialmente para converter dados brutos em uma saída totalmente processada e interpretável, e essas ferramentas requerem níveis variados de habilidades computacionais. Além disso, existem muitas opções de controle de qualidade que não são empregadas uniformemente durante o processamento de dados. Abordamos esses problemas com um ensaio completo para sequenciamento de cromatina acessível por transposase (ATAC-seq) e sequenciamento de imunoprecipitação de cromatina (ChIP-seq) pipeline upstream (CATCH-UP), um pipeline baseado em Python fácil de usar para a análise de conjuntos de dados ChIP-seq e ATAC-seq em massa de arquivos fastq brutos para faixas de figurão visualizáveis e chamadas de picos. Esse pipeline é simples de instalar e executar, exigindo conhecimento computacional mínimo. O pipeline é modular, escalável e paralelizável em várias infraestruturas de computação, permitindo fácil relatório de metodologia para permitir a análise reproduzível de conjuntos de dados novos ou publicados.
Introdução
A expressão gênica deve ser rigidamente regulada para que as células estabeleçam e mantenham sua função biológica correta. É bem sabido que a expressão gênica aberrante está subjacente à patogênese de muitas doenças e, portanto, um grande interesse de pesquisa reside na compreensão dos mecanismos de regulação gênica1. A expressão gênica é facilitada por elementos regulatórios, como promotores e intensificadores. Dentro de sua sequência, esses elementos contêm locais de ligação do fator de transcrição (TF), que, quando ativos, fornecem uma plataforma para a ligação do TF. A ligação de TFs nesses locais resulta em um deslocamento de nucleossomos, resultando em um aumento na acessibilidade do DNA e um subsequente aumento na permissibilidade à maquinaria transcricional. Como resultado dessa maior acessibilidade, essas regiões do DNA são mais sensíveis a nucleases e transposases como DNase e Tn5, uma propriedade bioquímica que tem sido explorada por pesquisadores que investigam a regulação transcricional 2,3.
DNase-seq e ATAC-seq permitem que os pesquisadores mapeiem regiões de cromatina aberta, locais de ligação de TF e posicionamento nucleossômico em todo o genoma. Dessas duas técnicas, o ATAC-seq cresceu em popularidade na última década devido ao protocolo simples de duas etapas e a um baixo requisito de número de células (50.000 células em comparação com 1 milhão por réplica para DNase-seq). Embora o ATAC-seq forneça uma visão geral da paisagem geral da cromatina em uma população de células, ele é amplamente agnóstico a quais proteínas específicas estão se ligando ao genoma 4,5. Para identificar os locais onde uma proteína específica está interagindo com o genoma, a técnica padrão-ouro é a imunoprecipitação da cromatina (ChIP)-seq. ChIP-seq envolve a fixação química de interações proteína-DNA em uma célula, seguida de imunoprecipitação ("pull-down") usando um anticorpo específico para a proteína de interesse para selecionar fragmentos de DNA ligados pela proteína de interesse (POI). Esses fragmentos de DNA podem ser sequenciados para revelar os locais de ligação genômica de proteínas específicas, como TFs, ou locais contendo modificações específicas de histonas1. Ao combinar conjuntos de dados ATAC-seq e ChIP-seq, uma imagem detalhada do cenário regulatório pode ser derivada para uma população de células.
O fluxo de trabalho básico necessário para a análise é o seguinte: as leituras de sequenciamento bruto devem ser controladas pela qualidade antes do alinhamento com um genoma de referência ("mapeamento"). As leituras mapeadas com sucesso podem então ser filtradas para remover leituras de baixa qualidade e duplicatas de PCR. Para visualizar essas leituras mapeadas e filtradas, é necessário calcular a "cobertura" dessas leituras em todo o genoma. Isso gera um arquivo que pode ser carregado em um navegador de genoma, como visualização multilocus (MLV) ou o navegador de genoma UCSC como uma "trilha" 6 , 7 . A identificação de picos, ou "chamadas de pico" dessas faixas de cobertura, é normalmente obtida usando ferramentas como LanceOtron ou MACS2 8,9. Finalmente, através da análise da localização do pico, podem ser feitas comparações de forma e tamanho entre amostras ou condições biológicas. A análise e integração desses conjuntos de dados é um processo complexo de várias etapas no qual diferentes combinações de ferramentas de bioinformática podem ser implementadas. Diferentes versões das ferramentas podem ser incompatíveis entre si e podem alterar a saída do processamento de dados. Há também uma grande variedade no poder computacional e na proficiência do usuário necessários para implementar diferentes partes do processamento de dados, conforme mostrado nos pipelines nf-core10, panpipes11, genpipes12, PEPATAC13 ou ChIP-AP14.
No geral, isso levou a inconsistências tanto na análise quanto no relato da análise, o que, por sua vez, levou a uma baixa reprodutibilidade, acessibilidade e conveniência para qualquer pessoa com conhecimento limitado de bioinformática. Abordamos todos esses problemas com o CATCH-UP (pipeline upstream completo ATAC-seq e ChIP-seq), um pipeline fácil de usar, flexível e modular para processar dados ChIP-seq e ATAC/DNase-seq. A implementação do CATCH-UP requer experiência mínima em bioinformática; Ele pode ser executado em várias infraestruturas de computação e permite a análise de dados reproduzíveis dentro e entre grupos de pesquisa.
CATCH-UP é um pipeline Snakemake baseado em Python construído para padronizar a análise de dados ChIP-seq e ATAC-seq. Ele usa dados brutos de sequenciamento (arquivos fastq.gz) como entrada e gera uma saída na forma de arquivos de pico (.bed) fornecendo o respectivo resultado para cada etapa. Disponibilizamos um arquivo de configuração em formato yaml (config.yaml), no qual o usuário pode editar os parâmetros de cada etapa de análise. O sistema de gestão implementado no snakemake permite o uso de diferentes infraestruturas computacionais (como servidores, clusters, sistemas em nuvem ou computadores pessoais) e em paralelo se o usuário fornecer uma grande quantidade de dados.
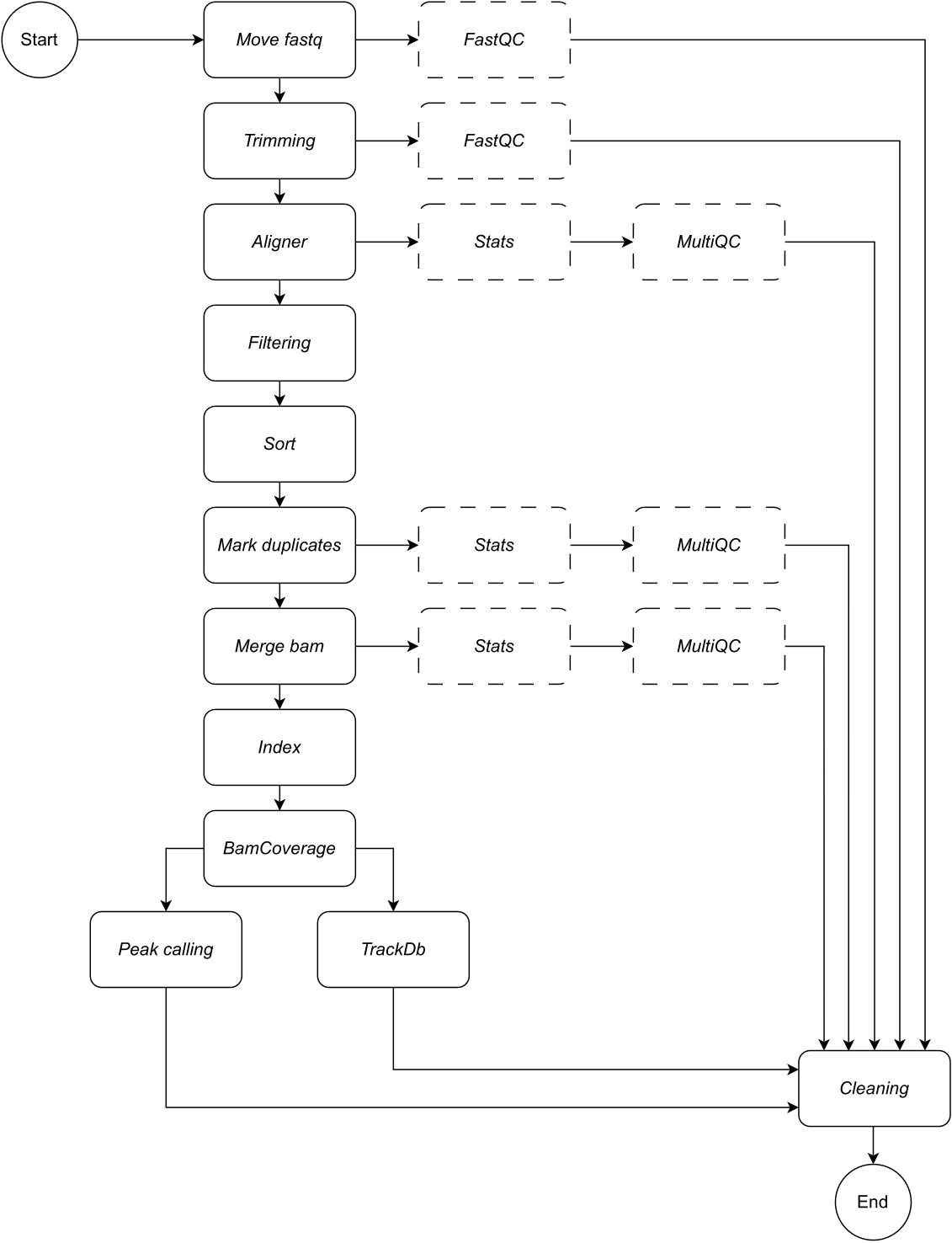
Abaixo, fornecemos uma descrição detalhada de cada etapa do fluxo de trabalho (consulte a Figura 1 para obter a ilustração do fluxo de trabalho). Esta explicação é essencial para seguir o passo a passo na seção de protocolo:
Mover fastq: a primeira etapa do pipeline é copiar os arquivos fastq brutos para o diretório de análise nomeado. Isso deixa os dados originais intocados para evitar corromper ou modificar os arquivos de dados brutos.
Concatenação: se os dados brutos de sequenciamento contiverem várias pistas, esta etapa será necessária para concatenar as pistas antes da análise. Por padrão, o pipeline lida com todos os arquivos fastq como exemplos únicos. Essa etapa de concatenação deve ser definida no arquivo de configuração.
Corte: etapa opcional de limpeza de dados. Isso permite o corte de leituras de baixa qualidade ou sequências de adaptadores usando trimmomatic15. O usuário pode fornecer arquivos fasta personalizados de sequências de adaptadores; Um exemplo é fornecido no diretório do adaptador. Parâmetros de corte adicionais podem ser definidos no arquivo de configuração. Por padrão, o fluxo de trabalho ignora essa regra.
Alinhador: para alinhamento, Gravata borboleta216 é aplicado por padrão; Ferramentas de alinhamento alternativas, como BWA-MEM217 , também podem ser especificadas. A ferramenta de alinhamento Bowtie2 é selecionada como padrão, pois é particularmente adequada para alinhar leituras relativamente curtas a genomas relativamente grandes e, portanto, é adequada para o alinhamento de dados ChIP-seq e ATAC-seq a genomas de mamíferos. Para evitar arquivos intermediários, o alinhador é canalizado para a visualização samtools para salvar o arquivo bam na saída. Para esta regra, o usuário deve especificar a construção do genoma preferida na qual mapear as leituras, por exemplo, hg19 / hg38 (humano), mm10 / mm39 (mouse).
Filtragem: as leituras mapeadas corretamente são retidas e as leituras com baixa qualidade são filtradas. Padrão: visualização samtools, com parâmetros: -bShuF 4 -f 3 -q 30.
Classificar: as leituras alinhadas são classificadas na ordem da coordenada mais à esquerda. Padrão: samtools sort (invólucro snakemake), com parâmetro: -m 4G.
Marcar duplicatas: todas as leituras duplicadas são identificadas e sinalizadas. O usuário pode decidir removê-los alterando o parâmetro do arquivo de configuração. Padrão: Picard MarkDuplicates (wrapper snakemake), com parâmetro: --REMOVE_DUPLICATES False para sinalizar e reter duplicatas.
Mesclar bam: se os dados de sequenciamento forem compostos de réplicas ou amostras, o usuário poderá querer mesclar em um único bam. Nesse caso, o usuário pode optar por mesclar os bams ou manter os arquivos bam separados durante toda a análise. Se o usuário optar por mesclar bams (empregando samtools merge), um prefixo comum deve ser especificado para os bams mesclados.
Índice: esta etapa indexa as coordenadas ordenadas. Padrão: índice samtools (wrapper snakemake), usando parâmetros padrão especificados por samtools.
BamCoverage: esta regra cria uma faixa de cobertura de figurão a partir de leituras alinhadas. A ferramenta bamCoverage do deepTools é aplicada e a cobertura é calculada como o número de leituras por compartimento, no qual o compartimento representa uma janela de um tamanho especificado. Nesse pipeline, bamCoverage é aplicado com os seguintes parâmetros definidos como padrão: -bs 1 -normalizeUsing RPKM -extendReads.
Chamada de pico: LanceOtron8 foi selecionado como o peakcaller padrão para este pipeline. Ao contrário dos chamadores de pico tradicionais, que são principalmente baseados em testes estatísticos, o LanceOtron é um chamador de pico baseado em aprendizado profundo, que incorpora medições de enriquecimento genômico e testes estatísticos e demonstrou superar o chamador de pico padrão da indústria, MACS29. Para que os figurões sejam compatíveis com o LanceOtron, a cobertura deve ser calculada por par de bases e o RPKM normalizado; isso é refletido nas configurações padrão da etapa BamCoverage. O MACS2 pode ser selecionado como um chamador de pico alternativo. A liberação de novos chamadores de pico será monitorada e incorporada conforme aplicável, a fim de manter e otimizar o desempenho desse pipeline de análise.
TrackDb: cria uma associação de pares chave-valor de arquivos bigwig para carregá-los e visualizá-los em ferramentas como as plataformas MLV6 ou UCSC Genome Browser18 .
Além dos dados de saída, cada etapa do pipeline gera um arquivo de log e as verificações de controle de qualidade apropriadas são fornecidas para que o usuário possa acompanhar o progresso da análise. O FastQC19 é aplicado a dados de sequenciamento brutos e aparados (se selecionados) (etapas 1 - Mover fastq e 2 - Aparamento). As estatísticas da Samtools mais o MultiQC20 são usadas para coletar, produzir e visualizar relatórios de controle de qualidade em arquivos bam na saída nas etapas 3 - Alinhador, 6 - Marcar duplicatas e 7 - Mesclar bam. Para obter mais informações sobre cada uma das ferramentas aplicadas nas etapas acima, consulte a Tabela 1.
Protocolo
1. Executando o pipeline CATCH-UP
- Clone o repositório UpStreamPipeline de https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Navegue até o diretório de trabalho escolhido, copie o seguinte código e execute na linha de comando:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Navegue dentro da pasta UpStreamPipeline baixada usando o comando: cd UpStreamPipeline
- Instale a distribuição anaconda (se necessário):
- Verifique se o anaconda já está instalado no sistema usando o comando which conda. Se o comando não mostrar nenhum caminho para nenhuma distribuição conda, baixe o mambaforge do https://github.com/conda-forge/miniforge#mambaforge e selecione a distribuição e a versão corretas para o sistema. Por exemplo, para usuários do Linux, use o wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Visite esta página da web para diferentes sistemas operacionais: https://github.com/conda-forge/miniforge/.
- Execute o instalador usando sh Mambaforge-Linux-x86_64.sh e inicialize o conda no sistema executando conda init.
- Instale e ative o ambiente conda upstream (os requisitos do ambiente conda upstream estão listados na Tabela 2):
- Instale o ambiente usando o comando mamba env create - file=envs/upstream.yml.
- Ative o ambiente usando o comando conda activate upstream.
- Depois que o ambiente conda upstream for instalado com êxito, ative o ambiente usando o comando conda activate upstream e navegue até a pasta CATCH-UP usando cd genetics/CATCH-UP.
- Edite o arquivo de configuração, que pode ser encontrado na pasta config usando o comando cd /config/analysis.yaml, e modifique-o de acordo com a especificação de análise usando um editor de texto. Siga as instruções linha por linha para editar cada parâmetro dentro do próprio arquivo. Este arquivo será retido após a análise e atuará para documentar os parâmetros de execução para ajudar na reprodutibilidade.
- Abra e edite os três arquivos a seguir em um editor de texto (por exemplo, Editor de Texto para Mac ou Bloco de Notas para Windows):
- Edite 1_fastqfile_home_dir.txt arquivo para conter uma lista de todos os arquivos fastq a serem analisados.
NOTA: Números de leitura e ramais (por exemplo, _R1/_R2 e .fastq.gz) devem ser excluídos. Por exemplo, se um projeto contiver esta lista de arquivos fastq:
Sample1_conditionA_L001_R1 . fastq . bom
Sample1_conditionA_L001_R2 . fastq . bom
Sample1_conditionA_L002_R1 . fastq . bom
Sample1_conditionA_L002_R2 . fastq . bom
Sample1_conditionB_L001_R1 . fastq . bom
Sample1_conditionB_L001_R2 . fastq . bom
Sample1_conditionB_L002_R1 . fastq . bom
Sample1_conditionB_L002_R2 . fastq . bom
Sample2_conditionA_L001_R1 . fastq . bom
Sample2_conditionA_L001_R2 . fastq . bom
Sample2_conditionA_L002_R1 . fastq . bom
Sample2_conditionA_L002_R2 . fastq . bom
Sample2_conditionB_L001_R1 . fastq . bom
Sample2_conditionB_L001_R2 . fastq . bom
Sample2_conditionB_L002_R1 . fastq . bom
Sample2_conditionB_L002_R2 . fastq . bom
Nesse caso, o 1_fastqfile_home_dir.txt é o seguinte:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Se os dados brutos contiverem pistas de sequenciamento que exijam concatenação, edite 2_fastq arquivo file_concat.txt para definir o prefixo dos nomes de arquivo a serem concatenados. Se não houver pistas de sequenciamento para concatenar, não edite 2_fastqfile_concat.txt. Certifique-se de que cada linha de 2_fastqfile_concat.txt contenha um prefixo de exemplo da seguinte maneira:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Se for necessário mesclar os dados de amostras diferentes, edite 3_merge_bams.txt arquivo com o prefixo dos nomes de arquivo a serem mesclados. Certifique-se de que cada linha contenha um prefixo de exemplo da seguinte maneira:
Amostra1
Amostra2
A Figura 2 mostra um resumo de como resumir esses três arquivos. O protocolo pode ser aplicado a dados de sequenciamento de extremidade única ou emparelhada. O pipeline usa como padrão a análise de extremidade emparelhada, a menos que especificado de outra forma; Isso pode ser modificado no arquivo de configuração (consulte a etapa 1.6).
- Edite 1_fastqfile_home_dir.txt arquivo para conter uma lista de todos os arquivos fastq a serem analisados.
- Depois que todos os arquivos necessários forem editados, use snakemake para executar CATCH-UP da seguinte maneira: snakemake --configfile=config/analysis_name.yaml all --cores 4.
NOTA: Para obter instruções e documentação mais detalhadas, consulte a pasta CATCH-UP no repositório GitHub UpStreamPipeline, disponível aqui. Isso inclui documentação detalhada sobre como modificar corretamente o arquivo de configuração, desde a alteração de caminhos para arquivos de dados de sequenciamento e armazenamento de resultados até a edição de parâmetros para cada etapa.
Resultados
O pipeline CATCH-UP produz um resultado, log e saída de controle de qualidade (QC) para cada etapa. No arquivo de configuração, o usuário pode optar por manter ou remover os arquivos de saída para reduzir a memória de armazenamento necessária. Todas as saídas são explicadas da seguinte forma:
00. fastq_home_dir: O arquivo de configuração, o le_home_dir.txt FastQFi e merge_bams.txt são copiados para esta pasta para referência e reprodutibilidade.
01. Leituras: Os arquivos FastQ são copiados para esta pasta para evitar alterações dos dados brutos originais durante o processo de fluxo de trabalho, as pistas podem ser concatenadas se especificadas.
02. Corte: Arquivos FastQ com leitura e adaptadores cortados, se especificado.
03. Alinhador: alinhamento com o genoma selecionado.
04. Filtragem: Filtragem de controle de qualidade.
05. Sorted: Classificação de arquivos BAM.
06. Duplicatas: Sinalizando duplicatas.
07. Merge: Mesclando arquivos BAM se isso foi especificado em config.yaml.
08. bam_coverages: arquivo bigwig da cobertura.
09. peak_calling: um arquivo de cama da saída de chamada de pico do LanceOtron.
10. track: produz um arquivo de texto formatado pronto para ser usado no Genome Browser, se necessário.
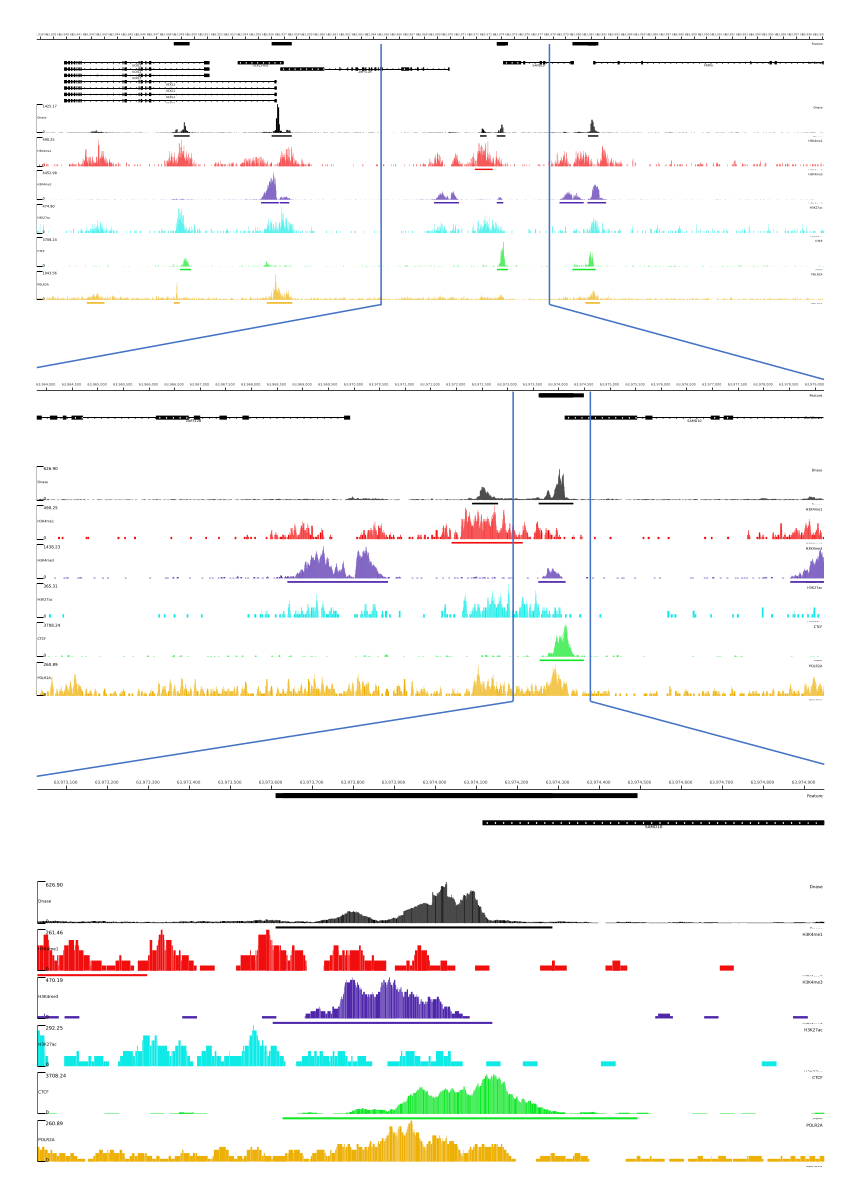
Para as saídas 01, 02, 03, 06 e 07, são fornecidas métricas de CQ e arquivos HTML. Além disso, na Figura 3, fornecemos um exemplo de dados processados usando CATCH-UP, visualizando a saída final por meio da plataforma MLV.

Figura 1: Fluxo de trabalho do CATCH-UP. Dada uma lista de arquivos fastq, o CATCH-UP processa em paralelo todas as amostras em todas as etapas upstream. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Representação ilustrativa explicando como 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt e 3_merge_bams.txt devem ser modificados corretamentepara executar o CATCH-UP. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Exemplo de saída do pipeline CATCH-UP. Os dados brutos de sequenciamento (arquivos fastq) foram baixados do ENCODE21. O pipeline CATCH-UP foi usado para processar os arquivos fastq para DNase-seq e 5 tipos de ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF e POLR2A). Os arquivos de saída do Bigwig foram carregados no Multi Locus View para visualização e identificação de elementos reguladores genômicos. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela 1: Recursos de documentação. Esta tabela mostra as ferramentas envolvidas no fluxo de trabalho CATCH-UP, o link para sua documentação e as respectivas referências. Clique aqui para baixar esta tabela.
Tabela 2: Lista de requisitos de canal e dependência para o ambiente conda upstream. Clique aqui para baixar esta tabela.
Tabela 3: Sistemas operacionais usados para testar o CATCH-UP. O Ubuntu foi testado em um cluster de alto desempenho e em uma máquina local. Clique aqui para baixar esta tabela.
Discussão
O aumento da aceitação e utilização de técnicas de NGS para gerar dados genômicos foi acompanhado por um aumento no desenvolvimento de ferramentas de bioinformática para a análise desses dados. Existem várias ferramentas que podem ser aplicadas para cada etapa da análise de dados, bem como muitos parâmetros diferentes que podem ser especificados em cada ferramenta 6,8,9,15,16,17,18,19,20,22,23,24 . Isso cria uma combinação muito diversificada de estratégias de análise que podem ser aplicadas, cada uma das quais pode produzir variações no resultado. Para comparar com precisão entre os experimentos, a padronização da análise bioinformática é essencial. Historicamente, os dados NGS são gerados por cientistas de laboratório úmido e os dados são analisados por bioinformáticos.
A análise de dados NGS pode ser dividida em pipelines "upstream" e "downstream", onde upstream inclui as etapas necessárias para ir da saída de dados brutos de uma máquina de sequenciamento para um formato que seja visualmente interpretável por um pesquisador. A análise a jusante inclui etapas adicionais que são personalizadas para a questão de pesquisa e o projeto experimental. Os dutos a montante são, portanto, generalizáveis e passíveis de padronização para melhorar a reprodutibilidade científica. Os dutos a jusante, por outro lado, são feitos sob medida, dependentes da questão biológica e exigem informações do investigador, tornando-os menos apropriados para padronização. Criamos um pipeline upstream fácil de usar que permite que os cientistas de laboratório úmido analisem de forma reprodutível seus próprios dados sem precisar de nenhum conhecimento prévio de bioinformática. Aqui, apresentamos o CATCH-UP, um pipeline construído usando a estrutura snakemake e projetado para ser amigável e combater o problema de reprodutibilidade na análise de dados ChIP-seq e ATAC-seq. Esse pipeline foi criado para lidar com dados ChIP-seq ou ATAC-seq. Depois que o usuário tiver baixado CATCH-UP, os parâmetros de análise e a nomenclatura de exemplo devem ser definidos antes de executar o pipeline na linha de comando usando uma única linha de código. Instruções passo a passo simples sobre como personalizar os parâmetros de análise para análise ChIP-seq ou ATAC-seq são fornecidas no próprio arquivo de configuração e em nosso guia passo a passo no repositório GitHub CATCH-UP.
Existem pipelines de análise existentes para dados ChIP-seq ou ATAC-seq, como PEPATAC e ChIP-AP. Embora esses pipelines tenham vantagens como a incorporação de análises upstream e downstream em um único fluxo de trabalho ou o uso de uma interface gráfica do usuário (GUI), essas ferramentas são direcionadas a bioinformáticos e cientistas com um nível moderado de treinamento computacional13,14. O CATCH-UP foi projetado para resolver dois problemas: permitir que cientistas de laboratório úmido sem treinamento em bioinformática realizem suas próprias análises upstream e permitir a padronização da análise upstream, facilitando relatórios fáceis e reprodutibilidade exata entre os laboratórios. O CATCH-UP é intencionalmente limitado à análise a montante, mas as saídas são compatíveis com ferramentas de análise a jusante, como as usadas para comparar estatisticamente conjuntos de dados ou inferir a ligação do fator de transcrição25,26.
Todas as etapas críticas necessárias para realizar uma análise upstream replicável são predefinidas no pipeline CATCH-UP para garantir a robustez. A natureza detalhada desse pipeline permite que o usuário siga a saída do pipeline passo a passo, o que é útil para solucionar problemas e permitir que o fluxo de trabalho analítico seja replicado. Dada a natureza em rápida evolução das técnicas de NGS, a natureza modular desse pipeline é benéfica, pois fornece a capacidade de ser facilmente adaptada para incorporar o lançamento de atualizações de versão de ferramentas e a implementação de novas ferramentas. O CATCH-UP foi testado com sucesso para os seguintes sistemas operacionais: Ubuntu, CentOS, macOS (CPU Intel) e Windows (Tabela 3). O pipeline foi construído para lidar com grandes experimentos contendo dezenas de amostras, paralelizando o fluxo de trabalho, tornando-o adaptável a diferentes projetos experimentais. No geral, a implementação do CATCH-UP na análise de dados ChIP-seq e ATAC-seq permite um fluxo de trabalho de análise amigável, reprodutível e altamente adaptável.
Divulgações
J.R.H. é cofundador e diretor da Nucleome Therapeutics e presta consultoria à empresa.
Agradecimentos
J.R.H. foi apoiado por doações do Wellcome Trust (225220/Z/22/Z e 106130/Z/14/Z) e do MRC (MC_UU_00029/3). M.B. foi apoiado pela doação do Wellcome Trust (225220/Z/22/Z). A ERGO foi apoiada pela bolsa de estudos do Ministério da Educação Nacional e Colocação de Candidatos Enviados ao Exterior para Educação de Pós-Graduação (YLSY), Ministério da Educação Nacional da República da Turquia. EG foi apoiado pelo Programa de Doutorado em Medicina Genômica e Estatística Wellcome (108861 / Z / 15 / Z). S.G.R. foi apoiado pela bolsa do Conselho de Pesquisa Médica (MRC) (MC_UU_00029/3).
Materiais
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Referências
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados