
Method Article
CATCH-UP: Eine Upstream-Pipeline mit hohem Durchsatz für Massen-ATAC-Seq- und ChIP-Seq-Daten
In diesem Artikel
Zusammenfassung
ATAC-seq und ChIP-seq ermöglichen eine detaillierte Untersuchung der Genregulation; Die Verarbeitung dieser Datentypen ist jedoch eine Herausforderung und zwischen den Forschungsgruppen oft inkonsistent. Wir stellen CATCH-UP vor: eine einfach zu bedienende Rechenpipeline, die eine standardisierte und reproduzierbare Datenverarbeitung und -analyse von neuen und veröffentlichten ATAC/ChIP-seq-Datensätzen ermöglicht.
Zusammenfassung
Assays für Transposase-zugängliches Chromatin (ATAC) und Chromatin-Immunpräzipitation (ChIP) in Verbindung mit Next-Generation-Sequencing (NGS) haben die Erforschung der Genregulation revolutioniert. Ein Mangel an Standardisierung bei der Analyse der hochdimensionalen Datensätze, die mit diesen Techniken erzeugt werden, hat es schwierig gemacht, Reproduzierbarkeit zu erreichen, was zu Diskrepanzen in den veröffentlichten, verarbeiteten Daten führt. Ein Teil dieses Problems ist auf die Vielfalt der bioinformatischen Werkzeuge zurückzuführen, die für die Analyse dieser Art von Daten zur Verfügung stehen. Zweitens sind eine Reihe verschiedener bioinformatischer Werkzeuge nacheinander erforderlich, um Rohdaten in eine vollständig verarbeitete und interpretierbare Ausgabe umzuwandeln, und diese Werkzeuge erfordern unterschiedliche Rechenfähigkeiten. Darüber hinaus gibt es viele Möglichkeiten der Qualitätskontrolle, die bei der Datenverarbeitung nicht einheitlich eingesetzt werden. Wir adressieren diese Probleme mit einem vollständigen Assay für die Transposase-zugängliche Chromatinsequenzierung (ATAC-seq) und die Chromatin-Immunpräzipitationssequenzierung (ChIP-seq) Upstream-Pipeline (CATCH-UP), eine einfach zu bedienende, Python-basierte Pipeline für die Analyse von ChIP-seq- und ATAC-seq-Massendatensätzen von Fastq-Rohdateien bis hin zu visualisierbaren Bigwig-Tracks und Peaks-Aufrufen. Diese Pipeline ist einfach zu installieren und auszuführen und erfordert nur minimale Rechenkenntnisse. Die Pipeline ist modular, skalierbar und parallelisierbar auf verschiedenen Recheninfrastrukturen und ermöglicht eine einfache Berichterstattung über die Methodik, um eine reproduzierbare Analyse neuartiger oder veröffentlichter Datensätze zu ermöglichen.
Einleitung
Die Genexpression muss streng reguliert werden, damit die Zellen ihre korrekte biologische Funktion etablieren und aufrechterhalten können. Es ist allgemein bekannt, dass die aberrante Genexpression der Pathogenese vieler Krankheiten zugrunde liegt, und daher liegt ein großer Teil des Forschungsinteresses auf dem Verständnis der Mechanismen der Genregulation1. Die Genexpression wird durch regulatorische Elemente wie Promotoren und Enhancer erleichtert. Innerhalb ihrer Sequenz enthalten diese Elemente Bindungsstellen des Transkriptionsfaktors (TF), die, wenn sie aktiv sind, eine Plattform für die TF-Bindung bieten. Die Bindung von TFs an diesen Stellen führt zu einer Verschiebung der Nukleosomen, was zu einer Erhöhung der DNA-Zugänglichkeit und damit zu einer Erhöhung der Zulässigkeit für die Transkriptionsmaschinerie führt. Infolge dieser verbesserten Zugänglichkeit sind diese DNA-Regionen empfindlicher für Nukleasen und Transposasen wie DNase und Tn5, eine biochemische Eigenschaft, die von Forschern zur Untersuchung der transkriptionellen Regulation ausgenutzt wurde 2,3.
DNase-seq und ATAC-seq ermöglichen es Forschern, Regionen mit offenem Chromatin, TF-Bindungsstellen und nukleosomaler Positionierung im gesamten Genom zu kartieren. Von diesen beiden Techniken hat ATAC-seq in den letzten zehn Jahren aufgrund des einfachen zweistufigen Protokolls und der geringen Anforderungen an die Zellzahl (50.000 Zellen im Vergleich zu 1 Million pro Replikat für DNase-seq) an Popularität gewonnen. Während ATAC-seq einen Überblick über die allgemeine Chromatinlandschaft in einer Zellpopulation bietet, ist es weitgehend agnostisch, welche spezifischen Proteine an das Genom binden 4,5. Um die Stellen zu identifizieren, an denen ein bestimmtes Protein mit dem Genom interagiert, ist die Goldstandardtechnik die Chromatin-Immunpräzipitation (ChIP)-seq. ChIP-seq beinhaltet die chemische Fixierung von Protein-DNA-Wechselwirkungen in einer Zelle, gefolgt von einer Immunpräzipitation ("Pull-down") unter Verwendung eines Antikörpers, der für das interessierende Protein spezifisch ist, um DNA-Fragmente zu selektieren, die durch das Protein of Interest (POI) gebunden sind. Diese DNA-Fragmente können sequenziert werden, um die genomischen Bindungsstellen spezifischer Proteine wie TFs oder Stellen, die spezifische Histonmodifikationen enthalten, aufzudecken1. Durch die Kombination von ATAC-seq- und ChIP-seq-Datensätzen kann ein detailliertes Bild der regulatorischen Landschaft für eine Zellpopulation abgeleitet werden.
Der grundlegende Arbeitsablauf, der für die Analyse erforderlich ist, ist wie folgt: Rohsequenzierungs-Reads müssen vor dem Abgleich mit einem Referenzgenom ("Mapping") qualitätskontrolliert werden. Die erfolgreich zugeordneten Lesevorgänge können dann gefiltert werden, um sowohl Lesevorgänge mit geringer Qualität als auch PCR-Duplikate zu entfernen. Um diese kartierten und gefilterten Reads zu visualisieren, ist es notwendig, die "Abdeckung" dieser Reads über das Genom zu berechnen. Dadurch wird eine Datei erzeugt, die als "Track" in einen Genombrowser wie z.B. Multi-Locus View (MLV) oder den UCSC-Genombrowser hochgeladen werden kann6,7. Die Peak-Identifizierung oder das "Peak-Calling" dieser Abdeckungsspuren wird in der Regel mit Tools wie LanceOtron oder MACS2 erreicht 8,9. Schließlich können durch die Analyse der Peaklage, -form und -größe Vergleiche zwischen Proben oder biologischen Bedingungen angestellt werden. Die Analyse und Integration dieser Datensätze ist ein komplexer mehrstufiger Prozess, in dem verschiedene Kombinationen von bioinformatischen Werkzeugen implementiert werden können. Verschiedene Versionen der Tools können nicht miteinander kompatibel sein und die Ausgabe der Datenverarbeitung verändern. Es gibt auch eine große Vielfalt an Rechenleistung und Benutzerkenntnissen, die erforderlich sind, um verschiedene Teile der Datenverarbeitung zu implementieren, wie in den Pipelines nf-core10, panpipes11, genpipes12, PEPATAC13 oder ChIP-AP14 gezeigt.
Insgesamt hat dies zu Inkonsistenzen sowohl in der Analyse als auch in der Berichterstattung der Analyse geführt, was wiederum zu einer schlechten Reproduzierbarkeit, Zugänglichkeit und Bequemlichkeit für alle mit begrenzten Kenntnissen in der Bioinformatik geführt hat. All diese Probleme lösen wir mit CATCH-UP (complete ATAC-seq and ChIP-seq upstream pipeline), einer einfach zu bedienenden, flexiblen und modularen Pipeline für die Verarbeitung von ChIP-seq- und ATAC/DNase-seq-Daten. Die Implementierung von CATCH-UP erfordert nur minimale Erfahrung in der Bioinformatik; Es kann auf verschiedenen Recheninfrastrukturen betrieben werden und ermöglicht eine reproduzierbare Datenanalyse innerhalb und zwischen Forschungsgruppen.
CATCH-UP ist eine Python-basierte Snakemake-Pipeline, die entwickelt wurde, um die Analyse von ChIP-seq- und ATAC-seq-Daten zu standardisieren. Es nimmt Rohsequenzierungsdaten (fastq.gz Dateien) als Eingabe und generiert eine Ausgabe in Form von Peak-Dateien (.bed), die das jeweilige Ergebnis für jeden Schritt liefern. Wir stellen eine Konfigurationsdatei im YAML-Format (config.yaml) zur Verfügung, in der der Benutzer die Parameter jedes Analyseschritts bearbeiten kann. Das in snakemake implementierte Managementsystem ermöglicht die Nutzung verschiedener Rechnerinfrastrukturen (wie Server, Cluster, Cloud-Systeme oder PCs) und parallel, wenn der Benutzer eine große Datenmenge zur Verfügung stellt.
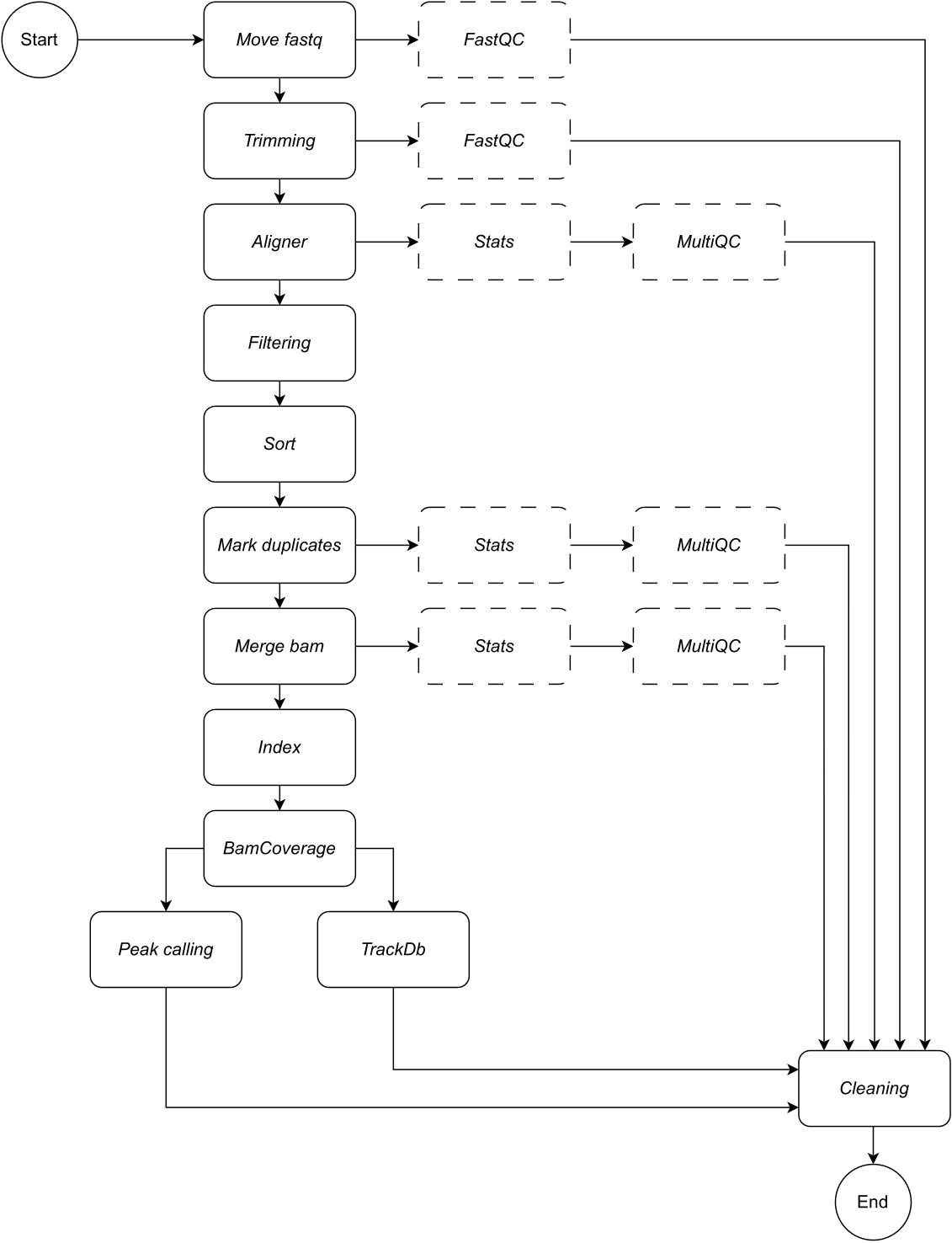
Im Folgenden finden Sie eine detaillierte Beschreibung der einzelnen Schritte des Workflows (siehe Abbildung 1 für die Abbildung des Workflows). Diese Erklärung ist unerlässlich, um der Schritt-für-Schritt-Anleitung im Protokollteil folgen zu können:
Verschieben von fastq: Der erste Schritt der Pipeline besteht darin, die unformatierten fastq-Dateien in das benannte Analyseverzeichnis zu kopieren. Dadurch bleiben die Originaldaten unberührt, um eine Beschädigung oder Änderung der Rohdatendateien zu vermeiden.
Verketten: Wenn Rohsequenzierungsdaten mehrere Lanes enthalten, ist dieser Schritt erforderlich, um die Lanes vor der Analyse zu verketten. Standardmäßig behandelt die Pipeline alle fastq-Dateien als einzelne Beispiele. Dieser Verkettungsschritt muss in der Konfigurationsdatei definiert werden.
Trimming: optionaler Schritt zur Datenbereinigung. Dies ermöglicht das Trimmen von Lesevorgängen oder Adaptersequenzen mit geringer Qualität durch den Einsatz von trimmomatic15. Der Benutzer kann benutzerdefinierte Fasta-Dateien von Adaptersequenzen bereitstellen. Ein Beispiel finden Sie im Verzeichnis Adapter. In der Konfigurationsdatei können zusätzliche Trimmparameter definiert werden. Standardmäßig überspringt der Workflow diese Regel.
Aligner: Für die Ausrichtung wird standardmäßig Bowtie216 angewendet; Alternative Ausrichtungswerkzeuge wie BWA-MEM217 können ebenfalls angegeben werden. Das Bowtie2-Alignment-Tool ist standardmäßig ausgewählt, da es besonders gut darin ist, relativ kurze Reads an relativ große Genome anzupassen und daher gut für die Ausrichtung von ChIP-seq- und ATAC-seq-Daten an Säugetiergenomen geeignet ist. Um Zwischendateien zu vermeiden, wird der Aligner in die samtools-Ansicht geleitet, um die BAM-Datei in der Ausgabe zu speichern. Für diese Regel muss der Benutzer den bevorzugten Genom-Build angeben, auf dem die Reads abgebildet werden sollen, z. B. hg19/hg38 (Mensch), mm10/mm39 (Maus).
Filterung: Korrekt zugeordnete Lesevorgänge werden beibehalten, und Lesevorgänge mit geringer Qualität werden herausgefiltert. Standard: samtools-Ansicht, mit den Parametern: -bShuF 4 -f 3 -q 30.
Sortieren: Ausgerichtete Lesevorgänge werden in der Reihenfolge der Koordinate ganz links sortiert. Standard: samtools sort (snakemake wrapper), mit dem Parameter: -m 4G.
Duplikate markieren: Alle doppelten Lesevorgänge werden identifiziert und gekennzeichnet. Der Benutzer kann entscheiden, ob er sie entfernen möchte, indem er den Parameter der Konfigurationsdatei ändert. Standard: Picard MarkDuplicates (snakemake wrapper), mit dem Parameter: --REMOVE_DUPLICATES False, um Duplikate zu markieren und beizubehalten.
Merge bam: Wenn die Sequenzierungsdaten aus Replikaten oder Stichproben bestehen, möchte der Benutzer möglicherweise in einem einzelnen bam zusammenführen. In diesem Fall kann der Benutzer wählen, ob er die BAMs zusammenführen oder die BAM-Dateien während der gesamten Analyse getrennt halten möchte. Wenn der Benutzer sich für das Zusammenführen von BAMs entscheidet (Verwendung von samtools merge), muss ein gemeinsames Präfix für die zusammengeführten BAMs angegeben werden.
Index: In diesem Schritt werden die sortierten Koordinaten indiziert. Standard: samtools-Index (snakemake-Wrapper), unter Verwendung von Standardparametern, die von samtools festgelegt wurden.
BamCoverage: Diese Regel erstellt eine Bigwig-Abdeckungsspur aus ausgerichteten Lesevorgängen. Das Werkzeug bamCoverage von deepTools wird angewendet, und die Abdeckung wird als die Anzahl der Lesevorgänge pro Bin berechnet, in der die Bin ein Fenster einer bestimmten Größe darstellt. In dieser Pipeline wird bamCoverage angewendet, wobei die folgenden Parameter als Standard festgelegt sind: -bs 1 -normalizeUsing RPKM -extendReads.
Peak Calling: LanceOtron8 wurde als Standard-Peakcaller für diese Pipeline ausgewählt. Im Gegensatz zu herkömmlichen Peak-Callern, die meist statistisch testbasiert sind, ist LanceOtron ein Deep-Learning-basierter Peak-Caller, der genomische Anreicherungsmessungen und statistische Tests umfasst und nachweislich den Industriestandard-Peak-Caller MACS29 übertrifft. Damit Bonzen mit LanceOtron kompatibel sind, muss die Abdeckung pro Basenpaar berechnet und RPKM normalisiert werden. Dies spiegelt sich in den Standardeinstellungen für den Schritt BamCoverage wider. MACS2 kann als alternativer Peak-Caller ausgewählt werden. Die Veröffentlichung neuer Peak-Caller wird überwacht und gegebenenfalls integriert, um die Leistung dieser Analysepipeline aufrechtzuerhalten und zu optimieren.
TrackDb: Hiermit wird eine Schlüssel-Wert-Paar-Assoziation von Bigwig-Dateien erstellt, um sie in Tools wie MLV6 oder UCSC Genome Browser18 zu laden und zu visualisieren.
Zusätzlich zu den Ausgabedaten wird für jeden Schritt der Pipeline eine Protokolldatei ausgegeben, und es werden entsprechende Qualitätskontrollen bereitgestellt, damit der Benutzer den Fortschritt der Analyse verfolgen kann. FastQC19 wird auf rohe und getrimmte (falls ausgewählt) Sequenzierungsdaten angewendet (Schritte 1 - Verschieben von fastq und 2 - Trimmen). Samtools stats plus MultiQC20 werden verwendet, um Qualitätskontrollberichte für bam-Dateien in der Ausgabe in den Schritten 3 - Aligner, 6 - Duplikate markieren und 7 - bam zusammenführen" zu sammeln, zu erstellen und zu visualisieren. Weitere Informationen zu den einzelnen Werkzeugen, die in den obigen Schritten angewendet wurden, finden Sie in Tabelle 1.
Protokoll
1. Ausführen der CATCH-UP-Pipeline
- Klonen Sie das UpStreamPipeline-Repository aus https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Navigieren Sie zum ausgewählten Arbeitsverzeichnis, kopieren Sie den folgenden Code, und führen Sie ihn auf der Befehlszeile aus:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Navigieren Sie in den UpStreamPipeline-Ordner, den Sie mit dem folgenden Befehl heruntergeladen haben: cd UpStreamPipeline
- Installieren Sie die Anaconda-Distribution (falls erforderlich):
- Überprüfen Sie, ob Anaconda bereits auf dem System installiert ist, indem Sie den Befehl which conda verwenden. Wenn der Befehl keinen Pfad zu einer Conda-Distribution anzeigt, laden Sie mambaforge von https://github.com/conda-forge/miniforge#mambaforge herunter und wählen Sie die richtige Distribution und Version für das System aus. Verwenden Sie beispielsweise für Linux-Benutzer wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Besuchen Sie diese Webseite für verschiedene Betriebssysteme: https://github.com/conda-forge/miniforge/.
- Führen Sie das Installationsprogramm mit sh Mambaforge-Linux-x86_64.sh aus und initialisieren Sie conda im System, indem Sie conda init ausführen.
- Installieren und aktivieren Sie die Upstream-Conda-Umgebung (Anforderungen an die Upstream-Conda-Umgebung sind in Tabelle 2 aufgeführt):
- Installieren Sie die Umgebung mit dem Befehl mamba env create - file=envs/upstream.yml.
- Aktivieren Sie die Umgebung mit dem Befehl conda activate upstream.
- Sobald die Upstream-Conda-Umgebung erfolgreich installiert wurde, aktivieren Sie die Umgebung mit dem Befehl conda activate upstream und navigieren Sie mit cd genetics/CATCH-UP zum CATCH-UP-Ordner.
- Bearbeiten Sie die Konfigurationsdatei, die sich im Konfigurationsordner befindet, mit dem Befehl cd /config/analysis.yaml und ändern Sie sie entsprechend der Analysespezifikation mit einem Texteditor. Befolgen Sie die zeilenweisen Anweisungen, um jeden Parameter in der Datei selbst zu bearbeiten. Diese Datei wird nach der Analyse aufbewahrt und dient zur Dokumentation der Laufparameter, um die Reproduzierbarkeit zu verbessern.
- Öffnen und bearbeiten Sie die folgenden drei Dateien in einem Texteditor (z. B. TextEdit für Mac oder Notepad für Windows):
- Bearbeiten Sie 1_fastqfile_home_dir.txt Datei so, dass sie eine Liste aller zu analysierenden Fastq-Dateien enthält.
HINWEIS: Lesenummern und Durchwahlen (z. B. _R1/_R2 und .fastq.gz) müssen ausgeschlossen werden. Wenn ein Projekt beispielsweise diese Liste von fastq-Dateien enthält:
Sample1_conditionA_L001_R1 . fastq . GZ
Sample1_conditionA_L001_R2 . fastq . GZ
Sample1_conditionA_L002_R1 . fastq . GZ
Sample1_conditionA_L002_R2 . fastq . GZ
Sample1_conditionB_L001_R1 . fastq . GZ
Sample1_conditionB_L001_R2 . fastq . GZ
Sample1_conditionB_L002_R1 . fastq . GZ
Sample1_conditionB_L002_R2 . fastq . GZ
Sample2_conditionA_L001_R1 . fastq . GZ
Sample2_conditionA_L001_R2 . fastq . GZ
Sample2_conditionA_L002_R1 . fastq . GZ
Sample2_conditionA_L002_R2 . fastq . GZ
Sample2_conditionB_L001_R1 . fastq . GZ
Sample2_conditionB_L001_R2 . fastq . GZ
Sample2_conditionB_L002_R1 . fastq . GZ
Sample2_conditionB_L002_R2 . fastq . GZ
In diesem Fall sieht die 1_fastqfile_home_dir.txt wie folgt aus:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Wenn Rohdaten Sequenzierungsspuren enthalten, die eine Verkettung erfordern, bearbeiten Sie 2_fastq Fi-le_concat.txt-Datei, um das Präfix der zu verkettenden Dateinamen zu definieren. Wenn keine Sequenzierungsspuren zum Verketten vorhanden sind, bearbeiten Sie 2_fastqfi-le_concat.txt nicht. Stellen Sie sicher, dass jede Zeile 2_fastqfi-le_concat.txt ein Beispielpräfix wie folgt enthält:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Wenn das Zusammenführen der Daten verschiedener Beispiele erforderlich ist, bearbeiten Sie 3_merge_bams.txt Datei mit dem Präfix der zusammenzuführenden Dateinamen. Stellen Sie sicher, dass jede Zeile ein Beispielpräfix wie folgt enthält:
Beispiel1
Beispiel2
Abbildung 2 zeigt eine Zusammenfassung der Zusammenfassung dieser drei Dateien. Das Protokoll kann entweder auf Single- oder Paired-End-Sequenzierungsdaten angewendet werden. Sofern nicht anders angegeben, wird für die Pipeline standardmäßig die Analyse mit gekoppeltem Ende verwendet. Dies kann in der Konfigurationsdatei geändert werden (siehe Schritt 1.6).
- Bearbeiten Sie 1_fastqfile_home_dir.txt Datei so, dass sie eine Liste aller zu analysierenden Fastq-Dateien enthält.
- Nachdem alle erforderlichen Dateien bearbeitet wurden, verwenden Sie snakemake, um CATCH-UP wie folgt auszuführen: snakemake --configfile=config/analysis_name.yaml all --cores 4.
HINWEIS: Ausführlichere Anweisungen und Dokumentationen finden Sie im Ordner CATCH-UP im UpStreamPipeline-GitHub-Repository, das hier verfügbar ist. Dazu gehört eine detaillierte Dokumentation zum korrekten Ändern der Konfigurationsdatei, vom Ändern von Pfaden für die Sequenzierung von Datendateien über das Speichern von Ergebnissen bis hin zum Bearbeiten von Parametern für jeden Schritt.
Ergebnisse
Die CATCH-UP-Pipeline erzeugt für jeden Schritt ein Ergebnis, ein Protokoll und eine Qualitätskontrollausgabe (QC). Innerhalb der Konfigurationsdatei kann der Benutzer wählen, ob er Ausgabedateien behalten oder entfernen möchte, um den erforderlichen Speicherplatz zu reduzieren. Alle Ausgaben werden wie folgt erklärt:
00. fastq_home_dir: Konfigurationsdatei, FastQFi-le_home_dir.txt und merge_bams.txt werden zur Referenz und Reproduzierbarkeit in diesen Ordner kopiert.
01. Lesevorgänge: FastQ-Dateien werden in diesen Ordner kopiert, um Änderungen an den ursprünglichen Rohdaten während des Workflow-Prozesses zu vermeiden, Lanes können verkettet werden, falls angegeben.
02. Trimmen: FastQ-Dateien mit Lese- und Adaptern getrimmt, falls angegeben.
03. Aligner: Ausrichtung auf das ausgewählte Genom.
04. Filtern: Qualitätskontrolle Filtern.
05. Sortiert: Sortierung von BAM-Dateien.
06. Duplikate: Kennzeichnung von Duplikaten.
07. Merge: Zusammenführen von BAM-Dateien, wenn dies in config.yaml angegeben wurde.
08. bam_coverages: Große Akte der Berichterstattung.
09. peak_calling: eine Bettdatei mit dem Ausgang des LanceOtron-Peak-Aufrufs.
10. Track: Erstellt eine formatierte Textdatei, die bei Bedarf im Genome Browser verwendet werden kann.
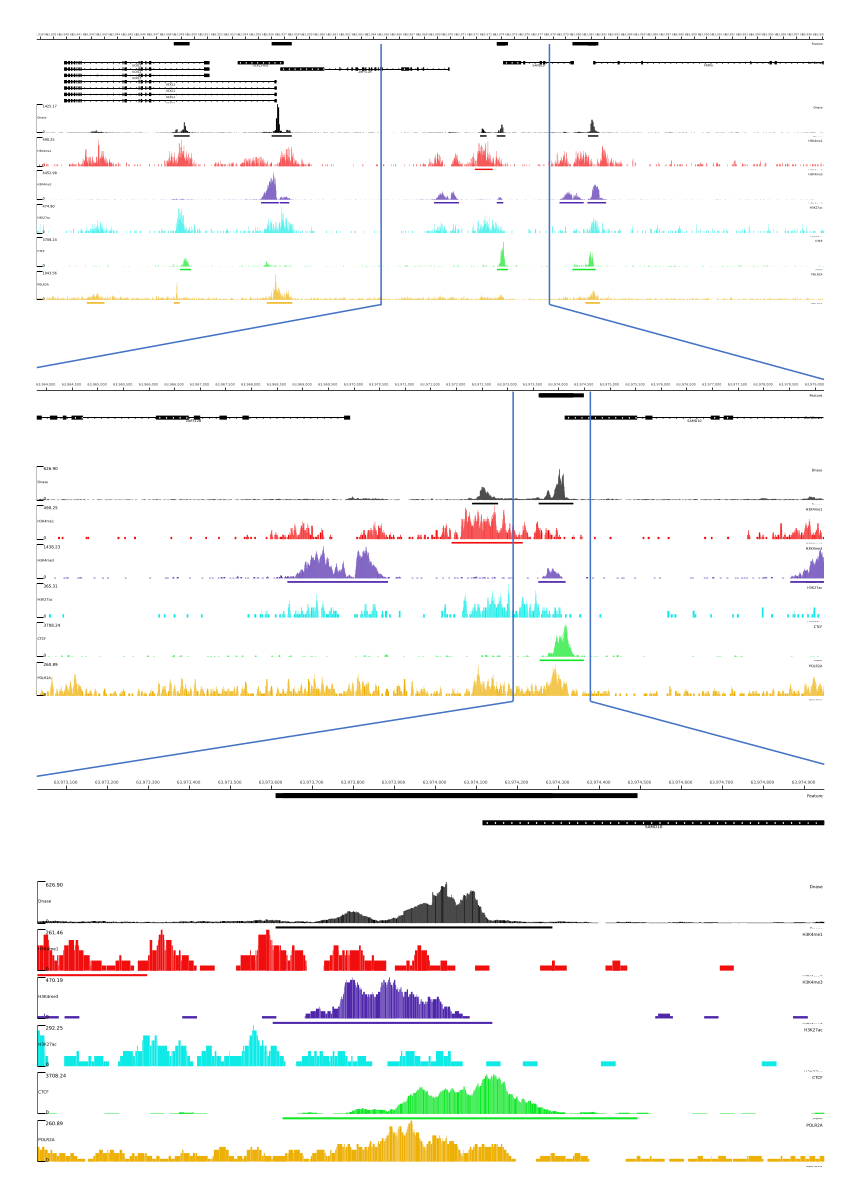
Für die Ausgaben 01, 02, 03, 06 und 07 werden QC-Metriken und HTML-Dateien bereitgestellt. Darüber hinaus zeigen wir in Abbildung 3 ein Beispiel für die Verarbeitung von Daten mit CATCH-UP, wobei die endgültige Ausgabe über die MLV-Plattform visualisiert wird.

Abbildung 1: Ablauf von CATCH-UP. Anhand einer Liste von fastq-Dateien verarbeitet CATCH-UP parallel alle Samples durch alle Upstream-Schritte. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Illustrative Darstellung, die erklärt, wie 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt und 3_merge_bams.txt korrekt geändert werden müssen, um CATCH-UP durchzuführen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Beispielausgabe der CATCH-UP-Pipeline. Rohsequenzierungsdaten (fastq-Dateien) wurden von ENCODE21 heruntergeladen. Die CATCH-UP-Pipeline wurde verwendet, um die fastq-Dateien für DNase-seq und 5 Typen von ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF und POLR2A) zu verarbeiten. Bigwig-Ausgabedateien wurden zur Visualisierung und Identifizierung genomischer regulatorischer Elemente in Multi Locus View hochgeladen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Tabelle 1: Dokumentationsressourcen. Diese Tabelle zeigt die am CATCH-UP-Workflow beteiligten Werkzeuge, den Link zu ihrer Dokumentation und die entsprechenden Referenzen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 2: Liste der Kanal- und Abhängigkeitsanforderungen für die Upstream-Conda-Umgebung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 3: Betriebssysteme, die zum Testen von CATCH-UP verwendet wurden. Ubuntu wurde auf einem Hochleistungscluster und einem lokalen Rechner getestet. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Die zunehmende Akzeptanz und Nutzung von NGS-Techniken zur Generierung genomischer Daten ging einher mit einer zunehmenden Entwicklung von Bioinformatik-Tools für die Analyse dieser Daten. Es gibt mehrere Werkzeuge, die für jeden Schritt der Datenanalyse angewendet werden können, sowie viele verschiedene Parameter, die in jedem Werkzeug angegeben werden können 6,8,9,15,16,17,18,19,20,22,23,24 . Dies führt zu einer sehr vielfältigen Kombination von Analysestrategien, die angewendet werden können, von denen jede zu Variationen im Ergebnis führen kann. Um einen genauen Vergleich zwischen Experimenten zu ermöglichen, ist eine Standardisierung der bioinformatischen Analyse unerlässlich. In der Vergangenheit wurden NGS-Daten von Nasslaborwissenschaftlern generiert und von Bioinformatikern analysiert.
Die NGS-Datenanalyse kann in "Upstream"- und "Downstream"-Pipelines unterteilt werden, wobei Upstream die notwendigen Schritte umfasst, um von der Rohdatenausgabe einer Sequenziermaschine zu einem Format zu gelangen, das von einem Forscher visuell interpretiert werden kann. Die nachgelagerte Analyse umfasst zusätzliche Schritte, die auf die Forschungsfrage und das Versuchsdesign zugeschnitten sind. Vorgelagerte Pipelines sind daher verallgemeinerbar und können für eine verbesserte wissenschaftliche Reproduzierbarkeit standardisiert werden. Downstream-Pipelines hingegen sind maßgeschneidert, abhängig von der biologischen Fragestellung und erfordern Einblicke des Forschers, was sie für eine Standardisierung weniger geeignet macht. Wir haben eine benutzerfreundliche Upstream-Pipeline geschaffen, die es Nasslaborwissenschaftlern ermöglicht, ihre eigenen Daten reproduzierbar zu analysieren, ohne dass Vorkenntnisse in der Bioinformatik erforderlich sind. Hier stellen wir CATCH-UP vor, eine Pipeline, die auf dem snakemake-Framework basiert und sowohl benutzerfreundlich ist als auch das Problem der Reproduzierbarkeit bei der ChIP-seq- und ATAC-seq-Datenanalyse bekämpft. Diese Pipeline wurde für die Verarbeitung von ChIP-seq- oder ATAC-seq-Daten erstellt. Nachdem der Benutzer CATCH-UP heruntergeladen hat, müssen zunächst die Analyseparameter und die Benennung der Stichprobe definiert werden, bevor die Pipeline auf der Befehlszeile mit einer einzigen Codezeile ausgeführt wird. Eine einfache Schritt-für-Schritt-Anleitung zum Anpassen der Analyseparameter für die ChIP-seq- oder ATAC-seq-Analyse finden Sie in der Konfigurationsdatei selbst und in unserer Schritt-für-Schritt-Anleitung im CATCH-UP GitHub-Repository.
Es gibt bereits Analysepipelines für ChIP-seq- oder ATAC-seq-Daten, wie z. B. PEPATAC und ChIP-AP. Während diese Pipelines Vorteile bieten, wie z. B. die Integration von vor- und nachgelagerten Analysen in einen einzigen Workflow oder die Verwendung einer grafischen Benutzeroberfläche (GUI), richten sich diese Tools an Bioinformatiker und Wissenschaftler mit einem moderaten Maß an rechnerischer Ausbildung13,14. CATCH-UP wurde entwickelt, um zwei Probleme zu lösen: Nasslaborwissenschaftlern ohne bioinformatische Ausbildung die Durchführung ihrer eigenen vorgelagerten Analysen zu ermöglichen und die Standardisierung der vorgelagerten Analyse zu ermöglichen, indem eine einfache Berichterstattung und exakte Reproduzierbarkeit über Labore hinweg ermöglicht wird. CATCH-UP ist absichtlich auf die vorgelagerte Analyse beschränkt, aber die Ergebnisse sind kompatibel mit nachgelagerten Analysewerkzeugen, wie z. B. denen, die zum statistischen Vergleich von Datensätzen oder zur Ableitung der Transkriptionsfaktorbindung verwendet werden25,26.
Alle kritischen Schritte, die für die Durchführung einer replizierbaren Upstream-Analyse erforderlich sind, sind innerhalb der CATCH-UP-Pipeline vordefiniert, um die Robustheit zu gewährleisten. Die ausführliche Natur dieser Pipeline ermöglicht es dem Benutzer, die Ausgabe der Pipeline Schritt für Schritt zu verfolgen, was sowohl für die Problembehandlung als auch für die Replikation des analytischen Workflows nützlich ist. Angesichts der sich schnell entwickelnden Natur von NGS-Techniken ist der modulare Charakter dieser Pipeline von Vorteil, da sie die Möglichkeit bietet, leicht angepasst zu werden, um sowohl die Veröffentlichung von Tool-Versionsupdates als auch die Implementierung neuer Tools zu berücksichtigen. CATCH-UP wurde erfolgreich für die folgenden Betriebssysteme getestet: Ubuntu, CentOS, macOS (Intel-CPU) und Windows (Tabelle 3). Die Pipeline wurde entwickelt, um große Experimente mit Dutzenden von Proben zu bewältigen, indem der Arbeitsablauf parallelisiert und an verschiedene Versuchsdesigns angepasst werden kann. Insgesamt ermöglicht die Implementierung von CATCH-UP bei der Analyse von ChIP-seq- und ATAC-seq-Daten einen benutzerfreundlichen, reproduzierbaren und hochgradig anpassungsfähigen Analyse-Workflow.
Offenlegungen
J.R.H. ist Mitbegründer und Direktor von Nucleome Therapeutics und berät das Unternehmen.
Danksagungen
J.R.H. wurde durch Zuschüsse des Wellcome Trust (225220/Z/22/Z und 106130/Z/14/Z) und des MRC (MC_UU_00029/3) unterstützt. M.B. wurde durch das Wellcome Trust Grant (225220/Z/22/Z) unterstützt. E.R.G. wurde unterstützt durch das Stipendium des Ministeriums für nationale Bildung, Auswahl und Vermittlung von Kandidaten, die für die postgraduale Ausbildung ins Ausland geschickt wurden (YLSY), Ministerium für nationale Bildung der Republik Türkiye. E.G. wurde durch das Wellcome Genomic Medicine and Statistics PhD Programme (108861/Z/15/Z) unterstützt. S.G.R. wurde durch das Medical Research Council (MRC) Grant (MC_UU_00029/3) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Referenzen
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623 (2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521 (2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037 (2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101 (2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537 (2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008 (2021).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten