
Method Article
CATCH-UP: высокопроизводительный восходящий конвейер для больших объемов данных ATAC-seq и ChIP-seq
В этой статье
Резюме
ATAC-seq и ChIP-seq позволяют детально исследовать регуляцию генов; Однако обработка этих типов данных является сложной задачей и часто не согласуется между исследовательскими группами. Мы представляем CATCH-UP: простой в использовании вычислительный конвейер, который обеспечивает стандартизированную и воспроизводимую обработку данных и анализ новых и опубликованных наборов данных ATAC/ChIP-seq.
Аннотация
Анализ на доступный для транспозазы хроматин (ATAC) и иммунопреципитация хроматина (ChIP) в сочетании с секвенированием нового поколения (NGS) произвели революцию в изучении регуляции генов. Отсутствие стандартизации в анализе многомерных наборов данных, полученных с помощью этих методов, затруднило достижение воспроизводимости, что привело к расхождениям в опубликованных, обработанных данных. Отчасти эта проблема связана с разнообразием биоинформационных инструментов, доступных для анализа этих типов данных. Во-вторых, для последовательного преобразования исходных данных в полностью обработанный и интерпретируемый результат требуется ряд различных биоинформационных инструментов, и эти инструменты требуют разного уровня вычислительных навыков. Кроме того, существует множество вариантов контроля качества, которые не всегда используются при обработке данных. Мы решаем эти проблемы с помощью полного анализа для транспозазного доступного секвенирования хроматина (ATAC-seq) и иммунопреципитационного секвенирования хроматина (ChIP-seq) восходящего конвейера (CATCH-UP), простого в использовании конвейера на основе Python для анализа массивных наборов данных ChIP-seq и ATAC-seq от необработанных файлов fastq до визуализируемых треков больших шишек и пиковых вызовов. Этот конвейер прост в установке и запуске, требуя минимальных вычислительных знаний. Конвейер является модульным, масштабируемым и распараллеливаемым в различных вычислительных инфраструктурах, что позволяет легко сообщать о методологии для воспроизводимого анализа новых или опубликованных наборов данных.
Введение
Экспрессия генов должна строго регулироваться для того, чтобы клетки могли установить и поддерживать свою правильную биологическую функцию. Хорошо известно, что аберрантная экспрессия генов лежит в основе патогенеза многих заболеваний, и поэтому большой исследовательский интерес заключается в пониманиимеханизмов регуляции генов. Экспрессия генов облегчается регуляторными элементами, такими как промоторы и энхансеры. В своей последовательности эти элементы содержат сайты связывания фактора транскрипции (TF), которые при активации обеспечивают платформу для связывания TF. Связывание ТФ в этих сайтах приводит к смещению нуклеосом, что приводит к увеличению доступности ДНК и последующему увеличению допустимости для транскрипционного механизма. В результате этой повышенной доступности эти участки ДНК более чувствительны к нуклеазам и транспозазам, таким как ДНКаза и Tn5, биохимическое свойство, которое было использовано исследователями, изучающими транскрипционную регуляцию 2,3.
DNase-seq и ATAC-seq позволяют исследователям картировать области открытого хроматина, сайты связывания TF и нуклеосомное расположение по всему геному. Из этих двух методов популярность ATAC-seq выросла за последнее десятилетие благодаря простому двухэтапному протоколу и низкому требованию к количеству клеток (50 000 клеток по сравнению с 1 миллионом на репликацию для DNase-seq). В то время как ATAC-seq дает представление об общем ландшафте хроматина в популяции клеток, он в значительной степени не имеет значения, с какими специфическими белками связываются с геномом 4,5. Для того, чтобы определить места, где конкретный белок взаимодействует с геномом, золотым стандартом является иммунопреципитация хроматина (ChIP)-секвенирование. ChIP-seq включает в себя химическую фиксацию белок-ДНК-взаимодействий в клетке с последующей иммунопреципитацией («pull-down») с использованием антитела, специфичного к интересующему белку, для отбора фрагментов ДНК, связанных с интересующим белком (POI). Эти фрагменты ДНК могут быть секвенированы для выявления мест геномного связывания специфических белков, таких как TF, или сайтов, содержащих специфические модификации гистонов1. Комбинируя наборы данных ATAC-seq и ChIP-seq, можно получить подробную картину регуляторного ландшафта для популяции клеток.
Основной рабочий процесс, необходимый для анализа, заключается в следующем: качество необработанных прочитанных данных секвенирования должно контролироваться перед выравниванием с референсным геномом («картированием»). Успешно картированные чтения затем могут быть отфильтрованы для удаления как некачественных прочтений, так и дубликатов ПЦР. Для того, чтобы визуализировать эти картированные и отфильтрованные чтения, необходимо рассчитать «покрытие» этих прочтений по всему геному. При этом генерируется файл, который может быть загружен в браузер генома, такой как мультилокусный просмотр (MLV) или браузер генома UCSC в качестве «трека»6,7. Идентификация пиков, или «вызов пиков» этих треков покрытия, обычно достигается с помощью таких инструментов, как LanceOtron или MACS2 8,9. Наконец, с помощью анализа расположения, формы и размера пика можно провести сравнение образцов или биологических условий. Анализ и интеграция этих наборов данных представляет собой сложный многоступенчатый процесс, в котором могут быть реализованы различные комбинации биоинформационных инструментов. Различные версии инструментов могут быть несовместимы друг с другом и могут изменить вывод обработки данных. Кроме того, существует большое разнообразие вычислительной мощности и квалификации пользователя, необходимых для реализации различных частей обработки данных, как показано в конвейерах nf-core10, panpipes11, genpipe12, PEPATAC13 или ChIP-AP14.
В целом, это привело к несоответствиям как в анализе, так и в отчетности по анализу, что, в свою очередь, привело к плохой воспроизводимости, доступности и удобству для тех, кто обладает ограниченными знаниями в области биоинформатики. Мы решаем все эти проблемы с помощью CATCH-UP (полный восходящий конвейер ATAC-seq и ChIP-seq), простого в использовании, гибкого и модульного конвейера для обработки данных ChIP-seq и ATAC/DNase-seq. Внедрение CATCH-UP требует минимального опыта в области биоинформатики; Он может быть запущен в различных вычислительных инфраструктурах и обеспечивает воспроизводимый анализ данных внутри и между исследовательскими группами.
CATCH-UP — это конвейер Snakemake на основе Python, созданный для стандартизации анализа данных ChIP-seq и ATAC-seq. Он принимает необработанные данные секвенирования (fastq.gz файлы) в качестве входных данных и генерирует выходные данные в виде файлов peak (.bed), предоставляющих соответствующий результат для каждого шага. Мы предоставляем конфигурационный файл в формате yaml (config.yaml), в котором пользователь может редактировать параметры каждого шага анализа. Система управления, реализованная в snakemake, позволяет использовать различные вычислительные инфраструктуры (например, серверы, кластеры, облачные системы или персональные компьютеры) и параллельно, если пользователь предоставляет большой объем данных.
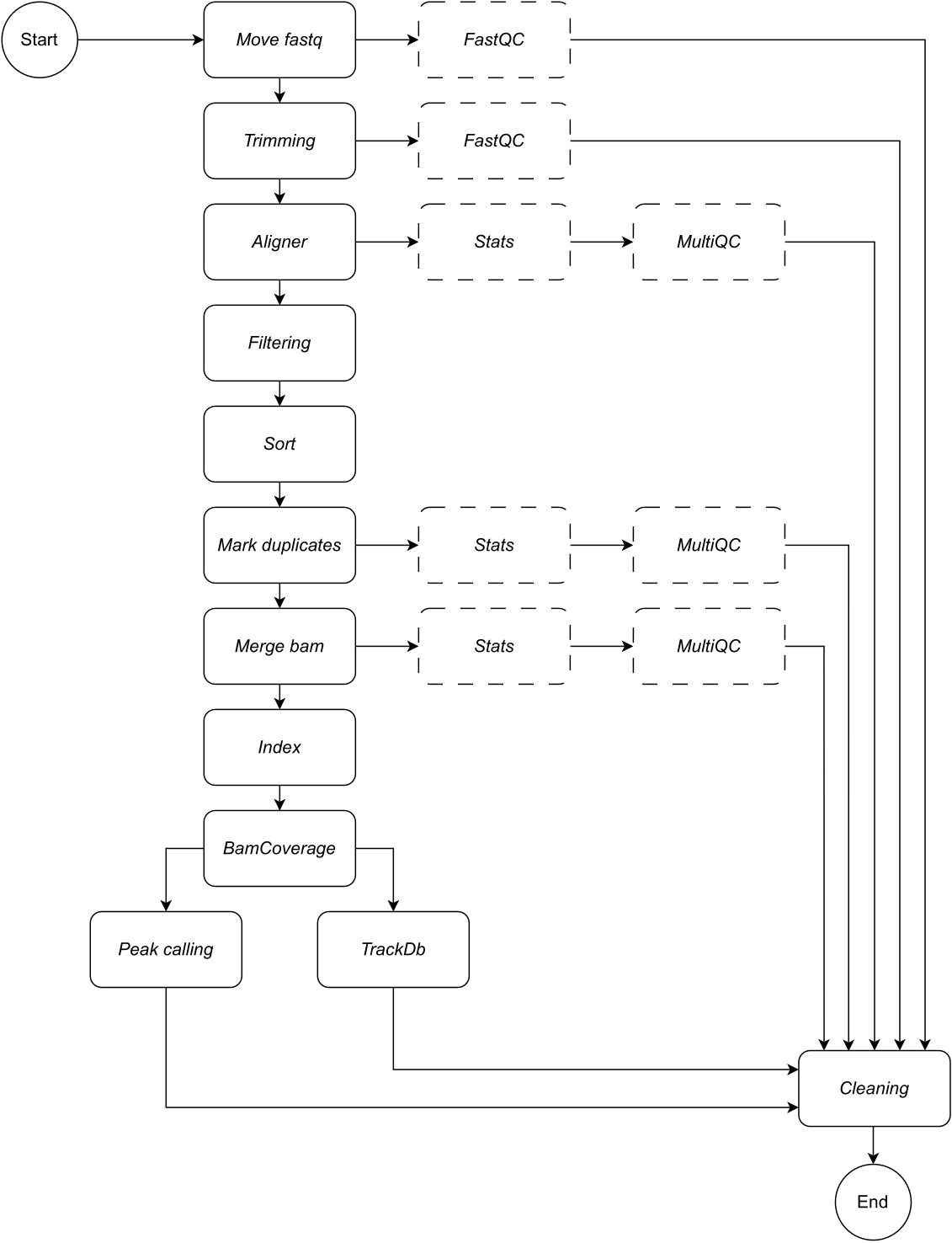
Ниже приведено подробное описание каждого шага рабочего процесса (иллюстрацию рабочего процесса см. на рисунке 1 ). Это объяснение необходимо для того, чтобы следовать пошаговым инструкциям в разделе протокола:
Перемещение fastq: первым шагом конвейера является копирование необработанных файлов fastq в указанный каталог анализа. При этом исходные данные остаются нетронутыми, чтобы избежать повреждения или изменения файлов необработанных данных.
Конкатенация: если исходные данные секвенирования содержат несколько полос движения, этот шаг необходим для объединения полос перед анализом. По умолчанию конвейер обрабатывает все fastq-файлы как отдельные выборки. Этот шаг конкатенации должен быть определен в файле конфигурации.
Обрезка: необязательный шаг очистки данных. Это позволяет обрезать некачественные чтения или последовательности адаптеров с помощью trimmomatic15. Пользователь может предоставить пользовательские fasta файлы последовательностей адаптеров; Пример приведен в каталоге адаптера. Дополнительные параметры обрезки могут быть определены в файле конфигурации. По умолчанию рабочий процесс пропускает это правило.
Aligner: для выравнивания по умолчанию применяется Bowtie216 ; Также можно указать альтернативные инструменты юстировки, такие как BWA-MEM217 . Инструмент выравнивания Bowtie2 выбран по умолчанию, поскольку он особенно хорошо подходит для выравнивания относительно коротких прочтений относительно больших геномов и, следовательно, хорошо подходит для выравнивания данных ChIP-seq и ATAC-seq с геномами млекопитающих. Чтобы избежать каких-либо промежуточных файлов, выравниватель передается по конвейеру в представление samtools для сохранения файла bam на выходе. Для этого правила пользователь должен указать предпочтительную сборку генома, на которую будут сопоставлены чтения, например, hg19/hg38 (человек), mm10/mm39 (мышь).
Фильтрация: правильно сопоставленные чтения сохраняются, а чтения с низким качеством отфильтровываются. По умолчанию: вид samtools, с параметрами: -bShuF 4 -f 3 -q 30.
Сортировка: выровненные чтения сортируются в порядке крайней левой координаты. По умолчанию: samtools sort (обертка snakemake), с параметром: -m 4G.
Пометить дубликаты: все дубликаты чтения идентифицируются и помечаются. Пользователь может принять решение об их удалении, изменив параметр файла конфигурации. По умолчанию: Picard MarkDuplicates (обёртка snakemake), с параметром: --REMOVE_DUPLICATES False для пометки и сохранения дубликатов.
Merge bam: Если данные секвенирования состоят из реплик или образцов, пользователь может захотеть объединить их в один bam. В этом случае пользователь может выбрать слияние bams или хранить файлы bam отдельно на протяжении всего анализа. Если пользователь выбирает слияние bams (с помощью samtools merge), для объединенного bams должен быть указан общий префикс.
Индекс: на этом шаге индексируются отсортированные координаты. По умолчанию: индекс samtools (оболочка snakemake), с использованием параметров по умолчанию, заданных samtools.
BamCoverage: это правило создает большой трек покрытия из выровненных прочтений. Применяется инструмент bamCoverage от deepTools, и покрытие рассчитывается как количество прочтений на ячейку, в которой ячейка представляет собой окно заданного размера. В этом конвейере bamCoverage применяется со следующими параметрами, установленными по умолчанию: -bs 1 -normalizeUsing RPKM -extendReads.
Пиковый вызов: LanceOtron8 был выбран в качестве пикового вызова по умолчанию для этого конвейера. В отличие от традиционных пиковых вызовов, которые в основном основаны на статистических тестах, LanceOtron — это пиковый вызов на основе глубокого обучения, который включает в себя измерения геномного обогащения и статистическое тестирование и, как было показано, превосходит стандартный отраслевой пиковый вызов MACS29. Для того, чтобы большие шишки были совместимы с LanceOtron, покрытие должно быть рассчитано по паре оснований, а RPKM нормализовано; это отражено в настройках по умолчанию для шага BamCoverage. MACS2 может быть выбран в качестве альтернативного пикового вызова. Высвобождение новых пиковых вызовов будет отслеживаться и учитываться по мере необходимости, чтобы поддерживать и оптимизировать производительность этого конвейера анализа.
TrackDb: создает ассоциацию пар «ключ-значение» больших файлов для их загрузки и визуализации в таких инструментах, как платформы MLV6 или UCSC Genome Browser18 .
В дополнение к выходным данным, на каждом этапе конвейера выводится файл журнала, и предоставляются соответствующие проверки качества, чтобы пользователь мог отслеживать ход анализа. FastQC19 применяется к необработанным и обрезанным (если выбранным) данным секвенирования (шаги 1 - Перемещение fastq и 2 - Обрезка). Статистика Samtools плюс MultiQC20 используются для сбора, создания и визуализации отчетов о контроле качества в файлах bam на выходе в шагах 3 - Выравнивание, 6 - Маркировка дубликатов и 7 - Объединение бам. Для получения дополнительной информации о каждом из инструментов, применяемых в вышеуказанных шагах, см. Таблицу 1.
протокол
1. Запуск конвейера CATCH-UP
- Клонируем репозиторий UpStreamPipeline из https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline:
Перейдите в выбранный рабочий каталог, скопируйте следующий код и выполните в командной строке:
git clone git@github.com:Genome-Function-Initiative-Oxford/UpStreamPipeline.git - Перейдите в загруженную папку UpStreamPipeline с помощью команды: cd UpStreamPipeline
- Установите дистрибутив анаконды (при необходимости):
- Проверьте, установлена ли уже анаконда в системе, с помощью команды which conda. Если команда не показывает путь к какому-либо дистрибутиву conda, скачайте mambaforge из https://github.com/conda-forge/miniforge#mambaforge и выберите правильный дистрибутив и версию для системы. Например, для пользователей Linux используйте wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh. Посетите эту веб-страницу для различных операционных систем: https://github.com/conda-forge/miniforge/.
- Запустите установщик с помощью sh Mambaforge-Linux-x86_64.sh и инициализируйте conda в системе, выполнив conda init.
- Установите и активируйте вышестоящую среду conda (требования вышестоящей среды conda перечислены в таблице 2):
- Установите окружение с помощью команды mamba env create - file=envs/upstream.yml.
- Активируйте окружение с помощью команды conda activate upstream.
- После успешной установки вышестоящей среды conda активируйте среду с помощью команды conda activate upstream и перейдите в папку CATCH-UP с помощью cd genetics/CATCH-UP.
- Отредактируйте файл конфигурации, который можно найти в папке config с помощью команды cd /config/analysis.yaml, и измените его в соответствии со спецификацией анализа с помощью текстового редактора. Следуйте построчным инструкциям, чтобы отредактировать каждый параметр в самом файле. Этот файл будет сохранен после анализа и будет использоваться для документирования параметров прогона для обеспечения воспроизводимости.
- Откройте и отредактируйте следующие три файла в текстовом редакторе (например, TextEdit для Mac или Notepad для Windows):
- Отредактируйте 1_fastqfile_home_dir.txt файл, чтобы он содержал список всех файлов fastq для анализа.
ПРИМЕЧАНИЕ: Считывание номеров и добавочных номеров (например, _R1/_R2 и .fastq.gz) должно быть исключено. Например, если проект содержит такой список fastq-файлов:
Sample1_conditionA_L001_R1 . ФастКв . ГЗ
Sample1_conditionA_L001_R2 . ФастКв . ГЗ
Sample1_conditionA_L002_R1 . ФастКв . ГЗ
Sample1_conditionA_L002_R2 . ФастКв . ГЗ
Sample1_conditionB_L001_R1 . ФастКв . ГЗ
Sample1_conditionB_L001_R2 . ФастКв . ГЗ
Sample1_conditionB_L002_R1 . ФастКв . ГЗ
Sample1_conditionB_L002_R2 . ФастКв . ГЗ
Sample2_conditionA_L001_R1 . ФастКв . ГЗ
Sample2_conditionA_L001_R2 . ФастКв . ГЗ
Sample2_conditionA_L002_R1 . ФастКв . ГЗ
Sample2_conditionA_L002_R2 . ФастКв . ГЗ
Sample2_conditionB_L001_R1 . ФастКв . ГЗ
Sample2_conditionB_L001_R2 . ФастКв . ГЗ
Sample2_conditionB_L002_R1 . ФастКв . ГЗ
Sample2_conditionB_L002_R2 . ФастКв . ГЗ
В этом случае 1_fastqfile_home_dir.txt заключается в следующем:
Sample1_conditionA_L001
Sample1_conditionA_L002
Sample1_conditionB_L001
Sample1_conditionB_L002
Sample2_conditionA_L001
Sample2_conditionA_L002
Sample2_conditionB_L001
Sample2_conditionB_L002 - Если необработанные данные содержат полосы секвенирования, требующие конкатенации, отредактируйте файл 2_fastqfile_concat.txt, чтобы определить префикс имен файлов, которые должны быть объединены. Если нет полос секвенирования для конкатенации, не редактируйте 2_fastqfile_concat.txt. Убедитесь, что каждая строка le_concat.txt 2_fastqfi содержит один образец префикса, как показано ниже:
Sample1_conditionA
Sample1_conditionB
Sample2_conditionA
Sample2_conditionB - Если требуется слияние данных разных образцов, то отредактируйте файл 3_merge_bams.txt префиксом имен файлов, которые нужно объединить. Убедитесь, что каждая строка содержит один образец префикса, как показано ниже:
Образец1
Образец2
На рисунке 2 показана сводка того, как суммировать эти три файла. Протокол может быть применен как к одноконцевым, так и к парным данным секвенирования. Конвейер по умолчанию использует парный анализ, если не указано иное; Это можно изменить в файле конфигурации (см. шаг 1.6).
- Отредактируйте 1_fastqfile_home_dir.txt файл, чтобы он содержал список всех файлов fastq для анализа.
- После того, как все необходимые файлы будут отредактированы, используйте snakemake для запуска CATCH-UP следующим образом: snakemake --configfile=config/analysis_name.yaml all --cores 4.
ПРИМЕЧАНИЕ: Более подробные инструкции и документацию см. в папке CATCH-UP в репозитории UpStreamPipeline GitHub, доступной здесь. Это включает в себя подробную документацию по правильному изменению файла конфигурации, от изменения путей для секвенирования файлов данных и хранения результатов до редактирования параметров для каждого шага.
Результаты
Конвейер CATCH-UP создает результат, журнал и выходные данные контроля качества (QC) для каждого шага. В файле конфигурации пользователь может сохранить или удалить выходные файлы, чтобы уменьшить требуемый объем памяти. Все выходные данные объясняются следующим образом:
00. fastq_home_dir: файл конфигурации, le_home_dir.txt fastqfi и merge_bams.txt копируются в эту папку для справки и воспроизводимости.
01. Чтение: файлы FastQ копируются в эту папку, чтобы избежать изменений исходных исходных данных в процессе рабочего процесса, полосы могут быть объединены, если указаны.
02. Обрезка: обрезаются fastq файлы с чтением и адаптерами, если указано.
03. Выравниватель: выравнивание по выбранному геному.
04. Фильтрация: фильтрация контроля качества.
05. Sorted: Сортировка файлов BAM.
06. Дубликаты: пометка дубликатов.
07. Merge: Объединение файлов BAM, если это было указано в config.yaml.
08. bam_coverages: Большой файл репортажа.
09. peak_calling: подшивка пикового вывода вызова LanceOtron.
10. track: создает отформатированный текстовый файл, готовый к использованию в Genome Browser при необходимости.
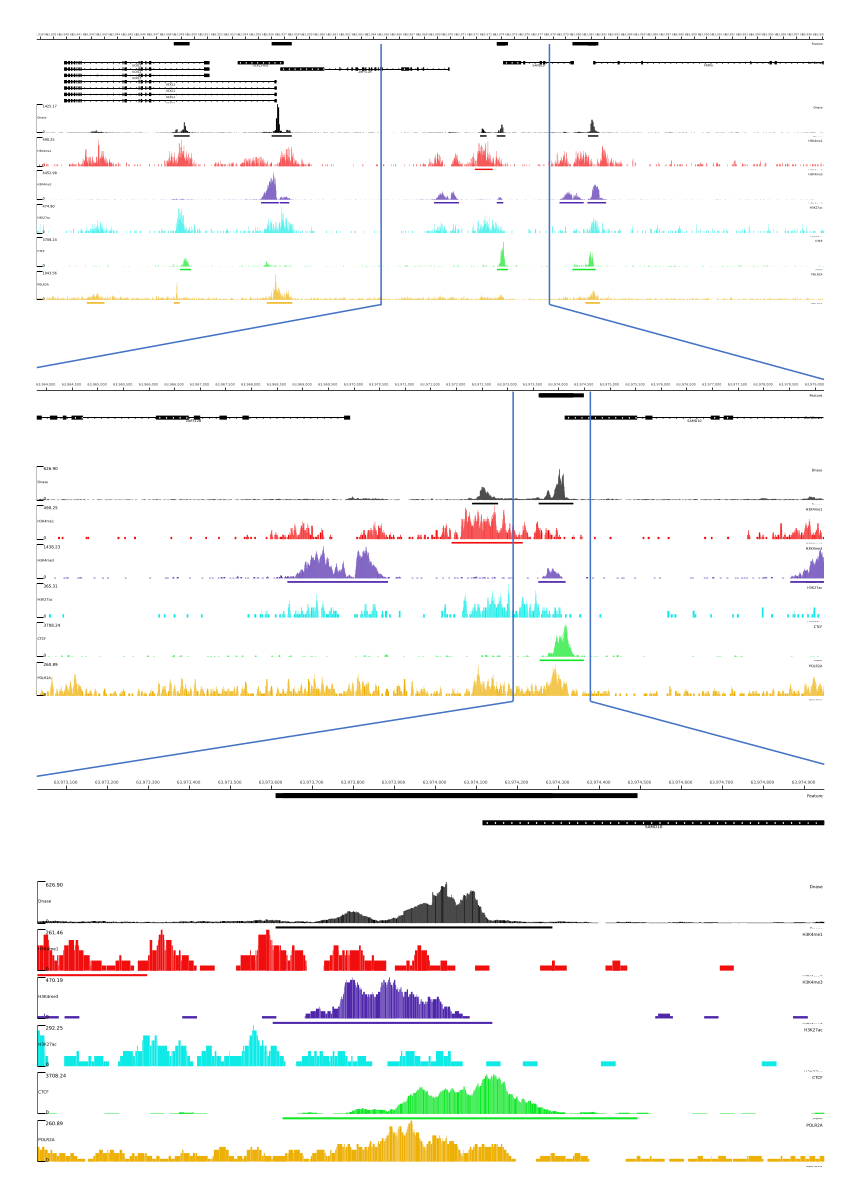
Для выходных данных 01, 02, 03, 06 и 07 предоставляются метрики контроля качества и HTML-файлы. Кроме того, на рисунке 3 мы приводим пример обработанных данных с помощью CATCH-UP, визуализируя конечный вывод через платформу MLV.

Рисунок 1: Рабочий процесс CATCH-UP. Имея список файлов fastq, CATCH-UP обрабатывает параллельно все сэмплы на всех этапах восходящего потока. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 2: Иллюстративное представление, объясняющее, как 1_fastqfile_home_dir.txt, 2_fastqfile_concat.txt и 3_merge_bams.txt должны быть правильно измененыдля запуска CATCH-UP. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 3: Пример вывода из конвейера CATCH-UP. Исходные данные секвенирования (файлы fastq) были загружены из ENCODE21. Конвейер CATCH-UP использовался для обработки fastq-файлов для DNase-seq и 5 типов ChIP-seq (H3K4me1, H3K4me3, H3K27ac, CTCF и POLR2A). Выходные файлы Bigwig были загружены в Multi Locus View для визуализации и идентификации геномных регуляторных элементов. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
Таблица 1: Ресурсы документации. В этой таблице показаны инструменты, задействованные в рабочем процессе CATCH-UP, ссылка на их документацию и соответствующие ссылки. Пожалуйста, нажмите здесь, чтобы скачать эту таблицу.
Таблица 2: Список требований к каналам и зависимостям для вышестоящей среды conda. Пожалуйста, нажмите здесь, чтобы скачать эту таблицу.
Таблица 3: Операционные системы, использованные для тестирования CATCH-UP. Ubuntu тестировалась на высокопроизводительном кластере и локальной машине. Пожалуйста, нажмите здесь, чтобы скачать эту таблицу.
Обсуждение
Более широкое внедрение и использование методов NGS для получения геномных данных сопровождалось расширением разработки биоинформационных инструментов для анализа этих данных. Существует множество инструментов, которые могут быть применены для каждого шага анализа данных, а также множество различных параметров, которые могут быть указаны в каждом инструменте 6,8,9,15,16,17,18,19,20,22,23,24 . Это создает чрезвычайно разнообразную комбинацию стратегий анализа, которые могут быть применены, каждая из которых может привести к вариациям в результате. Для точного сравнения между экспериментами необходима стандартизация биоинформатического анализа. Исторически сложилось так, что данные NGS генерируются учеными, работающими в «мокрой лаборатории», а данные анализируются биоинформатиками.
Анализ данных NGS можно разделить на «восходящие» и «нисходящие» конвейеры, где восходящий поток включает в себя необходимые шаги для перехода от необработанных данных, выведенных с помощью секвенционной машины, к формату, который может быть визуально интерпретирован исследователем. Последующий анализ включает в себя дополнительные шаги, которые зависят от исследовательского вопроса и плана эксперимента. Таким образом, трубопроводы вверх по добыче могут быть обобщены и поддаются стандартизации для улучшения научной воспроизводимости. С другой стороны, трубопроводы по технологической цепочке изготавливаются на заказ, зависят от биологического вопроса и требуют понимания со стороны исследователя, что делает их менее подходящими для стандартизации. Мы создали удобный для пользователя восходящий конвейер, который позволяет ученым, работающим в мокрых лабораториях, воспроизводимо анализировать свои собственные данные без необходимости каких-либо предварительных знаний в области биоинформатики. В этой статье мы представляем CATCH-UP, конвейер, созданный с использованием фреймворка snakemake и предназначенный как для удобства пользователя, так и для решения проблемы воспроизводимости при анализе данных ChIP-seq и ATAC-seq. Этот конвейер был создан для обработки данных ChIP-seq или ATAC-seq. После того, как пользователь загрузил CATCH-UP, необходимо сначала определить параметры анализа и именование образцов, прежде чем запускать конвейер в командной строке с использованием одной строки кода. Простые пошаговые инструкции о том, как настроить параметры анализа для анализа ChIP-seq или ATAC-seq, представлены в самом файле конфигурации и в нашем пошаговом руководстве в репозитории CATCH-UP GitHub.
Существуют конвейеры анализа данных ChIP-seq или ATAC-seq, такие как PEPATAC и ChIP-AP. Несмотря на то, что эти конвейеры имеют такие преимущества, как включение как восходящего, так и нисходящего анализа в единый рабочий процесс или использование графического пользовательского интерфейса (GUI), эти инструменты предназначены для биоинформатиков и ученых с умеренным уровнем вычислительной подготовки13,14. CATCH-UP был разработан для решения двух задач: позволить ученым, не имеющим биоинформатического образования, проводить собственный анализ на входе и стандартизировать анализ на входе за счет упрощения составления отчетов и точной воспроизводимости в лабораториях. CATCH-UP намеренно ограничен восходящим анализом, но его результаты совместимы с инструментами нисходящего анализа, такими как те, которые используются для статистического сравнения наборов данных или связывания фактора транскрипции25,26.
Все критические шаги, необходимые для выполнения воспроизводимого восходящего анализа, предварительно определены в конвейере CATCH-UP для обеспечения надежности. Подробный характер этого конвейера позволяет пользователю шаг за шагом следить за выходными данными конвейера, что полезно как для устранения неполадок, так и для репликации аналитического рабочего процесса. Учитывая быстро развивающийся характер методов секвенирования нового поколения, модульный характер этого конвейера является полезным, поскольку он обеспечивает возможность легкой адаптации для включения как выпуска обновлений версий инструментов, так и внедрения новых инструментов. CATCH-UP был успешно протестирован для следующих операционных систем: Ubuntu, CentOS, macOS (процессор Intel) и Windows (Таблица 3). Конвейер был построен для обработки больших экспериментов, содержащих десятки образцов, за счет распараллеливания рабочего процесса, что делает его адаптируемым к различным экспериментальным проектам. В целом, внедрение CATCH-UP в анализ данных ChIP-seq и ATAC-seq обеспечивает удобный, воспроизводимый и легко адаптируемый рабочий процесс анализа.
Раскрытие информации
Дж.Р.Х. является соучредителем и директором компании Nucleome Therapeutics и предоставляет консультационные услуги компании.
Благодарности
J.R.H. был поддержан грантами от Wellcome Trust (225220/Z/22/Z и 106130/Z/14/Z) и MRC (MC_UU_00029/3). М.Б. был поддержан грантом Wellcome Trust (225220/Z/22/Z). E.R.G была поддержана стипендией Министерства национального образования по отбору и трудоустройству кандидатов, отправленных за рубеж для получения последипломного образования (YLSY), Министерством национального образования Турецкой Республики. Э.Г. была поддержана докторской программой Wellcome по геномной медицине и статистике (108861/Z/15/Z). S.G.R. был поддержан грантом Совета по медицинским исследованиям (MRC) (MC_UU_00029/3).
Материалы
| Name | Company | Catalog Number | Comments |
| CATCH-UP | GitHub | https://github.com/Genome-Function-Initiative-Oxford/UpStreamPipeline/tree/main/genetics/CATCH-UP | |
| CentOS | Linux | Version 7 | Any of the operating systems listed here may be used |
| macOS | Apple | Version 13 Ventura | Any of the operating systems listed here may be used |
| Ubuntu | Ubuntu | Version 22.04 LTS | Any of the operating systems listed here may be used |
| Windows | Microsoft | Version 11 | Any of the operating systems listed here may be used |
Ссылки
- Downes, D. J., Hughes, J. R. Natural and experimental rewiring of gene regulatory regions. Annual Review of Genomics and Human Genetics. 23, 73-97 (2022).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Research. 16 (1), 123-131 (2006).
- Jin, W., et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 528 (7580), 142-146 (2015).
- Agbleke, A. A., et al. Advances in chromatin and chromosome research: Perspectives from multiple fields. Molecular Cell. 79 (6), 881-901 (2020).
- Sergeant, M. J., et al. Multi locus view: an extensible web-based tool for the analysis of genomic data. Communications Biology. 4 (1), 623(2021).
- Kuhn, R. M., Haussler, D., Kent, W. J. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 14 (2), 144-161 (2013).
- Hentges, L. D., et al. LanceOtron: a deep learning peak caller for genome sequencing experiments. Bioinformatics. 38 (18), 4255-4263 (2022).
- Gaspar, J. M. Improved peak-calling with MACS2. bioRxiv. , 496521(2018).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Rich-Griffin, C., et al. Panpipes: a pipeline for multiomic single-cell data analysis. bioRxiv. , (2023).
- Bourgey, M., et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience. 8 (6), giz037(2019).
- Smith, J. P., et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genomics and Bioinformatics. 3 (4), lqab101(2021).
- Suryatenggara, J., Yong, K. J., Tenen, D. E., Tenen, D. G., Bassal, M. A. ChIP-AP: an integrated analysis pipeline for unbiased ChIP-seq analysis. Briefings in Bioinform. 23 (1), bbab537(2022).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (5), 2114-2120 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Vasimuddin, M., Misra, S., Li, H., Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). , 314-324 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. , https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32 (19), 3047-3048 (2016).
- Luo, Y., et al. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Research. 48 (D1), D882-D889 (2020).
- Danecek, P., et al. Twelve years of SAMtools and BCFtools. Gigascience. 10 (2), giab008(2021).
- Picard Toolkit. , http://broadinstitute.github.io/picard/ (2019).
- Ramírez, F., et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 44 (W1), W160-W165 (2016).
- Stark, R., Brown, G. DiffBind:Differential binding analysis of ChIP-Seq peak data. Bioconductor. , https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2016).
- Schep, A. N., et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 25 (11), 1757-1170 (2015).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены