
Method Article
使用硫酸二甲酯探测RNA结构 体 外 和细胞测序突变分析
摘要
该协议提供了用硫酸二甲酯修饰RNA以进行突变分析实验的说明。它包括使用两种替代文库制备方法进行的 体外 和 体内 探测。
摘要
RNA结构在几乎任何生物过程中的作用都变得越来越明显,特别是在过去十年中。然而,解决RNA结构的经典方法,如RNA晶体学或冷冻电镜,未能跟上快速发展的领域和对高通量解决方案的需求。使用硫酸二甲酯 (DMS) 进行测序的突变分析 MaPseq 是一种基于测序的方法,用于从碱基与 DMS 的反应性推断 RNA 结构。当碱基未配对时,DMS在其Watson-Crick面上甲基化腺苷中的N1氮和胞嘧啶中的N3。用热稳定的II组内含子逆转录酶(TGIRT-III)逆转录修饰的RNA导致甲基化碱基作为突变掺入cDNA。当对所得cDNA进行测序并将其映射回参考转录本时,每个碱基的相对突变率表明碱基的"状态"为配对或未配对。尽管DMS反应性在 体外 和细胞中都具有很高的信噪比,但这种方法对处理程序中的偏差很敏感。为了减少这种偏差,本文提供了一种在细胞中使用DMS和 体外 转录RNA进行RNA处理的方案。
引言
自从发现RNA同时具有结构1,2和催化3性质以来,RNA的重要性及其在众多生物过程中的调节功能逐渐被揭示。事实上,RNA结构对基因调控的影响越来越受到关注4。与蛋白质一样,RNA具有一级,二级和三级结构,分别指核苷酸的序列,碱基配对相互作用的2D映射以及这些碱基配对结构的3D折叠。虽然确定三级结构是理解RNA依赖性过程背后的确切机制的关键,但二级结构在RNA功能方面也提供了大量信息,并且是进一步3D折叠的基础5。
然而,使用传统方法确定RNA结构具有固有的挑战性。虽然对于蛋白质,晶体学,核磁共振(NMR)和低温电子显微镜(cryo-EM)已经可以确定结构基序的多样性,允许仅从序列6进行结构预测,但这些方法并不广泛适用于RNA。事实上,RNA是具有构建块(核苷酸)的柔性分子,与氨基酸对应物相比,具有更大的构象和旋转自由度。此外,通过碱基配对的相互作用比氨基酸残基的相互作用更具动态性和多功能性。因此,经典方法仅对具有明确定义、高度紧凑结构的相对较小的RNA成功7。
确定RNA结构的另一种方法是通过化学探测结合二代测序(NGS)。该策略生成有关RNA序列中每个碱基的结合状态(即其二级结构)的信息。简而言之,RNA分子中不参与碱基配对的碱基被小化合物差异修饰。用专门的逆转录酶(RT)逆转录这些RNA将修饰作为突变整合到互补的脱氧核糖核酸(cDNA)中。然后通过聚合酶链反应(PCR)扩增这些cDNA分子并进行测序。为了获得有关其结合或未结合的"状态"的信息,计算感兴趣的RNA中每个碱基的突变频率,并将其作为约束输入结构预测软件8。基于最近邻规则9 和最小自由能计算10,该软件生成最适合所获得的实验数据11,12 的结构模型。
DMS-MaPseq使用DMS,其以高度特异性的方式甲基化沃森-克里克面腺苷中的N1氮和胞嘧啶中的N3氮13。在逆转录中使用热稳定的II组内含子逆转录酶(TGIRT-III)可创建具有前所未有的信噪比的突变谱,甚至允许对由两个或多个替代构象产生的重叠谱进行反卷积14,15。此外,DMS可以穿透细胞膜和整个组织,使得在生理环境中进行探测成为可能。然而,高质量数据的生成具有挑战性,因为处理程序的变化会影响结果。因此,我们为 体外 和细胞内DMS-MaPseq提供了详细的方案,以减少偏差并指导新人通过他们可能遇到的困难。特别是鉴于最近的SARS-CoV2大流行,RNA病毒的高质量数据是研究基因表达和寻找可能治疗方法的重要工具。
研究方案
注意:有关本协议中使用的所有材料、软件、试剂、仪器和细胞的详细信息,请参阅 材料表 。
1. 基因特异性 体外 DMS-MaP
- RNA 体外 转录
- 获取目标 RNA 的双链 (ds)DNA 序列(例如,作为 DNA 片段、质粒或来自预先存在/基因组 DNA 的 PCR)。如果 DNA 序列包含聚合酶启动子,请跳至步骤 3。
- 执行重叠PCR以将RNA聚合酶促进子附着在所需DNA片段的上游(T7聚合酶的正向引物:5'TAATACGACTCACTATAGG +靶序列3'的第一碱基)。
- 体外将DNA片段转录成RNA。始终将RNA放在冰上。
- 使用脱氧核糖核酸酶消化DNA。
- 使用基于柱的方法(步骤2.4)或通过乙醇沉淀(步骤2.5)分离RNA。以适当的体积洗脱,预期产率为~50μg。
- 通过在琼脂糖凝胶上运行来确保RNA的完整性;在运行前在70°C下使RNA变性2-3分钟。
注意:缓冲液和琼脂糖可能含有降解RNA并可能污染RNA样品的RNA酶。该实验室以前曾使用过预制琼脂糖凝胶;结果(尤其是RNA)有时是模棱两可的。使用琼脂糖或PAGE凝胶获得最佳结果。 - 直接使用将RNA在-80°C下储存数月,除非解冻后可见降解。
- 体外 DMS 修改(在 105 mM DMS 下)
- 制备足够量的复性缓冲液(0.4M二甲胂酸钠,pH 7.2,含有6mM MgCl2)。
注意:对于每个反应(最终体积为 100 μL),加入 89 μL 复性缓冲液。 - 对于每个反应,将 89 μL 复性缓冲液转移到指定的 1.5 mL 管中,并在放置在化学罩下方的恒温摇床中预热 37 °C。
注意:DMS具有剧毒,必须始终保存在化学罩下,直到被还原剂淬灭。 - 在 10 μL 无核酸酶水 (NFH 2O) 中洗脱 1-10 pmol的 RNA;转移到PCR管中。
- 在95°C的热循环仪中孵育1分钟以使RNA变性。
- 立即放在冰块上,以免折叠错误。
- 将RNA样品加入带有37°C复性缓冲液的指定管中,充分混合,孵育10-20分钟以重新溶解RNA。
注意:大多数RNA将以毫秒到秒的顺序折叠,尽管存在例外16。 - 向 RNA 样品中加入 1 μL 100% (10.5 M) DMS,并在以每分钟 800-1,400 转 (rpm) 振荡的同时孵育 5 分钟。
注意:此步骤中的摇晃(或其他混合方式)至关重要,因为DMS是疏水性的,可能无法完全溶解在复性缓冲液中。反应时间的偏差可能会影响DMS反应性的重现性。为了最大限度地减少移液误差,如果DMS的终浓度保持在1%(105 mM)的浓度,则可以在将其添加到样品中之前将其溶解在100%乙醇中。对于未经处理的对照,DMS可以用二甲基亚砜(DMSO)或水代替。 - 反应时间 5 分钟后,用 60 μL 100% β-巯基乙醇 (BME) 淬灭,充分混合,并立即将 RNA 置于冰上。
注意:用BME淬灭反应以清理后,可以安全地将RNA从引擎盖中取出。然而,由于其强烈的气味和刺激性,仍应避免将BME直接暴露于周围环境中。 - 通过乙酸钠-乙醇沉淀(参见步骤2.5)或基于柱的方法(参见步骤2.6)纯化RNA,并在10μL水中洗脱。
- 使用分光光度计定量RNA。
- 直接使用将修饰的RNA储存在-80°C。
注意:应避免长期储存,因为DMS处理后RNA不太稳定。
- 制备足够量的复性缓冲液(0.4M二甲胂酸钠,pH 7.2,含有6mM MgCl2)。
- 修饰RNA的基因特异性RT-PCR检测
注意:有关DMS处理片段的RT-PCR设置,请参见 图1 。- 在 10 μL 无核酸酶 (NF) H2O 中洗脱 100 ng 修饰的 RNA。 转移到 PCR 管中。
- 向试管中加入 4 μL 5x 第一链缓冲液 (FSB)、1 μL dNTP 混合物(每个 10 mM)、1 μL 0.1 M 二硫苏糖醇 (DTT)(避免冻融循环)、1 μL RNase 抑制剂、1 μL 10 μM 反向引物(单引物或引物池)和 1 μL TGIRT III。
注意:对于一组引物,请勿将 1 μL 的 10 μM 每种引物直接添加到室温中;相反,先混合引物,并从混合物中加入 1 μL(总引物浓度为 10 μM)。 - 在热循环仪中在57°C孵育30分钟至1.5小时(通常,30分钟足以制成500nt产物)。
- 加入 1 μL 4 M NaOH,通过移液混合,并在 95 °C 下孵育 3 分钟以降解 RNA。
注意:此步骤至关重要,因为它通过降解RNA从cDNA中释放TGIRT。如果跳过,下游PCR可能会受到影响。 - 使用基于色谱柱的方法(参见步骤2.6)进行净化,充分去除引物,并在10 μL NFH 2O中洗脱。
- PCR扩增cDNA,方法是使用旨在平衡产量和保真度的PCR试剂盒,每25 μL反应使用1 μL逆转录产物。
注意:引物的熔解温度应为~60°C。 - 在琼脂糖凝胶或预制琼脂糖凝胶上运行 2 μL PCR 产物以验证 PCR 成功。
- 理想情况下,PCR 后只应显示一个条带。如果是这样,请使用基于柱的方法清理反应。如果存在替代条带,请使用剩余的PCR反应从凝胶中切除正确的条带。以足够小的体积(例如 10 μL)洗脱。
- 使用分光光度计定量提取的片段。
- 使用适合所需测序平台的方法对dsDNA片段进行索引以进行测序。

图 1:大型 DMS 处理片段的 RT-PCR 实验装置。 当对修饰的RNA进行逆转录时,不会记录引物退火序列上的修饰。因此,当片段长度超过400-500 bp时,需要设计在引物区域中重叠的片段,如此处的示例所示。片段的长度取决于测序需求。当使用配对末端150循环测序时,片段不应超过300 bp。缩写:RT-PCR = 逆转录聚合酶链反应;DMS = 硫酸二甲酯。 请点击此处查看此图的大图。
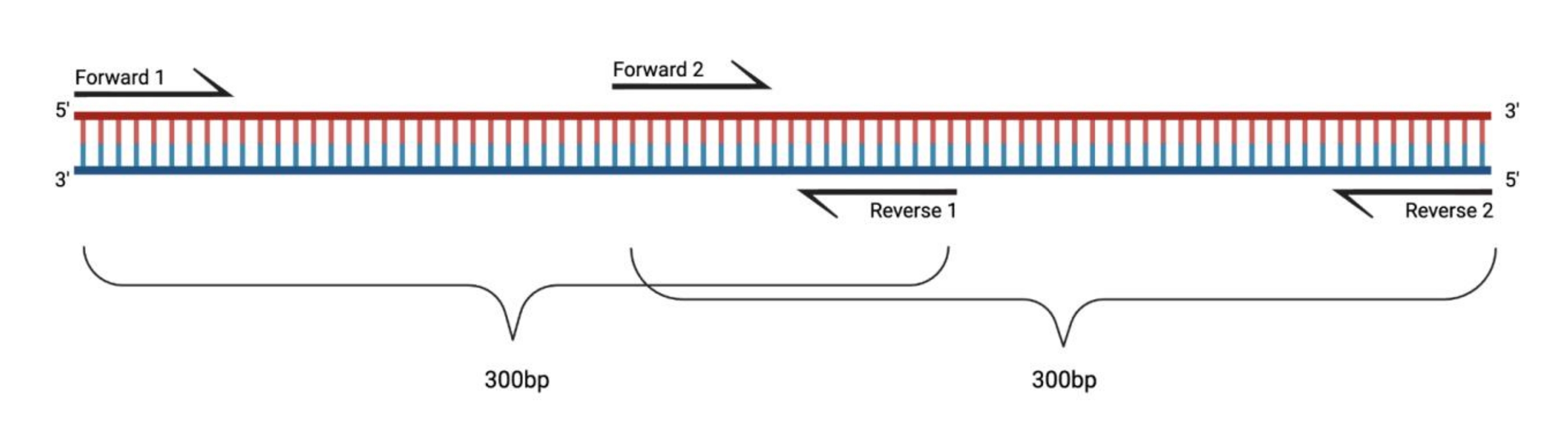{kind=link}
2. 使用病毒感染细胞的全基因组DMS-MaP
注意:在细胞中,DMS处理也可以与上述基因特异性扩增方法结合使用。全基因组文库需要巨大的测序深度才能实现对单个基因的完全覆盖。然而,如果病毒RNA在提取后占核糖耗尽RNA的很大一部分,则全基因组测序将是合适的。此外,其他富集方法可以与全基因组文库生成方法结合使用。
- 糖尿病治疗
- 培养感染病毒的细胞,直到所需的感染阶段。
- 将细胞容器转移到专用通风橱中,该通风橱适用于处理所需生物安全水平的病毒和DMS等试剂产生的化学烟雾。
- 向培养基中加入2.5%体积的DMS,并用封口膜密封容器(通常为10厘米板)。
注意:使用 DMS 很容易修改不足和过度修改。将DMS直接添加到细胞中时,充分混合非常重要。或者,将新培养基预热在37°C的50mL锥形管中,并直接剧烈摇动加入DMS。将用过的培养基倒在细胞上,然后在含有DMS的培养基中缓慢移液。 - 转移到37°C培养箱中5分钟。
注意:根据在培养箱外处理DMS所需的时间,5分钟可能会导致过度修饰。保持从加入DMS到孵育的时间≤1分钟。如果是第一次进行实验,建议进行DMS滴定并改变孵育时间(3分钟至10分钟之间),以找到最佳修饰率,并确保结果在浓度窗口内具有稳定性。 - 小心地移出含DMS的培养基(到适当的化学废物中),并轻轻加入10 mL终止缓冲液(含30%BME的PBS [例如,3 mL BME和7 mL PBS])。
注意:如果细胞没有强烈粘附,则添加DMS和BME可以将细胞从平板中取出。如果细胞正在提升,则可以将它们视为悬浮细胞 - 而不是去除含有DMS的培养基,直接添加终止缓冲液,并将带有DMS和BME的细胞刮出到50 mL锥形管中。通过以3,000× g离心3分钟来沉淀细胞;确保去除任何残留的DMS,这些DMS可以在细胞下沉淀成大液滴。如果最初无法去除DMS培养基,建议在30%BME中进行额外的洗涤步骤。 - 刮取细胞,并将其转移到 15 mL 锥形管中。
- 通过以3,000 ×g 离心3分钟来沉淀。
- 除去上清液并用 10 mL PBS 洗涤 2 次。
- 小心地去除尽可能多的残留PBS。
- 将沉淀溶解在适量的RNA分离试剂中(例如,T75培养瓶为3 mL,10 cm板为1 mL)。
注意:试剂量不足可能会影响RNA产量。
- RNA提取和核糖体RNA(rRNA)去除
- 向 RNA 分离试剂中的 1 mL 匀浆细胞中加入 200 μL 氯仿,涡旋 15-20 秒直至亮粉红色,然后孵育长达 3 分钟直至可见相分离。
注意:粉红色脂质相应稳定在底部。如果不是这种情况,涡旋时间可能不足。 - 在4°C下以最大速度(~20,000× g)旋转15分钟。
- 将上层水相转移到新管中。
- 通过乙酸钠 - 乙醇沉淀(参见步骤2.5)或基于柱的方法(参见步骤2.6)纯化RNA,并在足够体积的NF H2O中洗脱。
- 检查琼脂糖凝胶上的RNA完整性。寻找对应于两个核糖体亚基的两个条带。
- 使用首选方法去除rRNA,并在足够体积(通常为20-50μL)的NFH 2O中洗脱。
注意:对于下游应用,建议在 8 μL 的体积中加入 ~500 ng 的总 RNA。 非核糖体 RNA 通常仅占总 RNA 的 5%-10%。 - 使用分光光度计进行定量。
- 向 RNA 分离试剂中的 1 mL 匀浆细胞中加入 200 μL 氯仿,涡旋 15-20 秒直至亮粉红色,然后孵育长达 3 分钟直至可见相分离。
- 库生成
- 使用基因特异性 RT-PCR 或其他方法生成文库15.如果使用随机六聚体进行底漆,请在低 Tm (37-42 °C) 下添加孵育步骤以允许六聚体退火。
注意:标准文库生成试剂盒也可以通过用TGIRT替换RT酶并将RT温度更改为57°C来使用。
- 使用基因特异性 RT-PCR 或其他方法生成文库15.如果使用随机六聚体进行底漆,请在低 Tm (37-42 °C) 下添加孵育步骤以允许六聚体退火。
- 使用RNA Clean & Concentrator色谱柱进行基于色谱柱的RNA净化
注意:所有步骤都应在室温下进行。- 向样品管中加入 NF H2O,使其体积达到 50 μL。
- 向样品中加入 100 μL 结合缓冲液和 150 μL 100% 乙醇。
- 混合并转移到离心柱中。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- 加入 400 μL RNA 制备缓冲液。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- 加入 700 μL RNA 洗涤缓冲液。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- 加入 400 μL RNA 洗涤缓冲液。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- (可选)将色谱柱转移到新的收集管中,并以10,000-16,000× g 旋转2分钟。
- 将色谱柱转移到干净的无RNA酶管中,并加入适量的NF H2O。
- 以 10,000-16,000 × g 旋转 1 分钟。
- 酸性苯酚-氯仿RNA提取。
- 加入等体积的酸性苯酚:氯仿:异戊醇。
- 彻底涡旋,并以14,000× g 离心5分钟。
- 如果没有相分离,则加入 20 μL 2 M NaCl,然后重复离心。
- 将水相转移到新管中。
- 加入 500 μL 异丙醇和 2 μL 助沉淀剂。
- 混合并在室温下孵育3分钟;然后,在-80°C孵育过夜。
- 通过在4°C下以最大速度(~20,000× g)离心30分钟来沉淀RNA。
- 用 200 μL 冰冷的 70% 乙醇洗涤沉淀。
- 以最大速度(~20,000 × g)旋转5分钟;丢弃流出。
- 将沉淀重悬于适量的NF H2O中。
- 使用寡核苷酸清洁柱和浓缩器柱进行基于色谱柱的cDNA纯化
注意:所有步骤都应在室温下进行。- 向样品管中加入 NF H2O,使其体积达到 50 μL。
- 加入 100 μL 结合缓冲液和 400 μL 100% 乙醇。
- 混合并转移到离心柱中。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- 加入 750 μL DNA 洗涤缓冲液。
- 以 10,000-16,000 × g 旋转 30 秒;丢弃流出。
- (可选)将色谱柱转移到新的收集管中,并以10,000-16,000× g 旋转2分钟。
- 将色谱柱转移到干净的无RNA酶管中,并加入适量的NF H2O。
- 以 10,000-16,000 × g 旋转 1 分钟。
3. 测序数据分析
注意:要从DMS-MaP测序数据创建RNA二级结构模型,必须通过几个不同的步骤处理生成的.fastq文件。这些步骤可以使用
- 使用 TrimGalore 或 Cutadapt 修剪适配器序列。
- 使用 Bowtie2 将读取映射到参考序列(.fasta 格式)。
- 使用专门的 RNA 结构软件(例如 DREEM14、RNA-Framework17 或类似软件)对读数进行计数,并创建反应性图谱。
- (可选)聚类读取以使用DREEM14,DRACO 17,DANCE-MaP18或类似物找到替代RNA构象。
- 使用RNAStructure12,ViennaRNA或类似方法根据反应性曲线预测最小自由能结构。
- 使用VARNA(https://varna.lri.fr/)或类似方法可视化RNA11 结构。
注意:为了实用性,DREEM (www.rnadreem.org) 和 RNA-Framework19 等软件在其管道中大量包含步骤 1--5,从而简化了分析过程。然而,任何结构预测都应该小心处理(例如,通过验证结构与数据的一致性20。
结果
基因特异性 体外 DMS-MaP
为了研究SARS2的5'UTR,病毒的前300 bp作为gBlock序列与三个引物一起订购。其中包括两个通过PCR 繁殖 片段的引物("FW"和"RV"),以及一个用于连接T7启动子("FW-T7")的引物。这些序列如 表1所示。
| 名字 | 序列 (5'->3') |
| 固件 | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| 房车 | GCAAACTGAGTTGGACGTGT |
| 固件-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
表1:SARS-CoV2 5'UTR的DMS-MaP RT-PCR引物序列。 在这里,需要FW-T7和RV来生成用于 体外 转录的DNA模板,RV用于逆转录,FW-RV引物对用于随后的cDNAPCR扩增。引物退火到SARS-CoV2基因组(FW)的起点和感兴趣区域下游的序列。缩写:DMS-MaP = 使用硫酸二甲酯测序的突变分析;RT-PCR = 逆转录聚合酶链反应;SARS-CoV2 = 严重急性呼吸综合征-冠状病毒 2;UTR = 未翻译区域;RV = 反向底漆;FW = 正向引物。
为了从gBlock片段生成RNA,根据 图2A所示的方案,使用PCR预混料使用重叠PCR连接T7聚合酶启动子的序列。从细长的片段中,使用T7转录试剂盒生成RNA。随后使用DNase和RNA Clean&Concentrator柱分离的RNA消化DNA模板。
体外转录的质量控制是通过在1%琼脂糖凝胶和ssRNA分子量标准上运行RNA产物来完成的。由于只有一个可见的条带,因此进行了体外DMS探测和RT-PCR(见图2B)。
为了验证PCR反应的成功,使用dsDNA分子量标准在2%琼脂糖凝胶上运行样品。索引后,条带在同一凝胶上的运行速度应高出~150 bp,考虑到索引引物的大小。

图 2:DNA 模板的 体外 转录。 (一)为了在 体外 转录尚未具有内在RNA聚合酶启动因子的DNA模板,必须首先通过重叠PCR连接模板。这是通过使用正向引物来完成的,该引物包括与所需片段重叠的第一碱基上游的序列TAATACGACTCACTATAGG(在T7 RNA聚合酶的情况下)。此处带下划线的碱基象征聚合酶的转录起始位点。一旦启动子附着在dsDNA片段上,它就可以被T7聚合酶转录。重要的是,聚合酶使用与上述启动子序列相对的链作为模板(蓝色),有效地产生与指示启动子序列下游的序列相同的RNA(红色)。(B)具有ssRNA分子量标准(泳道1)和300 nt(泳道2)的 体外 转录RNA产物的1%琼脂糖凝胶。(C) 含有 GeneRuler 1 kb 加分子量标准(泳道 1)的 2% 琼脂糖凝胶,RT-PCR 后的 PCR 产物以 300 bp 运行(泳道 2),文库制备后的索引片段以 470 bp 运行(泳道 3)。缩写:RT-PCR = 逆转录聚合酶链反应;DMS = 硫酸二甲酯;nt = 核苷酸;dsDNA = 双链 DNA;ssRNA = 单链RNA。 请点击此处查看此图的大图。
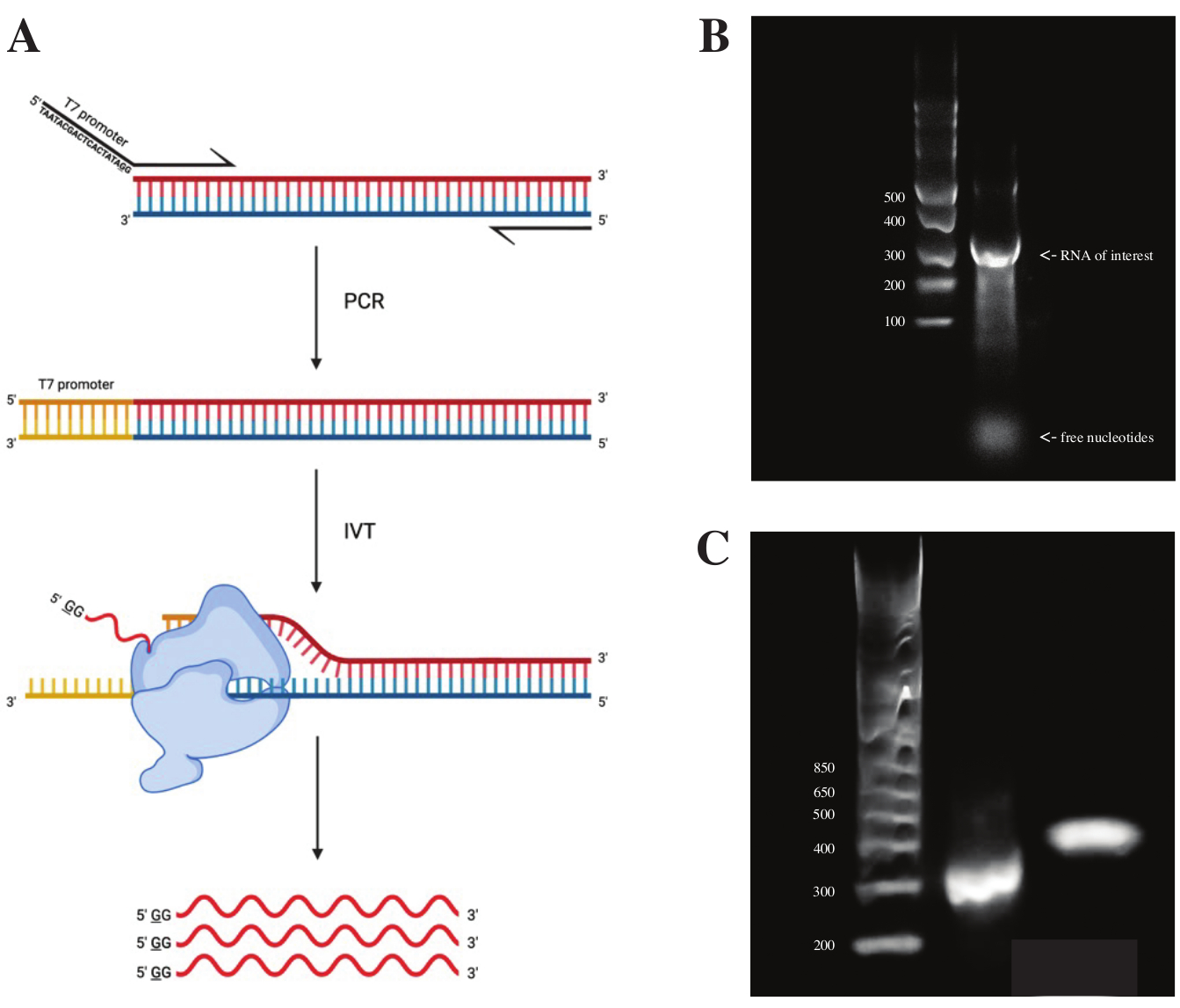{kind=link}
使用病毒感染细胞的全基因组 体内 DMS-MaP
在DMS治疗之前,HCT-8细胞感染了OC43。当在感染后4天观察到细胞病变效应(CPE)(如图 3A所示)时,处理这些细胞,提取RNA并使其核糖耗尽。当在琼脂糖凝胶上运行总RNA时,可以看到两条明亮的条带,占核糖体的40S和60S亚基,约占总RNA质量的95%(见 图3B)。当RNA提取不成功或被降解(例如,通过多次冻融循环)时,RNA降解产物在凝胶底部可见(参见 图3C,第二泳道)。此外,在rRNA耗竭后,两条亮条带消失,在泳道中留下涂片(参见 图3C,第三泳道)。最后,在文库制备后,样品具有不同的大小分布,并在最终的PAGE凝胶上显示为涂片。切除200个核苷酸(nt)和500 nt之间的条带,与计划用于分析这些文库的150 x 150配对末端测序运行一致。最重要的是,分离出以~150 nt运行的适配器二聚体(见 图3D)。

图3:病毒感染细胞体内DMS-MaP的检查点。 (A)病毒感染的HCT-8细胞的光学显微镜图像,4天dpi。为了从总RNA中获得尽可能高的病毒RNA产量,同时最大限度地减少细胞死亡引起的不利影响,应在CPE开始时甚至在此之前添加DMS,如图所示。(B) 含有 1 μg 总 RNA 的六个样品的 1% 琼脂糖凝胶。在每个泳道中,可以看到两条明亮的条带,分别代表40S和60S亚基,因为核糖体RNA占总RNA的~95%。注意:细胞内DMS处理会导致一些RNA片段化和拖尾,但两个rRNA条带仍然可见。轻度片段化后是可以容忍的,因为在细胞仍然活着时,在DMS孵育期间生成包含甲基化标记的信息并报告RNA结构。(C) GeneRuler 1 kb 的 1% 琼脂糖凝胶加上分子量标准 DNA 标记物(泳道 1)总 RNA,先前在 -80 °C 下储存 6 个月(泳道 2)和核糖耗尽 RNA(泳道 3)。当长时间储存RNA并经过几次冻融循环时,RNA开始降解,可能不应用于探测实验。此外,在核糖耗尽总RNA后,占核糖体40S和60S亚基的两个明亮条带褪色,残留RNA的涂片开始显示。(D) GeneRuler 1 kb 的 PAGE 凝胶加分子量标准 DNA 标记物(泳道 1)和全基因组制备的 RNA 文库样品。应根据测序需要切除凝胶。对于从两侧跨越150个循环的配对末端测序运行,应在300 bp至500 bp之间切除凝胶。应分离出适配器二聚体(以 170 bp 的速度运行)。缩写:DMS-MaP = 使用硫酸二甲酯测序的突变分析;DPI = 感染后天数;CPE = 细胞病变效应。请点击此处查看此图的大图。
{kind=link}
排序后,通过向DREEM网络服务器(http://rnadreem.org/)提交作业以及.fasta参考文件来分析.fastq文件。服务器生成的输出包括由fastqc(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)和TrimGalore(https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)生成的质量控制文件,以及包含群体平均突变频率的其他输出文件。除了显示具有交互式.html(见图4A)格式的突变频率的图表和一个包含每个碱基的原始反应物的.csv文件和一个可由多个RNA结构预测软件读取的struct_constraint.txt文件外,还包括一个位向量.txt文件报告通过读取突变。据此,通过将.fasta和struct_constraint.txt文件提交给RNAfold网络服务器(http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)来计算总体平均结构。它使用ViennaRNA软件根据最小自由能生成结构预测,可以在线查看或以ct或Vienna格式下载。为了生成RNA结构模型,将这些可下载文件提交给VARNA(https://varna.lri.fr/,见图4B)。最后,稳定版本的DREEM(https://codeocean.com/capsule/6175523/tree/v1)可以使用bitvector.txt文件来搜索替代的RNA构象。为了使用DREEM获得良好的结构模型,应实现每个碱基10,000个读数的覆盖率;对于群集,每个碱基最多可能需要 100,000 次读取。整个工作流程的概述如图4C所示。

图 4:从 SARS-CoV2 5'UTR 的化学探测实验中获得的示例性数据。 (A) 按碱基着色的 SARS-CoV2 基因组前 300 个碱基的反应性图谱(A:红色、C:蓝色、U:绿色、G:黄色)。原始反应性计算为绝对突变频率除以覆盖率。具有开放构象的碱具有高反应性值;参与碱基配对的碱基反应性值较低。U和G未被DMS修饰,反应性值低,源于聚合酶不忠。预测是用DREEM网络服务器进行的。(B) 根据瓦尔纳的反应性值预测的 SARS-CoV2 5'UTR 的结构模型。具有高反应性值的碱以红色着色;反应性值低的碱基以白色着色。(C)DMS-MaP分析的工作流程,从测序获得的.fastq文件开始。这些可以使用fastqc进行质量控制;适配器序列使用 TrimGalore 进行修剪,然后使用 Bowtie2 映射回参考序列。从获得的 .bam 文件中,DREEM 对每次读取中的突变进行计数,从而创建突变图或 .bitvector.txt 文件。这些以位置依赖的方式报告每个读取的突变,在此基础上可以创建群体平均反应性曲线。或者,可以使用DREEM对位载体进行聚类,以搜索替代的RNA构象。最后,使用软件(例如VARNA)对获得的结构模型进行可视化。缩写:DMS-MaP = 使用硫酸二甲酯测序的突变分析;SARS-CoV2 = 严重急性呼吸系统综合症-冠状病毒 2。请点击此处查看此图的大图。
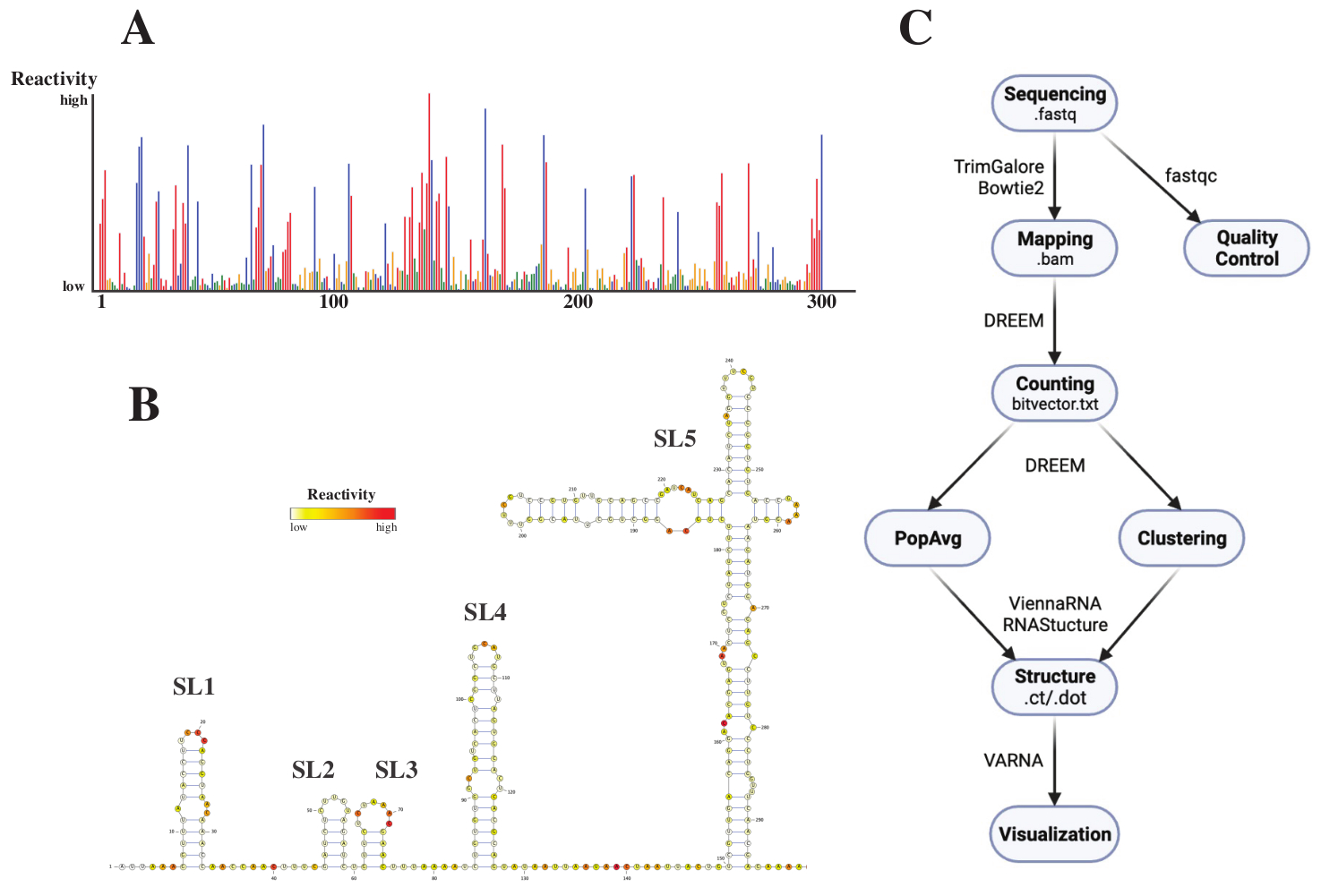{kind=link}
讨论
此处的协议描述了如何使用DMS突变分析实验在 体外 和细胞中探测RNA。此外,它还提供了有关如何为Illumina测序准备文库以生成基因特异性数据并分析获得的.fastq文件的说明。此外,可以使用全基因组文库方法。然而,基因特异性RT-PCR可产生最高质量和最可靠的数据。因此,如果比较样品,重要的是要确保使用相同的测序策略制备它们,因为文库生成会导致一些偏差。重现性应始终使用重复进行测量。
几点注意事项
RNA是一种不稳定的分子,对高温和RNA酶的降解都很敏感。因此,建议采取特殊措施,即使用个人防护装备(PPE)、无RNA酶材料和RNA酶抑制剂。最重要的是,RNA应尽可能保存在冰上。这尤其适用于甲基化RNA,它对高温更加敏感。
重要的是要确认目标RNA结构对DMS浓度和缓冲条件不敏感。pH 7-7.5 时的 100 mM Tris、100 mM MOPS 和 100 mM HEPES 等缓冲液可提供高信号,但可能不足以在反应过程中维持 pH值 21。由于DMS在水中水解会降低pH值,因此强缓冲液对于在改性反应过程中保持中性pH值至关重要。添加bicine已被证明有助于将pH维持为略微碱性21 ,但导致Gs和Us上的DMS修饰较低,这可能是有益的,但由于产生的信号比As和Cs低得多,因此应单独分析,并且本协议中不再进一步讨论。
在基因特异性RT-PCR中,修饰的RNA被逆转录到DNA中,并通过PCR扩增成片段。虽然RNA的大小理论上是无限的,但这些PCR片段的长度不应超过400-500个碱基对(bp),以防止逆转录反应过程中的偏差。理想情况下,片段应在测序运行范围内(即,如果使用150 x 150循环配对末端测序程序进行测序,则单个片段不应超过300 bp)。当使用循环次数较少的测序程序时,可以使用dsDNA酶对PCR产物进行片段化。此外,由于引物序列中的序列不包含任何结构信息,因此当探针的RNA包含>1片段时,片段必须重叠。RT反应可以包含用于不同片段的多个RT引物(最多10种不同的RT引物)。根据序列的不同,合并RT引物会使逆转录效率降低,但通常效果很好。每个PCR反应应单独进行。
当用DMS探测RNA时,实验条件起着额外的作用,因为许多RNA在热力学上不稳定,并且根据温度等环境因素改变其构象。为避免不规则,实验条件应尽可能保持恒定,在反应时间方面也是如此。当保持碱性条件(缓冲能力和一价(Na)和二价离子(Mg)的存在)以确保RNA24的适当折叠时,缓冲条件似乎可以在一定程度上交换17,20,22,23。
关于修饰RNA的文库制备,必须考虑几个方面。首先,如前所述,修饰的RNA不如未修饰的RNA稳定,这意味着它们可能需要优化片段时间以获得最佳片段大小分布。此外,某些RNA文库制备试剂盒以及许多其他RNAseq方法在逆转录试剂盒中使用随机引物。这可能会导致参考文献的覆盖率降低,特别是在基因的3'中,并最终导致覆盖深度不足。如果某个区域的覆盖率太低,则可能需要从结构预测中删除这些碱基。除了RT-PCR和全基因组RNAseq试剂盒外,还可以使用其他文库制备方法。当使用RNA的小片段或必须避免引物区域中探测信息的丢失时,包括将3'和/或5'接头连接到RNA的方案是有利的。
最后,必须始终仔细解释化学探测实验的分析。目前,还没有软件可以仅从序列中高精度地预测任何RNA的RNA结构。尽管化学探测约束大大提高了准确性,但为长RNA(>500 nt)生成良好的模型仍然具有挑战性。这些模型应通过其他方法和/或诱变进一步测试。RNA预测软件针对最大碱基对数进行优化,从而显着惩罚开放构象,这些构象可能无法准确代表RNA折叠5。因此,应通过量化与基础化学探测数据(例如,通过AUROC)和重复之间的预测一致性(例如,通过mFMI)来测试获得的结构模型,如Lan等人20所示。
理想情况下,应该使用不同系统中的几个实验来挑战获得的结构模型来加强一个人的假设。这些可能包括体 外 和细胞内方法的使用、代偿突变以及不同的细胞系和物种。此外,原始反应性通常与结构预测一样,甚至更具信息性,因为它们记录了RNA折叠系综的"基本事实"快照。因此,原始反应性非常适合比较不同条件之间的结构变化,并且非常有用。重要的是,使用化学探测约束和计算预测计算的最低自由能结构只能用作完整结构模型的起始假设。
披露声明
作者没有利益冲突需要声明。
致谢
没有
材料
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
参考文献
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。