
Method Article
CATCH-UP: Una canalización ascendente de alto rendimiento para datos masivos ATAC-Seq y ChIP-Seq
En este artículo
Resumen
ATAC-seq y ChIP-seq permiten una investigación detallada de la regulación génica; Sin embargo, el procesamiento de estos tipos de datos es desafiante y, a menudo, inconsistente entre los grupos de investigación. Presentamos CATCH-UP: un canal computacional fácil de usar que permite el procesamiento y análisis de datos estandarizados y reproducibles de conjuntos de datos ATAC/ChIP-seq nuevos y publicados.
Resumen
El ensayo de cromatina accesible a la transposasa (ATAC) y la inmunoprecipitación de cromatina (ChIP), junto con la secuenciación de próxima generación (NGS), han revolucionado el estudio de la regulación génica. La falta de estandarización en el análisis de los conjuntos de datos altamente dimensionales generados por estas técnicas ha dificultado la reproducibilidad, lo que ha dado lugar a discrepancias en los datos publicados y procesados. Parte de este problema se debe a la diversa gama de herramientas bioinformáticas disponibles para el análisis de este tipo de datos. En segundo lugar, se requieren secuencialmente una serie de diferentes herramientas bioinformáticas para convertir los datos brutos en un resultado totalmente procesado e interpretable, y estas herramientas requieren diversos niveles de habilidades computacionales. Además, hay muchas opciones para el control de calidad que no se emplean de manera uniforme durante el procesamiento de datos. Abordamos estos problemas con un ensayo completo para la secuenciación de cromatina accesible a la transposasa (ATAC-seq) y la secuenciación de inmunoprecipitación de cromatina (ChIP-seq) (CATCH-UP), una tubería fácil de usar basada en Python para el análisis de conjuntos de datos masivos de ChIP-seq y ATAC-seq, desde archivos fastq sin procesar hasta pistas de peces gordos visualizables y llamadas de picos. Esta canalización es fácil de instalar y ejecutar, y requiere un conocimiento informático mínimo. La canalización es modular, escalable y paralelizable en varias infraestructuras informáticas, lo que permite una fácil generación de informes de la metodología para permitir el análisis reproducible de conjuntos de datos nuevos o publicados.
Introducción
La expresión génica debe estar estrictamente regulada para que las células establezcan y mantengan su correcta función biológica. Es bien sabido que la expresión génica aberrante subyace en la patogénesis de muchas enfermedades y, por lo tanto, gran parte del interés de la investigación radica en la comprensión de los mecanismos de la regulación génica1. La expresión génica se ve facilitada por elementos reguladores como promotores y potenciadores. Dentro de su secuencia, estos elementos contienen sitios de unión al factor de transcripción (TF), que, cuando están activos, proporcionan una plataforma para la unión de TF. La unión de TFs en estos sitios da como resultado un desplazamiento de los nucleosomas, lo que resulta en un aumento en la accesibilidad al ADN y un aumento posterior en la permisibilidad a la maquinaria transcripcional. Como resultado de esta mayor accesibilidad, estas regiones del ADN son más sensibles a las nucleasas y transposasas como la DNasa y la Tn5, una propiedad bioquímica que ha sido explotada por los investigadores que investigan la regulación transcripcional 2,3.
DNase-seq y ATAC-seq permiten a los investigadores mapear regiones de cromatina abierta, sitios de unión de TF y posicionamiento nucleosomal en todo el genoma. De estas dos técnicas, ATAC-seq ha crecido en popularidad en la última década debido al sencillo protocolo de dos pasos y a un bajo requisito de número de células (50.000 células en comparación con 1 millón por réplica para DNase-seq). Si bien ATAC-seq proporciona una visión general del panorama general de la cromatina en una población de células, es en gran medida independiente de qué proteínas específicas se unen al genoma 4,5. Con el fin de identificar los lugares donde una proteína específica está interactuando con el genoma, la técnica de referencia es la inmunoprecipitación de cromatina (ChIP). ChIP-seq implica la fijación química de las interacciones proteína-ADN en una célula, seguida de inmunoprecipitación ("pull-down") utilizando un anticuerpo específico de la proteína de interés para seleccionar fragmentos de ADN unidos a la proteína de interés (POI). Estos fragmentos de ADN se pueden secuenciar para revelar las ubicaciones de unión genómica de proteínas específicas, como TF, o sitios que contienen modificaciones específicas de histonas1. Mediante la combinación de conjuntos de datos ATAC-seq y ChIP-seq, se puede obtener una imagen detallada del panorama regulatorio para una población de células.
El flujo de trabajo básico requerido para el análisis es el siguiente: las lecturas de secuenciación en bruto deben ser controladas antes de la alineación con un genoma de referencia ("mapeo"). A continuación, las lecturas asignadas correctamente se pueden filtrar para eliminar tanto las lecturas de baja calidad como los duplicados de PCR. Para visualizar estas lecturas mapeadas y filtradas, es necesario calcular la "cobertura" de estas lecturas en todo el genoma. Esto genera un archivo que se puede cargar en un navegador de genoma, como la vista multi-locus (MLV) o el navegador de genoma UCSC como una "pista"6,7. La identificación de picos, o "llamada de picos" de estas pistas de cobertura se logra típicamente utilizando herramientas como LanceOtron o MACS2 8,9. Finalmente, a través del análisis de la ubicación de los picos, se pueden hacer comparaciones de forma y tamaño entre muestras o condiciones biológicas. El análisis e integración de estos conjuntos de datos es un proceso complejo de varios pasos en el que se pueden implementar diferentes combinaciones de herramientas bioinformáticas. Las diferentes versiones de las herramientas pueden ser incompatibles entre sí y pueden cambiar el resultado del procesamiento de datos. También existe una amplia variedad en la potencia computacional y la competencia del usuario requerida para implementar diferentes partes del procesamiento de datos, como se muestra en las tuberías nf-core10, panpipes11, genpipes12, PEPATAC13 o ChIP-AP14.
En general, esto ha dado lugar a inconsistencias tanto en el análisis como en el informe del análisis, lo que, a su vez, ha dado lugar a una redunda en reproducibilidad, accesibilidad y conveniencia para cualquier persona con conocimientos limitados de bioinformática. Abordamos todos estos problemas con CATCH-UP (canalización ascendente completa de ATAC-seq y ChIP-seq), una canalización fácil de usar, flexible y modular para procesar datos de ChIP-seq y ATAC/DNase-seq. La implementación de CATCH-UP requiere una experiencia mínima en bioinformática; Se puede ejecutar en varias infraestructuras informáticas y permite el análisis de datos reproducibles dentro de los grupos de investigación y entre ellos.
CATCH-UP es una canalización de Snakemake basada en Python creada para estandarizar el análisis de datos ChIP-seq y ATAC-seq. Toma datos de secuenciación sin procesar (archivos fastq.gz) como entrada y genera una salida en forma de archivos de pico (.bed) que proporcionan el resultado respectivo para cada paso. Proporcionamos un archivo de configuración en formato yaml (config.yaml), en el que el usuario puede editar los parámetros de cada paso de análisis. El sistema de gestión implementado dentro de snakemake permite el uso de diferentes infraestructuras informáticas (como servidores, clústeres, sistemas en la nube u ordenadores personales) y en paralelo si el usuario proporciona una gran cantidad de datos.
A continuación, proporcionamos una descripción detallada de cada paso del flujo de trabajo (consulte la Figura 1 para ver la ilustración del flujo de trabajo). Esta explicación es esencial para seguir el paso a paso en el apartado de protocolo:
Mover fastq: el primer paso de la canalización es copiar los archivos fastq sin procesar en el directorio de análisis con nombre. Esto deja los datos originales intactos para evitar corromper o modificar los archivos de datos sin procesar.
Concatenación: si los datos de secuenciación sin procesar contienen varios carriles, este paso es necesario para concatenar los carriles antes del análisis. De forma predeterminada, la canalización maneja todos los archivos fastq como muestras individuales. Este paso de concatenación debe definirse en el archivo de configuración.
Recorte: paso opcional de limpieza de datos. Esto permite el recorte de lecturas de baja calidad o secuencias de adaptadores mediante el uso de trimmomatic15. El usuario puede proporcionar archivos fasta personalizados de secuencias de adaptadores; Se proporciona un ejemplo en el directorio del adaptador. Los parámetros de recorte adicionales se pueden definir en el archivo de configuración. De forma predeterminada, el flujo de trabajo omite esta regla.
Alineador: para la alineación, Bowtie216 se aplica de forma predeterminada; También se pueden especificar herramientas de alineación alternativas como BWA-MEM217 . La herramienta de alineación Bowtie2 se selecciona de forma predeterminada, ya que es particularmente hábil para alinear lecturas relativamente cortas con genomas relativamente grandes y, por lo tanto, es muy adecuada para la alineación de datos ChIP-seq y ATAC-seq con genomas de mamíferos. Para evitar cualquier archivo intermedio, el alineador se canaliza a la vista samtools para guardar el archivo bam en la salida. Para esta regla, el usuario debe especificar la compilación del genoma preferida en la que mapear las lecturas, por ejemplo, hg19/hg38 (humano), mm10/mm39 (ratón).
Filtrado: se conservan las lecturas asignadas correctamente y se filtran las lecturas de baja calidad. Predeterminado: vista samtools, con parámetros: -bShuF 4 -f 3 -q 30.
Ordenar: las lecturas alineadas se ordenan en orden de la coordenada más a la izquierda. Predeterminado: samtools sort (envoltorio de serpiente), con parámetro: -m 4G.
Marcar duplicados: todas las lecturas duplicadas se identifican y marcan. El usuario puede decidir eliminarlos cambiando el parámetro del archivo de configuración. Predeterminado: Picard MarkDuplicates (envoltorio de snakemade), con el parámetro: --REMOVE_DUPLICATES False para marcar y retener duplicados.
Fusionar bahía: si los datos de secuenciación se componen de réplicas o muestras, es posible que el usuario desee fusionarlos en una sola mezcla. En este caso, el usuario puede optar por fusionar los archivos bams o mantener los archivos bam separados durante todo el análisis. Si el usuario elige fusionar bams (empleando samtools merge), se debe especificar un prefijo común para los bams combinados.
Índice: este paso indexa las coordenadas ordenadas. Valor predeterminado: índice de samtools (contenedor de snakemake), utilizando los parámetros predeterminados especificados por samtools.
BamCoverage: esta regla crea una pista de cobertura de peluche grande a partir de lecturas alineadas. Se aplica la herramienta bamCoverage de deepTools y la cobertura se calcula como el número de lecturas por bin, en el que el bin representa una ventana de un tamaño especificado. En esta canalización, bamCoverage se aplica con los siguientes parámetros establecidos como predeterminados: -bs 1 -normalizeUsing RPKM -extendReads.
Llamada de pico: se seleccionó LanceOtron8 como el llamador de pico predeterminado para esta canalización. A diferencia de los llamadores de picos tradicionales, que en su mayoría se basan en pruebas estadísticas, LanceOtron es un llamador de picos basado en el aprendizaje profundo, que incorpora mediciones de enriquecimiento genómico y pruebas estadísticas y se ha demostrado que supera al llamador de picos estándar de la industria, MACS29. Para que los peces gordos sean compatibles con LanceOtron, la cobertura debe calcularse por par de bases y normalizarse en RPKM; esto se refleja en la configuración predeterminada del paso BamCoverage. MACS2 se puede seleccionar como un llamador de pico alternativo. La versión de los nuevos llamadores de picos se supervisará e incorporará según corresponda para mantener y optimizar el rendimiento de esta canalización de análisis.
TrackDb: crea una asociación de pares clave-valor de archivos bigwig para cargarlos y visualizarlos en herramientas como las plataformas MLV6 o UCSC Genome Browser18 .
Además de los datos de salida, cada paso de la canalización genera un archivo de registro y se proporcionan las comprobaciones de control de calidad adecuadas para que el usuario pueda realizar un seguimiento del progreso del análisis. FastQC19 se aplica a los datos de secuenciación sin procesar y recortados (si se selecciona) (pasos 1 - Mover fastq y 2- Recortar). Las estadísticas de Samtools más MultiQC20 se utilizan para recopilar, producir y visualizar informes de control de calidad en archivos bam en la salida en los pasos 3 - Alineador, 6 - Marcar duplicados y 7 - Fusionar bam. Para obtener más información sobre cada una de las herramientas aplicadas en los pasos anteriores, consulte la Tabla 1.
Protocolo
1. Ejecución de la canalización CATCH-UP
- Clona el repositorio UpStreamPipeline desde https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Navegue hasta el directorio de trabajo elegido, copie el siguiente código y ejecútelo en la línea de comandos:
clon de git git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Navegue dentro de la carpeta UpStreamPipeline descargada mediante el comando: cd UpStreamPipeline
- Instale la distribución anaconda (si es necesario):
- Compruebe si anaconda ya está instalado en el sistema utilizando el comando que conda. Si el comando no muestra ninguna ruta a ninguna distribución de Conda, descargue mambaforge de https://github.com/conda-forge/miniforge#mambaforge y seleccione la distribución y la versión correctas para el sistema. Por ejemplo, para los usuarios de Linux, use wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Visite esta página web para diferentes Sistemas Operativos: https://github.com/conda-forge/miniforge/.
- Ejecute el instalador mediante sh Mambaforge-Linux-x86_64.sh e inicialice conda en el sistema mediante la ejecución de conda init.
- Instale y active el entorno de Conda ascendente (los requisitos del entorno de Conda ascendente se enumeran en la Tabla 2):
- Instale el entorno usando el comando mamba env create - file=envs/upstream.yml.
- Active el entorno mediante el comando conda activate upstream.
- Una vez que el entorno de Conda ascendente se haya instalado correctamente, active el entorno mediante el comando conda activate upstream y navegue hasta la carpeta CATCH-UP mediante cd genetics/CATCH-UP.
- Edite el archivo de configuración, que se puede encontrar en la carpeta de configuración utilizando el comando cd /config/analysis.yaml, y modifíquelo en consecuencia con la especificación de análisis utilizando un editor de texto. Siga las instrucciones línea por línea para editar cada parámetro dentro del propio archivo. Este archivo se conservará después del análisis y actuará para documentar los parámetros de ejecución para ayudar a la reproducibilidad.
- Abra y edite los tres archivos siguientes en un editor de texto (por ejemplo, TextEdit para Mac o Bloc de notas para Windows):
- Edite 1_fastqfile_home_dir.txt archivo para que contenga una lista de todos los archivos fastq que se van a analizar.
NOTA: Se deben excluir los números de lectura y las extensiones (por ejemplo, _R1/_R2 y .fastq.gz). Por ejemplo, si un proyecto contiene esta lista de archivos fastq:
Sample1_conditionA_L001_R1 . fastq . GZ
Sample1_conditionA_L001_R2 . fastq . GZ
Sample1_conditionA_L002_R1 . fastq . GZ
Sample1_conditionA_L002_R2 . fastq . GZ
Sample1_conditionB_L001_R1 . fastq . GZ
Sample1_conditionB_L001_R2 . fastq . GZ
Sample1_conditionB_L002_R1 . fastq . GZ
Sample1_conditionB_L002_R2 . fastq . GZ
Sample2_conditionA_L001_R1 . fastq . GZ
Sample2_conditionA_L001_R2 . fastq . GZ
Sample2_conditionA_L002_R1 . fastq . GZ
Sample2_conditionA_L002_R2 . fastq . GZ
Sample2_conditionB_L001_R1 . fastq . GZ
Sample2_conditionB_L001_R2 . fastq . GZ
Sample2_conditionB_L002_R1 . fastq . GZ
Sample2_conditionB_L002_R2 . fastq . GZ
En este caso, el 1_fastqfile_home_dir.txt es el siguiente:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Si los datos sin procesar contienen carriles de secuenciación que requieren concatenación, edite 2_fastq archivo de file_concat.txt para definir el prefijo de los nombres de archivo que se van a concatenar. Si no hay carriles de secuenciación para concatenar, no edite 2_fastqfile_concat.txt. Asegúrese de que cada línea de 2_fastqfi le_concat.txt contenga un prefijo de muestra de la siguiente manera:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Si es necesario fusionar los datos de diferentes muestras, edite 3_merge_bams.txt archivo con el prefijo de los nombres de archivo que se van a combinar. Asegúrese de que cada línea contenga un prefijo de ejemplo de la siguiente manera:
Ejemplo1
Muestra2
En la figura 2 se muestra un resumen de cómo resumir estos tres archivos. El protocolo se puede aplicar a datos de secuenciación de extremo único o emparejados. El valor predeterminado de la canalización es el análisis de extremos emparejados, a menos que se especifique lo contrario; Esto se puede modificar en el archivo de configuración (consulte el paso 1.6).
- Edite 1_fastqfile_home_dir.txt archivo para que contenga una lista de todos los archivos fastq que se van a analizar.
- Una vez que se hayan editado todos los archivos necesarios, use snakemake para ejecutar CATCH-UP de la siguiente manera: snakemake --configfile=config/analysis_name.yaml all --cores 4.
NOTA: Para obtener instrucciones y documentación más detalladas, consulte la carpeta CATCH-UP dentro del repositorio de GitHub UpStreamPipeline, disponible aquí. Esto incluye documentación detallada sobre cómo modificar correctamente el archivo de configuración, desde el cambio de rutas para secuenciar archivos de datos y almacenar resultados hasta editar los parámetros para cada paso.
Resultados
La canalización CATCH-UP genera un resultado, un registro y una salida de control de calidad (QC) para cada paso. Dentro del archivo de configuración, el usuario puede elegir mantener o eliminar los archivos de salida para reducir la memoria de almacenamiento necesaria. Todos los resultados se explican de la siguiente manera:
00. fastq_home_dir: El archivo de configuración, elle_home_dir.txt FastQ Fi y merge_bams.txt se copian en esta carpeta para referencia y reproducibilidad.
01. Lecturas: Los archivos FASTQ se copian en esta carpeta para evitar alteraciones de los datos RAW originales durante el proceso de flujo de trabajo, los carriles se pueden concatenar si se especifica.
02. Recorte: Archivos FastQ con lectura y adaptadores recortados si se especifica.
03. Alineador: alineamiento con el genoma seleccionado.
04. Filtrado: filtrado de control de calidad.
05. Ordenado: Clasificación de archivos BAM.
06. Duplicados: Marcando duplicados.
07 . merge: Fusionar archivos BAM si esto se especificó en config.yaml.
08. bam_coverages: Ficha de la cobertura.
09. peak_calling: un archivo de cama de la salida de llamadas máximas de LanceAtron.
10. track: produce un archivo de texto formateado listo para ser utilizado en Genome Browser si es necesario.
Para las salidas 01, 02, 03, 06 y 07, se proporcionan métricas de control de calidad y archivos HTML. Además, en la Figura 3, proporcionamos un ejemplo de datos procesados utilizando CATCH-UP, visualizando el resultado final a través de la plataforma MLV.

Figura 1: Flujo de trabajo de CATCH-UP. Dada una lista de archivos fastq, CATCH-UP procesa en paralelo todas las muestras a través de todos los pasos ascendentes. Haga clic aquí para ver una versión más grande de esta figura.
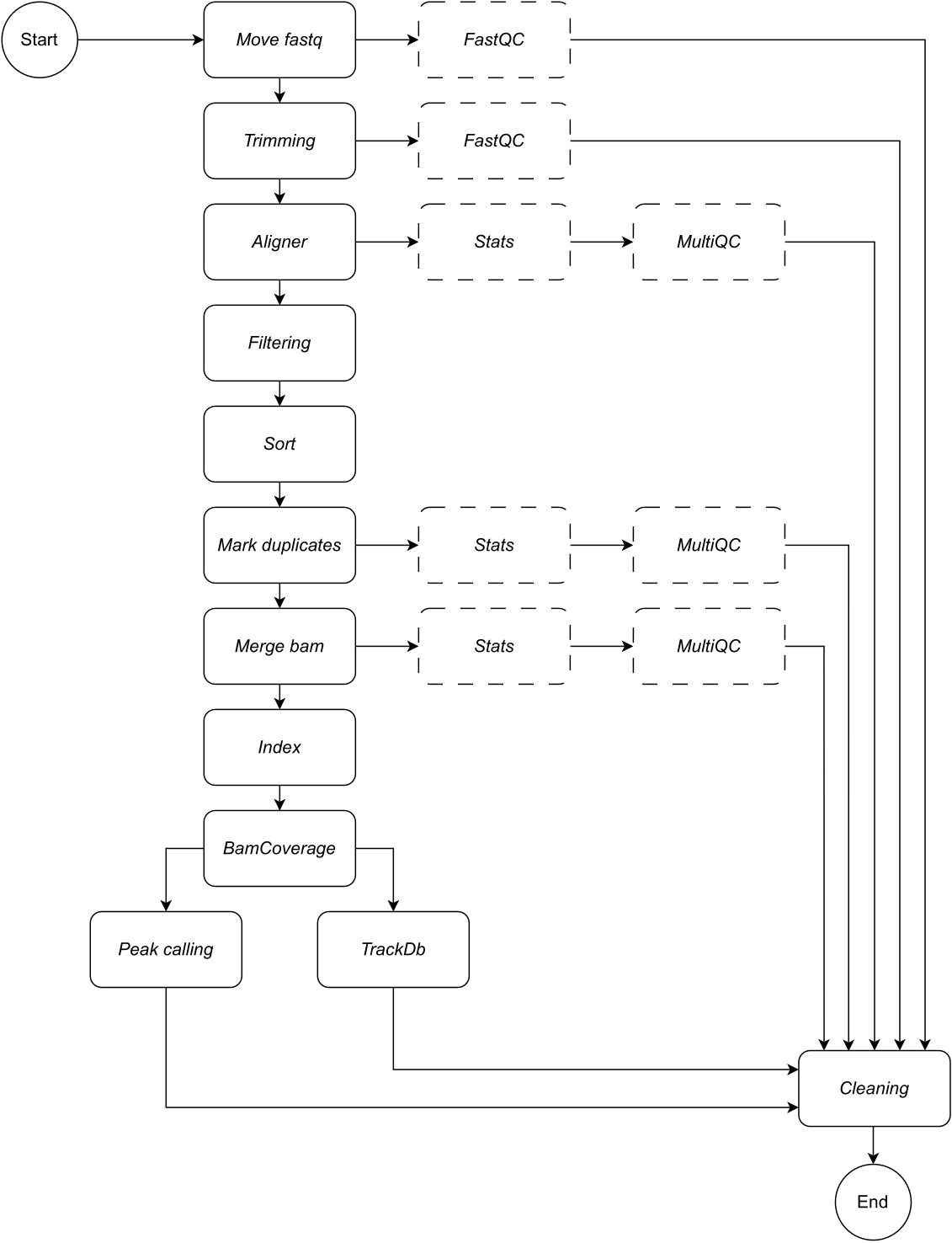{kind=link}

Figura 2: Representación ilustrativa que explica cómo 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt y 3_merge_bams.txt deben modificarse correctamentepara ejecutar CATCH-UP. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Ejemplo de salida de la canalización CATCH-UP. Los datos brutos de secuenciación (archivos fastq) se descargaron de ENCODE21. La canalización CATCH-UP se utilizó para procesar los archivos fastq para DNase-seq y 5 tipos de ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF y POLR2A). Los archivos de salida de Bigwig se cargaron en Multi Locus View para la visualización e identificación de elementos reguladores genómicos. Haga clic aquí para ver una versión más grande de esta figura.
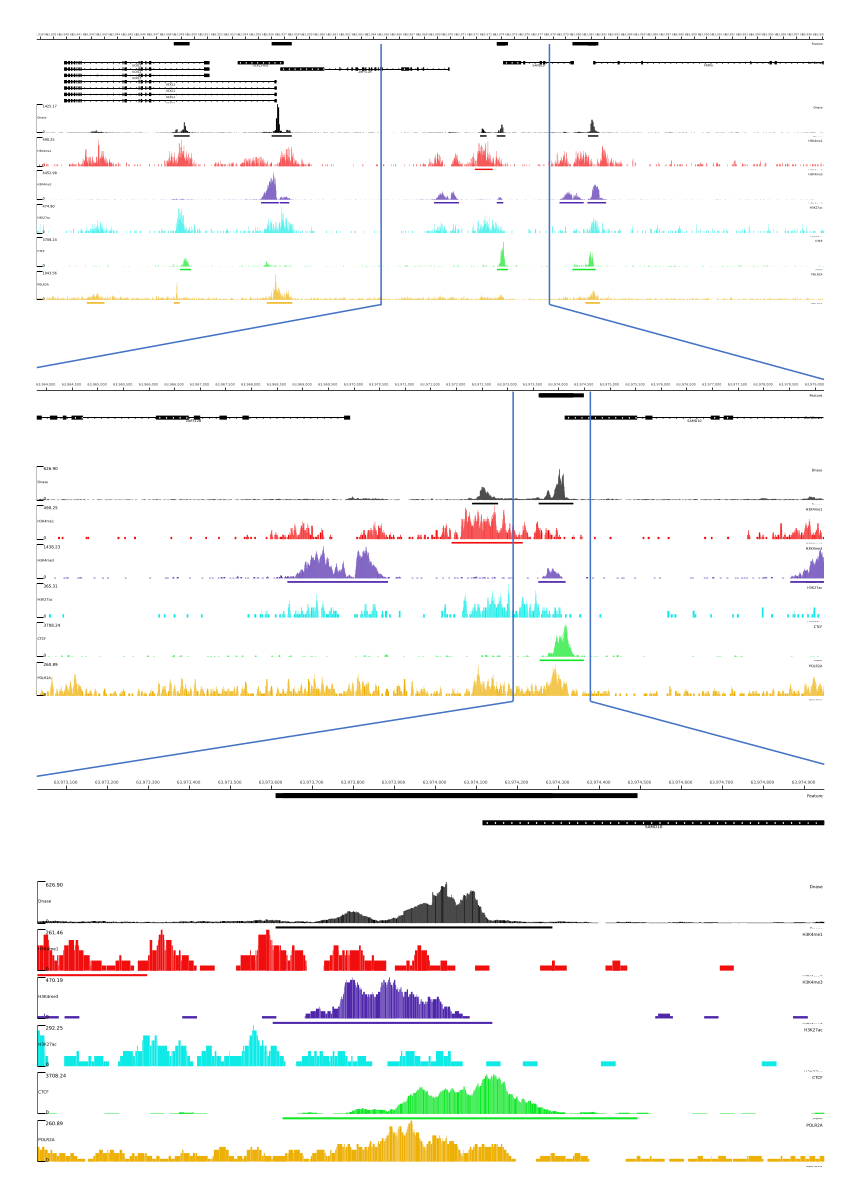{kind=link}
Tabla 1: Recursos de documentación. En esta tabla se muestran las herramientas implicadas en el flujo de trabajo de CATCH-UP, el enlace a su documentación y las referencias respectivas. Haga clic aquí para descargar esta tabla.
Tabla 2: Lista de requisitos de canal y dependencia para el entorno de Conda ascendente. Haga clic aquí para descargar esta tabla.
Tabla 3: Sistemas operativos utilizados para probar CATCH-UP. Ubuntu se probó en un clúster de alto rendimiento y en una máquina local. Haga clic aquí para descargar esta tabla.
Discusión
El aumento de la adopción y utilización de las técnicas de NGS para generar datos genómicos ha ido acompañado de un aumento en el desarrollo de herramientas bioinformáticas para el análisis de estos datos. Existen múltiples herramientas que se pueden aplicar para cada paso del análisis de datos, así como muchos parámetros diferentes que se pueden especificar dentro de cada herramienta 6,8,9,15,16,17,18,19,20,22,23,24 . Esto da lugar a una combinación muy diversa de estrategias de análisis que podrían aplicarse, cada una de las cuales podría producir variaciones en el resultado. Con el fin de comparar con precisión entre experimentos, la estandarización del análisis bioinformático es esencial. Históricamente, los datos de NGS son generados por científicos de laboratorio húmedo y los datos son analizados por bioinformáticos.
El análisis de datos NGS se puede dividir en canalizaciones "ascendentes" y "descendentes", donde las aguas arriba incluyen los pasos necesarios para pasar de la salida de datos sin procesar de una máquina de secuenciación a un formato que sea visualmente interpretable por un investigador. El análisis posterior incluye pasos adicionales que se adaptan a la pregunta de investigación y al diseño experimental. Por lo tanto, los gasoductos aguas arriba son generalizables y susceptibles de estandarización para mejorar la reproducibilidad científica. Por otro lado, los gasoductos aguas abajo están hechos a medida, dependen de la cuestión biológica y requieren la visión del investigador, lo que los hace menos apropiados para la estandarización. Hemos creado una tubería ascendente fácil de usar que permite a los científicos de laboratorio húmedo analizar de forma reproducible sus propios datos sin necesidad de ningún conocimiento previo de bioinformática. Aquí, presentamos CATCH-UP, una tubería construida utilizando el marco snakemake y diseñada para ser fácil de usar y combatir el problema de la reproducibilidad en el análisis de datos ChIP-seq y ATAC-seq. Esta canalización se ha creado para controlar datos ChIP-seq o ATAC-seq. Una vez que el usuario ha descargado CATCH-UP, primero se deben definir los parámetros de análisis y la nomenclatura de la muestra antes de ejecutar la canalización en la línea de comandos mediante una sola línea de código. En el propio archivo de configuración y en nuestra guía paso a paso en el repositorio de GitHub de CATCH-UP se proporcionan instrucciones sencillas paso a paso sobre cómo personalizar los parámetros de análisis para el análisis ChIP-seq o ATAC-seq.
Existen canalizaciones de análisis para los datos de ChIP-seq o ATAC-seq, como PEPATAC y ChIP-AP. Si bien estas canalizaciones tienen ventajas como la incorporación de análisis ascendentes y descendentes en un solo flujo de trabajo o el uso de una interfaz gráfica de usuario (GUI), estas herramientas están dirigidas a bioinformáticos y científicos con un nivel moderado de formación computacional13,14. CATCH-UP ha sido diseñado para resolver dos problemas: permitir que los científicos de laboratorio húmedo sin formación bioinformática realicen sus propios análisis previos y permitir la estandarización de los análisis ascendentes al facilitar la generación de informes y la reproducibilidad exacta en todos los laboratorios. CATCH-UP se limita intencionadamente al análisis ascendente, pero los resultados son compatibles con las herramientas de análisis posteriores, como las que se utilizan para comparar estadísticamente conjuntos de datos o inferir la unión de factores de transcripción25,26.
Todos los pasos críticos necesarios para realizar un análisis ascendente replicable están predefinidos dentro de la canalización CATCH-UP para garantizar la solidez. La naturaleza detallada de esta canalización permite al usuario seguir la salida de la canalización paso a paso, lo que resulta útil tanto para solucionar problemas como para permitir que se replique el flujo de trabajo analítico. Dada la naturaleza en rápida evolución de las técnicas de NGS, la naturaleza modular de esta canalización es beneficiosa, ya que proporciona la capacidad de adaptarse fácilmente para incorporar tanto la publicación de actualizaciones de versiones de herramientas como la implementación de nuevas herramientas. CATCH-UP se ha probado con éxito para los siguientes sistemas operativos: Ubuntu, CentOS, macOS (CPU Intel) y Windows (Tabla 3). La canalización se ha construido para manejar grandes experimentos que contienen decenas de muestras mediante la paralelización del flujo de trabajo, lo que la hace adaptable a diferentes diseños experimentales. En general, la implementación de CATCH-UP en el análisis de datos ChIP-seq y ATAC-seq permite un flujo de trabajo de análisis fácil de usar, reproducible y altamente adaptable.
Divulgaciones
J.R.H. es cofundador y director de Nucleome Therapeutics y asesora a la empresa.
Agradecimientos
J.R.H. contó con el apoyo de subvenciones del Wellcome Trust (225220/Z/22/Z y 106130/Z/14/Z) y del MRC (MC_UU_00029/3). M.B. fue apoyado por la subvención de Wellcome Trust (225220/Z/22/Z). E.R.G contó con el apoyo de la beca del Ministerio de Educación Nacional para la Selección y Colocación de Candidatos Enviados al Extranjero para la Educación de Posgrado (YLSY) del Ministerio de Educación Nacional de la República de Türkiye. E.G. contó con el apoyo del Programa de Doctorado en Medicina Genómica y Estadística de Wellcome (108861/Z/15/Z). S.G.R. contó con el apoyo de la subvención del Consejo de Investigación Médica (MRC) (MC_UU_00029/3).
Materiales
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Referencias
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623(2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521(2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037(2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101(2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537(2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008(2021).
- Picard Toolkit. , http://broadinstitute.github.io/picard/ (2019).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados