
Method Article
Sondare la struttura dell'RNA con profilo mutazionale del dimetilsolfato con sequenziamento in vitro e nelle cellule
In questo articolo
Riepilogo
Il protocollo fornisce istruzioni per modificare l'RNA con dimetilsolfato per esperimenti di profilazione mutazionale. Include il sondaggio in vitro e in vivo con due metodi alternativi di preparazione della libreria.
Abstract
Il ruolo della struttura dell'RNA praticamente in qualsiasi processo biologico è diventato sempre più evidente, specialmente nell'ultimo decennio. Tuttavia, gli approcci classici per risolvere la struttura dell'RNA, come la cristallografia dell'RNA o la crio-EM, non sono riusciti a tenere il passo con il campo in rapida evoluzione e la necessità di soluzioni ad alto rendimento. Profilo mutazionale con sequenziamento mediante dimetilsolfato (DMS) MaPseq è un approccio basato sul sequenziamento per dedurre la struttura dell'RNA dalla reattività di una base con DMS. Il DMS metila l'azoto N1 nelle adenosine e l'N3 nelle citosine sulla loro faccia di Watson-Crick quando la base non è spaiata. La trascrizione inversa dell'RNA modificato con la trascrittasi inversa dell'introne termostabile di gruppo II (TGIRT-III) porta all'incorporazione delle basi metilate come mutazioni nel cDNA. Quando si sequenzia il cDNA risultante e lo si rimappone a una trascrizione di riferimento, i tassi di mutazione relativi per ciascuna base sono indicativi dello "stato" della base come accoppiata o non accoppiata. Anche se le reattività del DMS hanno un elevato rapporto segnale-rumore sia in vitro che nelle cellule, questo metodo è sensibile alla distorsione nelle procedure di manipolazione. Per ridurre questo pregiudizio, questo articolo fornisce un protocollo per il trattamento dell'RNA con DMS nelle cellule e con RNA trascritto in vitro .
Introduzione
Dalla scoperta che l'RNA ha proprietà sia strutturali1,2 che catalitiche3, l'importanza dell'RNA e la sua funzione regolatrice in una pletora di processi biologici sono stati gradualmente scoperti. In effetti, l'effetto della struttura dell'RNA sulla regolazione genica ha guadagnato crescente attenzione4. Come le proteine, l'RNA ha strutture primarie, secondarie e terziarie, riferendosi rispettivamente alla sequenza di nucleotidi, alla mappatura 2D delle interazioni di accoppiamento di basi e al ripiegamento 3D di queste strutture accoppiate di basi. Mentre determinare la struttura terziaria è la chiave per comprendere i meccanismi esatti alla base dei processi dipendenti dall'RNA, la struttura secondaria è anche altamente informativa sulla funzione dell'RNA ed è la base per un ulteriore ripiegamento 3D5.
Tuttavia, determinare la struttura dell'RNA è stato intrinsecamente impegnativo con gli approcci convenzionali. Mentre per le proteine, la cristallografia, la risonanza magnetica nucleare (NMR) e la microscopia elettronica criogenica (crio-EM) hanno permesso di determinare la diversità dei motivi strutturali, consentendo la previsione della struttura dalla sola sequenza6, questi approcci non sono ampiamente applicabili agli RNA. In effetti, gli RNA sono molecole flessibili con elementi costitutivi (nucleotidi) che hanno molta più libertà conformazionale e rotazionale rispetto alle loro controparti aminoacidiche. Inoltre, le interazioni attraverso l'accoppiamento di basi sono più dinamiche e versatili di quelle dei residui di amminoacidi. Di conseguenza, gli approcci classici hanno avuto successo solo per RNA relativamente piccoli con strutture ben definite e altamente compatte7.
Un altro approccio per determinare la struttura dell'RNA è attraverso il sondaggio chimico combinato con il sequenziamento di nuova generazione (NGS). Questa strategia genera informazioni sullo stato di legame di ciascuna base in una sequenza di RNA (cioè la sua struttura secondaria). In breve, le basi in una molecola di RNA che non sono impegnate nell'accoppiamento di basi sono modificate in modo differenziale da piccoli composti chimici. La trascrizione inversa di questi RNA con trascrittasi inverse specializzate (RT) incorpora le modifiche in acido desossiribonucleico complementare (cDNA) come mutazioni. Queste molecole di cDNA vengono poi amplificate dalla reazione a catena della polimerasi (PCR) e sequenziate. Per ottenere informazioni sul loro "stato" come legato o non legato, le frequenze di mutazione in ciascuna base in un RNA di interesse sono calcolate e inserite nel software di previsione della struttura come vincoli8. Sulla base delle regole del vicino più vicino9 e dei calcoli dell'energia libera minima 10, questo software genera modelli di struttura che meglio si adattano ai dati sperimentali ottenuti11,12.
DMS-MaPseq utilizza DMS, che metila l'azoto N1 nelle adenosine e l'azoto N3 nelle citosine sulla loro faccia di Watson-Crick in modo altamente specifico13. L'uso della trascrittasi inversa dell'introne termostabile del gruppo II (TGIRT-III) nella trascrizione inversa crea profili mutazionali con rapporti segnale-rumore senza precedenti, consentendo anche la deconvoluzione di profili sovrapposti generati da due o più conformazioni alternative14,15. Inoltre, il DMS può penetrare nelle membrane cellulari e in interi tessuti, rendendo possibile il sondaggio all'interno di contesti fisiologici. Tuttavia, la generazione di dati di buona qualità è impegnativa, poiché le variazioni nella procedura di gestione possono influire sui risultati. Pertanto, forniamo un protocollo dettagliato per DMS-MaPseq sia in vitro che in cellula per ridurre i pregiudizi e guidare i nuovi arrivati al metodo attraverso le difficoltà che possono incontrare. Soprattutto alla luce della recente pandemia di SARS-CoV2, i dati di alta qualità sui virus a RNA sono uno strumento importante per studiare l'espressione genica e trovare possibili terapie.
Protocollo
NOTA: vedere la tabella dei materiali per i dettagli relativi a tutti i materiali, software, reagenti, strumenti e celle utilizzati in questo protocollo.
1. DMS-MaP in vitro gene-specifico
- Trascrizione dell'RNA in vitro
- Ottenere la sequenza dell'RNA di interesse come DNA a doppio filamento (ds) (ad esempio, come frammenti di DNA, plasmidi o PCR da DNA preesistente / genomico). Se la sequenza di DNA contiene un promotore della polimerasi, passare al punto 3.
- Eseguire la sovrapposizione PCR per attaccare un promotore di RNA polimerasi a monte del frammento di DNA desiderato (primer in avanti per la polimerasi T7: 5' TAATACGACTCACTATAGG + prime basi della sequenza target 3').
- Trascrivere in vitro il frammento di DNA in RNA. Mantenere sempre l'RNA sul ghiaccio.
- Digerire il DNA usando una DNasi.
- Isolare l'RNA utilizzando un approccio basato su colonne (fase 2.4) o mediante precipitazione di etanolo (fase 2.5). Eluire in un volume appropriato, aspettandosi una resa di ~50 μg.
- Garantire l'integrità dell'RNA eseguendolo su un gel di agarosio; denaturare l'RNA per 2-3 minuti a 70 °C prima di eseguirlo.
NOTA: Il tampone e l'agarosio possono contenere RNasi che degradano l'RNA e potrebbero contaminare il campione di RNA. I gel prefabbricati di agarosio sono stati precedentemente utilizzati in questo laboratorio; i risultati (specialmente con l'RNA) sono stati a volte ambigui. I migliori risultati sono stati ottenuti con gel di agarosio o PAGE. - Uso diretto di conservare l'RNA a -80 °C per diversi mesi, a meno che la degradazione non sia visibile dopo lo scongelamento.
- In vitro Modifica DMS (a 105 mM DMS)
- Preparare una quantità sufficiente di tampone ripiegabile (cacodilato di sodio 0,4 M, pH 7,2, contenente 6 mM MgCl2).
NOTA: Per ogni reazione (volume finale di 100 μL), aggiungere 89 μL di tampone ripiegabile. - Per ogni reazione, trasferire 89 μL di tampone ripiegabile in un tubo designato da 1,5 mL e preriscaldare a 37 °C in un termoshaker posto sotto una cappa chimica.
NOTA: Il DMS è altamente tossico e deve essere sempre tenuto sotto una cappa chimica fino a quando non viene spento da un agente riducente. - Eluire 1-10 pmol di RNA in 10 μL di acqua priva di nucleasi (NF H2O); trasferimento in un tubo PCR.
- Incubare in un termociclatore a 95 °C per 1 minuto per denaturare l'RNA.
- Posizionare immediatamente su un blocco di ghiaccio per evitare di piegarsi male.
- Aggiungere il campione di RNA alla provetta designata con tampone ripiegabile a 37 °C, mescolare bene e incubare per 10-20 minuti per ripiegare l'RNA.
NOTA: La maggior parte degli RNA si ripiega nell'ordine dei millisecondi ai secondi, sebbene esistano eccezioni16. - Aggiungere 1 μL di DMS al 100% (10,5 M) al campione di RNA e incubare per 5 minuti agitando a 800-1.400 rotazioni al minuto (rpm).
NOTA: L'agitazione (o altri mezzi di miscelazione) in questa fase è fondamentale poiché il DMS è idrofobo e potrebbe non dissolversi completamente nel tampone di ripiegatura. Deviazioni nei tempi di reazione potrebbero influenzare la riproducibilità delle reattività del DMS. Per ridurre al minimo l'errore di pipettaggio, il DMS può essere sciolto in etanolo al 100% prima di aggiungerlo al campione se viene mantenuta una concentrazione finale di DMS all'1% (105 mM). Per un controllo non trattato, il DMS può essere sostituito dal dimetilsolfossido (DMSO) o dall'acqua. - Dopo 5 minuti di tempo di reazione, temprare con 60 μL di β-mercaptoetanolo (BME) al 100%, mescolare bene e posizionare immediatamente l'RNA su ghiaccio.
NOTA: L'RNA può essere rimosso in modo sicuro dal cappuccio dopo aver spento la reazione con BME per pulirlo. Tuttavia, l'esposizione diretta di BME all'ambiente circostante dovrebbe comunque essere evitata a causa del suo forte odore e delle proprietà irritanti. - Pulire l'RNA mediante precipitazione di acetato di sodio ed etanolo (vedere punto 2.5) o un approccio basato su colonne (vedere punto 2.6) ed eluire in 10 μL di acqua.
- Quantificare l'RNA utilizzando uno spettrofotometro.
- Uso diretto di conservare l'RNA modificato a -80 °C.
NOTA: La conservazione a lungo termine deve essere evitata, poiché l'RNA è meno stabile dopo il trattamento con DMS.
- Preparare una quantità sufficiente di tampone ripiegabile (cacodilato di sodio 0,4 M, pH 7,2, contenente 6 mM MgCl2).
- RT-PCR gene-specifica di RNA modificato
NOTA: Vedere la Figura 1 per la configurazione RT-PCR dei frammenti trattati con DMS.- Eluire 100 ng di RNA modificato in 10 μL di H 2 O. Senza nucleasi (NF) H2O. Trasferimento in una provetta per PCR.
- Al tubo, aggiungere 4 μL di 5x tampone di primo filamento (FSB), 1 μL di miscela dNTP (10 mM ciascuno), 1 μL di 0,1 M ditiotreitolo (DTT) (evitare cicli di congelamento-disgelo), 1 μL di inibitore della RNasi, 1 μL di primer inverso da 10 μM (singolo primer o un pool di primer) e 1 μL di TGIRT III.
NOTA: Per un pool di primer, non aggiungere 1 μL di 10 μM di ciascun primer direttamente all'RT; invece, mescolare prima i primer e aggiungere 1 μL dalla miscela (a 10 μM di concentrazione totale del primer). - Incubare a 57 °C per 30 minuti a 1,5 h (in genere, 30 minuti sono sufficienti per ottenere un prodotto da 500 nt) in un termociclatore.
- Aggiungere 1 μL di 4 M NaOH, mescolare mediante pipettaggio e incubare a 95 °C per 3 minuti per degradare l'RNA.
NOTA: Questo passaggio è cruciale in quanto rilascia TGIRT dal cDNA degradando l'RNA. Se saltata, la PCR a valle potrebbe essere influenzata. - Pulire utilizzando un approccio basato su colonne (vedere punto 2.6) che rimuove sufficientemente i primer ed eluire in 10 μL di NF H2O.
- PCR-amplifica il cDNA utilizzando 1 μL del prodotto di trascrizione inversa per 25 μL della reazione con un kit PCR progettato per bilanciare resa e fedeltà.
NOTA: I primer devono avere una temperatura di fusione di ~60 °C. - Eseguire 2 μL del prodotto PCR su un gel di agarosio o un gel di agarosio prefabbricato per verificare il successo della PCR.
- Idealmente, solo una banda dovrebbe mostrare dopo la PCR. In tal caso, ripulisci la reazione utilizzando un approccio basato su colonne. Se sono presenti bande alternative, utilizzare la reazione PCR rimanente per asportare la fascia corretta dal gel. Eluire in un volume sufficientemente piccolo (ad esempio, 10 μL).
- Quantificare i frammenti estratti utilizzando uno spettrofotometro.
- Indicizzare i frammenti di dsDNA per il sequenziamento utilizzando un approccio adatto alla piattaforma di sequenziamento desiderata.

Figura 1: Messa a punto sperimentale per la RT-PCR di grandi frammenti trattati con DMS. Quando si esegue la trascrizione inversa su un RNA modificato, le modifiche sulla sequenza a cui il primer si rimargina non verranno registrate. Pertanto, quando i frammenti superano i 400-500 bp di lunghezza, è necessario progettare frammenti che si sovrappongono nelle regioni di primer, come esemplificato qui. La lunghezza dei frammenti dipende dalle esigenze di sequenziamento. Quando si utilizza il sequenziamento a 150 cicli accoppiati, i frammenti non devono superare i 300 bp. Abbreviazioni: RT-PCR = reazione a catena della polimerasi a trascrizione inversa; DMS = dimetilsolfato. Fare clic qui per visualizzare una versione ingrandita di questa figura.
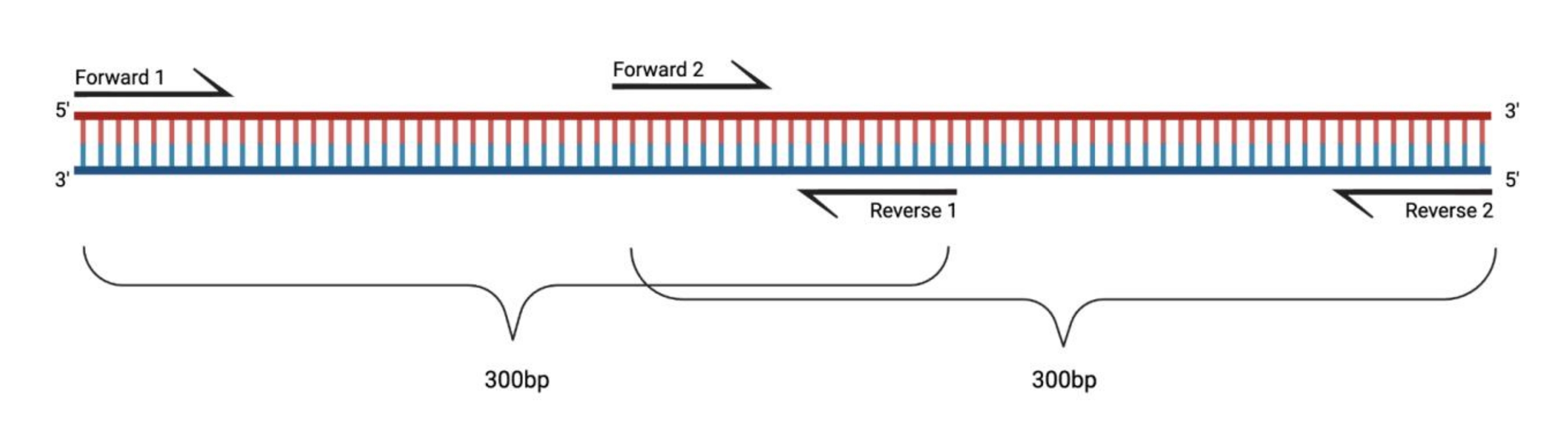{kind=link}
2. DMS-MaP dell'intero genoma utilizzando cellule infette da virus
NOTA: Nelle cellule, il trattamento DMS può anche essere combinato con l'approccio di amplificazione gene-specifico descritto sopra. La libreria dell'intero genoma richiede un'enorme profondità di sequenziamento per ottenere una copertura completa su un singolo gene. Tuttavia, se gli RNA virali costituiscono una frazione significativa dell'RNA ribodepleted dopo l'estrazione, il sequenziamento dell'intero genoma sarebbe appropriato. Inoltre, altri metodi di arricchimento possono essere combinati con il metodo di generazione della libreria dell'intero genoma.
- Trattamento DMS
- Crescere le cellule infette dal virus fino allo stadio desiderato dell'infezione.
- Trasferire il contenitore della cella in una cappa aspirante dedicata che sia appropriata per gestire sia i virus al livello di biosicurezza richiesto sia i fumi chimici generati da agenti come il DMS.
- Aggiungere un volume del 2,5% di DMS al terreno di coltura e sigillare il contenitore (tipicamente una piastra di 10 cm) con parafilm.
NOTA: è facile sotto-modificare e sovra-modificare con DMS. Quando si aggiunge DMS direttamente alle cellule, è molto importante mescolare bene. In alternativa, preriscaldare il nuovo mezzo in un tubo conico da 50 mL a 37 °C e aggiungere il DMS agitando vigorosamente direttamente. Decantare il mezzo esaurito sulle celle e pipettare lentamente nel mezzo contenente DMS. - Trasferire in un'incubatrice a 37 °C per 5 min.
NOTA: A seconda del tempo necessario per gestire il DMS all'esterno dell'incubatore, è possibile che 5 minuti portino a modifiche eccessive. Mantenere il tempo dall'aggiunta del DMS all'incubazione a ≤1 min. Se si esegue l'esperimento per la prima volta, si consiglia di eseguire una titolazione DMS e variare il tempo di incubazione (tra 3 minuti e 10 minuti) per trovare la velocità di modifica ottimale e garantire che i risultati siano robusti in una finestra di concentrazioni. - Pipettare con cautela il mezzo contenente DMS (in rifiuti chimici appropriati) e aggiungere delicatamente 10 ml di tampone di arresto (PBS con BME al 30% [ad esempio, 3 ml di BME e 7 ml di PBS]).
NOTA: L'aggiunta di DMS e BME può sollevare le cellule dalla piastra se le cellule non sono fortemente aderenti. Se le cellule si stanno sollevando, possono essere trattate come cellule di sospensione, invece di rimuovere il mezzo contenente DMS, aggiungere direttamente il tampone di arresto e raschiare le cellule con DMS e BME in un tubo conico da 50 ml. Pellettare le celle centrifugando per 3 minuti a 3.000 × g; assicurati di eliminare qualsiasi DMS residuo, che può pellet sotto le cellule in grandi goccioline. Si consiglia una fase di lavaggio supplementare con BME al 30% se il mezzo DMS non può essere rimosso inizialmente. - Raschiare le cellule e trasferirle in un tubo conico da 15 ml.
- Pellet per centrifugazione a 3.000 × g per 3 min.
- Rimuovere il surnatante e lavare 2 volte con 10 ml di PBS.
- Rimuovere con attenzione quanto più PBS residuo possibile.
- Sciogliere il pellet in una quantità appropriata del reagente di isolamento dell'RNA (ad esempio, 3 ml per un matraccio di coltura T75, 1 ml per una piastra di 10 cm).
NOTA: quantità insufficienti del reagente potrebbero influire sulla resa dell'RNA.
- Estrazione dell'RNA e deplezione dell'RNA ribosomiale (rRNA)
- A 1 mL di cellule omogeneizzate nel reagente di isolamento dell'RNA, aggiungere 200 μL di cloroformio, vortice per 15-20 s fino a rosa brillante, quindi incubare per un massimo di 3 minuti fino a quando la separazione di fase è visibile.
NOTA: La fase lipidica rosa dovrebbe depositarsi sul fondo. In caso contrario, il tempo di vortice era probabilmente insufficiente. - Centrifugare alla massima velocità (~ 20.000 × g) per 15 minuti a 4 °C.
- Trasferire la fase acquosa superiore in un nuovo tubo.
- Pulire l'RNA mediante precipitazione di acetato di sodio-etanolo (vedi fase 2.5) o un approccio basato su colonne (vedi punto 2.6) ed eluire in un volume sufficiente di NF H2O.
- Controllare l'integrità dell'RNA su un gel di agarosio. Cerca due bande corrispondenti alle due subunità ribosomiali.
- Esaurire gli rRNA usando l'approccio preferito ed eluire in un volume adeguato (tipicamente 20-50 μL) di NF H2O.
NOTA: Per le applicazioni a valle, ~ 500 ng di RNA totale è suggerito in un volume di 8 μL. Gli RNA non ribosomiali costituiscono tipicamente solo il 5% -10% dell'RNA totale. - Quantificare utilizzando uno spettrofotometro.
- A 1 mL di cellule omogeneizzate nel reagente di isolamento dell'RNA, aggiungere 200 μL di cloroformio, vortice per 15-20 s fino a rosa brillante, quindi incubare per un massimo di 3 minuti fino a quando la separazione di fase è visibile.
- Generazione di librerie
- Utilizzare RT-PCR gene-specifiche o altri approcci per generare librerie15. Se si utilizzano esameri casuali per l'adescamento, aggiungere una fase di incubazione a un basso Tm (37-42 °C) per consentire la ricottura dell'esamero.
NOTA: I kit di generazione della libreria standard possono essere utilizzati anche sostituendo l'enzima RT con TGIRT e modificando la temperatura RT a 57 °C.
- Utilizzare RT-PCR gene-specifiche o altri approcci per generare librerie15. Se si utilizzano esameri casuali per l'adescamento, aggiungere una fase di incubazione a un basso Tm (37-42 °C) per consentire la ricottura dell'esamero.
- Pulizia dell'RNA basata su colonne utilizzando le colonne RNA Clean & Concentrator
NOTA: tutte le fasi devono essere eseguite a temperatura ambiente.- Aggiungere NF H2O alla provetta per portarla ad un volume di 50 μL.
- Aggiungere 100 μL di tampone legante e 150 μL di etanolo al 100% al campione.
- Mescolare e trasferire in una colonna di rotazione.
- Girare a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- Aggiungere 400 μL di tampone di preparazione dell'RNA.
- Spin a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- Aggiungere 700 μL di tampone di lavaggio dell'RNA.
- Spin a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- Aggiungere 400 μL di tampone di lavaggio dell'RNA.
- Spin a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- (Facoltativo) Trasferire la colonna in un nuovo tubo di raccolta e ruotare a 10.000-16.000 × g per 2 minuti.
- Trasferire la colonna in un tubo pulito privo di RNAsi e aggiungere una quantità appropriata di NF H2O.
- Girare a 10.000-16.000 × g per 1 min.
- Estrazione di RNA acido fenolo-cloroformio.
- Aggiungere un volume uguale di acido fenolo:cloroformio:alcool isoamilico.
- Vortice accuratamente e centrifugare a 14.000 × g per 5 minuti.
- Se non c'è separazione di fase, aggiungere 20 μL di 2 M NaCl e ripetere la centrifugazione.
- Trasferire la fase acquosa in un nuovo tubo.
- Aggiungere 500 μL di isopropanolo e 2 μL di co-precipitante.
- Mescolare e incubare a RT per 3 minuti; quindi, incubare a -80 °C durante la notte.
- Pellettare l'RNA mediante centrifugazione alla massima velocità (~ 20.000 × g) per 30 minuti a 4 °C.
- Lavare il pellet con 200 μL di etanolo ghiacciato al 70%.
- Girare alla massima velocità (~ 20.000 × g) per 5 minuti; Eliminare il flusso through.
- Risospendere il pellet nella quantità appropriata di NF H2O.
- Pulizia del cDNA basata su colonne utilizzando le colonne Oligo Clean e Concentratore
NOTA: Tutti i passaggi devono essere condotti a temperatura ambiente.- Aggiungere NF H2O alla provetta per portarla ad un volume di 50 μL.
- Aggiungere 100 μL di tampone legante e 400 μL di etanolo al 100%.
- Mescolare e trasferire in una colonna di rotazione.
- Girare a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- Aggiungere 750 μL di tampone di lavaggio del DNA.
- Spin a 10.000-16.000 × g per 30 s; Eliminare il flusso through.
- (Facoltativo) Trasferire la colonna in un nuovo tubo di raccolta e ruotare a 10.000-16.000 × g per 2 minuti.
- Trasferire la colonna in un tubo pulito privo di RNAsi e aggiungere una quantità appropriata di NF H2O.
- Girare a 10.000-16.000 × g per 1 min.
3. Analisi dei dati di sequenziamento
NOTA: per creare modelli di struttura secondaria dell'RNA dai dati di sequenziamento DMS-MaP, i file .fastq risultanti devono essere elaborati in diversi passaggi. Questi passaggi possono essere eseguiti automaticamente utilizzando il comando
- Tagliare le sequenze dell'adattatore con TrimGalore o Cutadapt.
- Mappare le letture alle sequenze di riferimento (formato .fasta) utilizzando Bowtie2.
- Conta le letture con un software specializzato nella struttura dell'RNA (ad esempio, DREEM14, RNA-Framework17 o simili) e crea profili di reattività.
- (Facoltativo) Raggruppa le letture per trovare conformazioni alternative di RNA usando DREEM 14, DRACO17, DANCE-MaP18 o simili.
- Prevedere la struttura minima di energia libera in base ai profili di reattività utilizzando RNAStructure12, ViennaRNA o simili.
- Visualizza la struttura dell'RNA11 usando VARNA (https://varna.lri.fr/) o simili.
NOTA: Per praticità, software come DREEM (www.rnadreem.org) e RNA-Framework19 incorporano ampiamente i passaggi 1--5 nelle loro pipeline, semplificando il processo di analisi. Tuttavia, qualsiasi previsione della struttura deve essere gestita con cura (ad esempio, verificando l'accordo della struttura con i dati20.
Risultati
DMS-MaP in vitro gene-specifico
Per studiare il 5'UTR di SARS2, i primi 300 bp del virus sono stati ordinati come sequenza gBlock, insieme a tre primer. Questi includevano due primer per propagare il frammento ("FW" e "RV") tramite PCR, nonché uno per attaccare il promotore T7 ("FW-T7"). Queste sequenze possono essere viste nella Tabella 1.
| Nome | Sequenza (5'->3') |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCCCCCCAGGTAAC |
Tabella 1: Sequenza di primer per DMS-MaP RT-PCR di SARS-CoV2 5'UTR. Qui, FW-T7 e RV sono necessari per generare un modello di DNA per la trascrizione in vitro , il RV viene utilizzato nella trascrizione inversa e la coppia di primer FW-RV viene utilizzata nella successiva amplificazione PCR del cDNA. I primer risuonano fino all'inizio del genoma SARS-CoV2 (FW) e alla sequenza proprio a valle della regione di interesse. Abbreviazioni: DMS-MaP = Profilo mutazionale con sequenziamento mediante dimetilsolfato; RT-PCR = reazione a catena della polimerasi a trascrizione inversa; SARS-CoV2 = sindrome respiratoria acuta grave-coronavirus 2; UTR = regione non tradotta; RV = primer inverso; FW = primer in avanti.
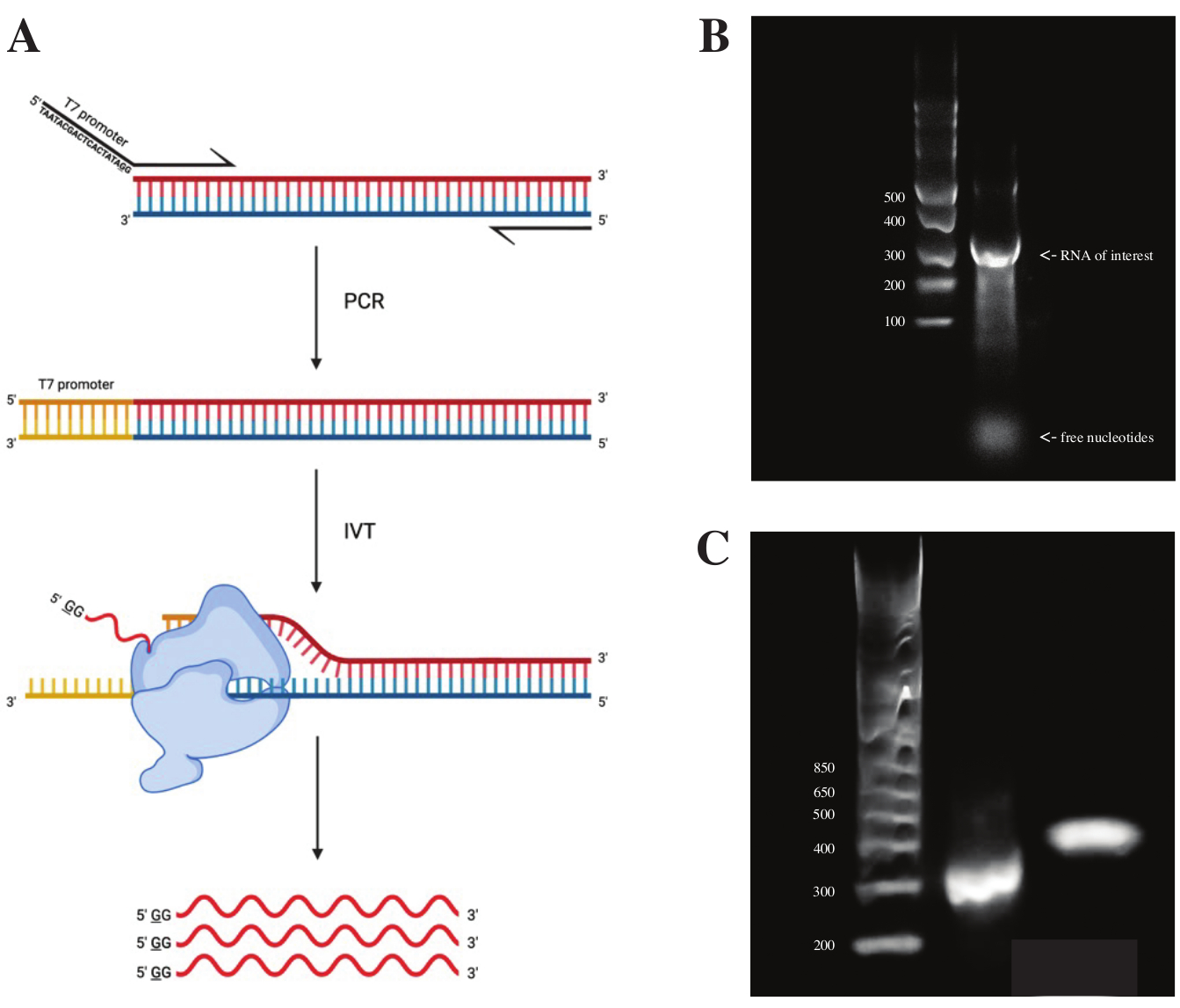
Per generare RNA dal frammento gBlock, la sequenza del promotore della polimerasi T7 è stata attaccata utilizzando la sovrapposizione PCR utilizzando la premiscela PCR secondo lo schema visto in Figura 2A. Dal frammento allungato, l'RNA è stato generato utilizzando il kit di trascrizione T7. Il modello di DNA è stato successivamente digerito utilizzando la DNasi e l'RNA isolati utilizzando colonne RNA Clean & Concentrator.
Il controllo di qualità della trascrizione in vitro è stato effettuato eseguendo il prodotto RNA su un gel di agarosio all'1% insieme a una scala di ssRNA. Poiché era visibile una sola banda, sono stati eseguiti il sondaggio DMS in vitro e la RT-PCR (vedi Figura 2B).
Per verificare il successo della reazione PCR, il campione è stato eseguito su un gel di agarosio al 2% utilizzando una scala dsDNA. Dopo l'indicizzazione, la banda dovrebbe funzionare ~ 150 bp più in alto sullo stesso gel, tenendo conto della dimensione dei primer di indicizzazione.

Figura 2: Trascrizione in vitro del modello di DNA. (A) Per trascrivere in vitro un modello di DNA che non ha ancora un promotore intrinseco RNA polimerasi, il modello deve essere prima allegato mediante sovrapposizione PCR. Questo viene fatto utilizzando un primer in avanti, che include la sequenza TAATACGACTCACTATAGG (nel caso della T7 RNA polimerasi) a monte delle prime basi sovrapposte al frammento desiderato. La base sottolineata qui simboleggia il sito di inizio della trascrizione della polimerasi. Una volta che il promotore si è attaccato al frammento di dsDNA, può essere trascritto dalla polimerasi T7. È importante sottolineare che la polimerasi utilizza il filamento opposto alla sequenza del promotore menzionata come modello (blu), creando effettivamente RNA identico alla sequenza immediatamente a valle della sequenza del promotore indicata (rosso). (B) Un gel di agarosio all'1% con una scala di ssRNA (corsia 1) e il prodotto di RNA trascritto in vitro a 300 nt (corsia 2). (C) Un gel di agarosio al 2% con GeneRuler 1 kb più Ladder (corsia 1), il prodotto PCR dopo RT-PCR funzionante a 300 bp (corsia 2) e il frammento indicizzato dopo la preparazione della libreria in esecuzione a 470 bp (corsia 3). Abbreviazioni: RT-PCR = reazione a catena della polimerasi a trascrizione inversa; DMS = dimetilsolfato; nt = nucleotidi; dsDNA = DNA a doppio filamento; ssRNA = RNA a singolo filamento. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Intero genoma in vivo DMS-MaP utilizzando cellule infettate da virus
Prima del trattamento DMS, le cellule HCT-8 erano infette da OC43. Quando è stato osservato un effetto citopatico (CPE) 4 giorni dopo l'infezione (dpi) (come si vede nella Figura 3A), queste cellule sono state trattate e l'RNA è stato estratto e ribodepleto. Durante l'esecuzione dell'RNA totale su un gel di agarosio, erano visibili due bande luminose, che rappresentavano le subunità 40S e 60S del ribosoma, che costituiscono circa il 95% della massa totale dell'RNA (vedi Figura 3B). Quando l'estrazione dell'RNA non ha avuto successo o è stata degradata (ad esempio, da più cicli di congelamento-scongelamento), i prodotti di degradazione dell'RNA erano visibili sul fondo del gel (vedi Figura 3C, seconda corsia). Inoltre, dopo la deplezione dell'rRNA, le due bande luminose sono scomparse, lasciando una macchia nella corsia (vedi Figura 3C, terza corsia). Infine, dopo la preparazione della libreria, i campioni avevano distribuzioni di dimensioni variabili e sono stati mostrati come uno striscio sul gel PAGE finale. La banda è stata asportata tra 200 nucleotidi (nt) e 500 nt, in accordo con il sequenziamento paired-end 150 x 150 pianificato per analizzare queste librerie. Ancora più importante, i dimeri dell'adattatore funzionanti a ~ 150 nt sono stati separati (vedi Figura 3D).

Figura 3: Checkpoint di DMS-MaP in vivo con cellule infette da virus. (A) Immagine al microscopio ottico di cellule HCT-8 infettate da virus, dpi di 4 giorni. Per ottenere la massima resa possibile di RNA virale dall'RNA totale riducendo al minimo gli effetti avversi dovuti alla morte cellulare, il DMS dovrebbe essere aggiunto all'inizio della CPE o anche prima, come si vede nell'immagine. (B) Un gel di agarosio all'1% con sei campioni di 1 μg di RNA totale. In ogni corsia sono visibili due bande luminose, che rappresentano le subunità 40S e 60S, poiché l'RNA ribosomiale costituisce ~ 95% dell'RNA totale. Nota: il trattamento DMS in-cell causa una certa frammentazione e sbavatura dell'RNA, ma le due bande di rRNA dovrebbero essere ancora visibili. La frammentazione lieve dopo la modifica è tollerata perché le informazioni contenenti il segno di metilazione vengono generate e riportate sulla struttura dell'RNA durante l'incubazione del DMS mentre le cellule sono ancora vive. (C) Un gel di agarosio all'1% di GeneRuler 1 kb più marcatore di DNA a scala (corsia 1) RNA totale precedentemente conservato a -80 °C per 6 mesi (corsia 2) e RNA ribodepletto (corsia 3). Quando si conserva l'RNA per lungo tempo con diversi cicli di congelamento-disgelo, l'RNA inizia a degradarsi e probabilmente non dovrebbe essere utilizzato per esperimenti di sondaggio. Inoltre, dopo aver ribodepletato l'RNA totale, le due bande luminose, che rappresentano le subunità 40S e 60S del ribosoma, svaniscono e inizia a mostrare uno striscio degli RNA residui. (D) Un gel PAGE di GeneRuler 1 kb più marcatore di DNA a scala (corsia 1) e un campione di libreria di RNA preparato per l'intero genoma. Il gel deve essere asportato in base alle esigenze di sequenziamento. Per un sequenziamento a estremità accoppiata che copre 150 cicli da entrambi i lati, il gel deve essere asportato tra 300 bp e 500 bp. I dimeri adattatori (funzionanti a 170 bp) devono essere separati. Abbreviazioni: DMS-MaP = Profilo mutazionale con sequenziamento mediante dimetilsolfato; dpi = giorni dopo l'infezione; CPE = effetto citopatico. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
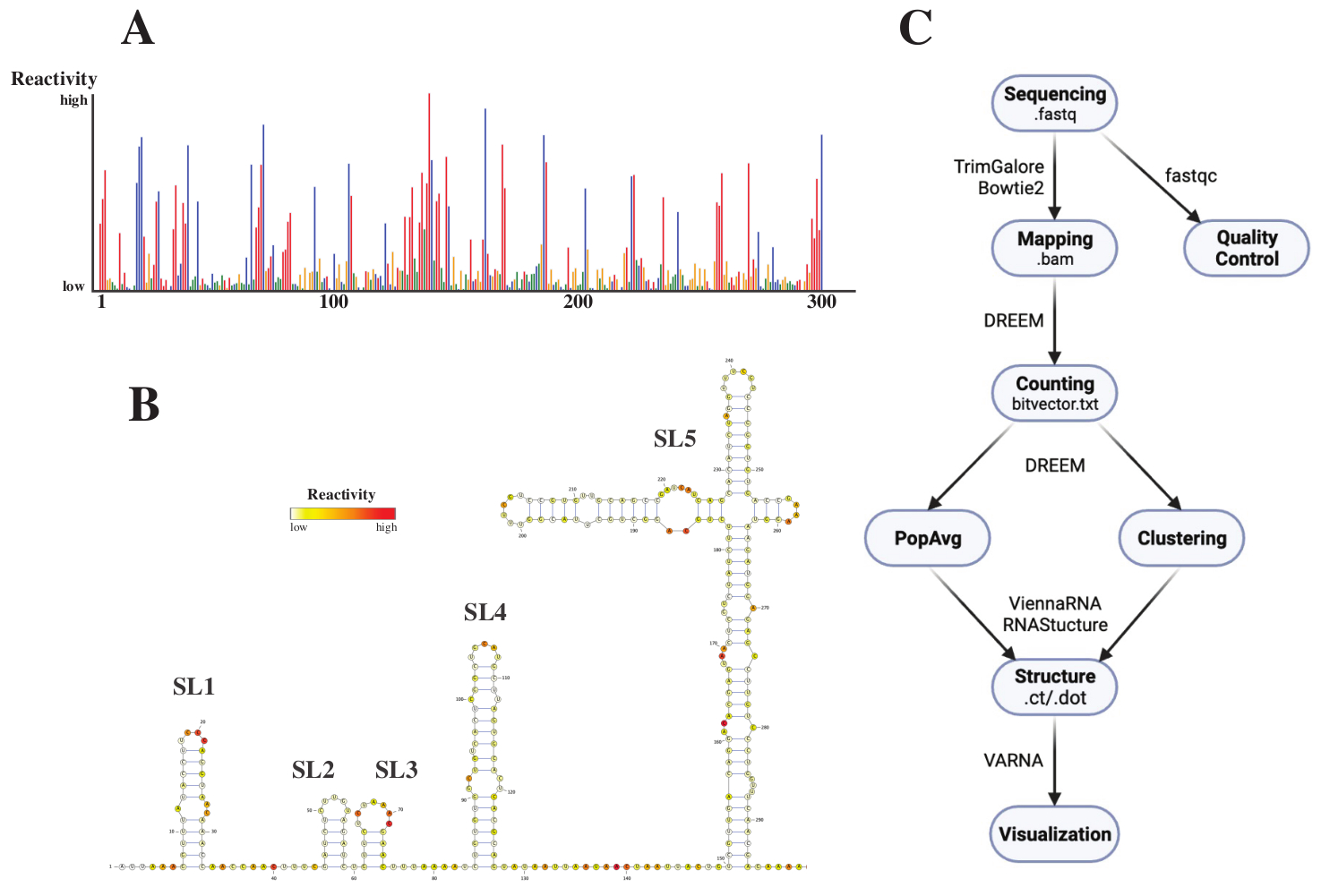
Dopo la sequenziazione, i file .fastq sono stati analizzati inviando un lavoro al server web DREEM (http://rnadreem.org/), insieme a un file di riferimento .fasta. L'output generato dal server include file di controllo qualità generati da fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) e TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), nonché altri file di output contenenti le frequenze medie di mutazione della popolazione. Oltre al diagramma che mostra le frequenze delle mutazioni con un formato .html interattivo (vedi Figura 4A) e un file .csv con le riattivazioni grezze per base e un file struct_constraint.txt, leggibile da diversi software di previsione della struttura dell'RNA, questo include anche un file bitvector.txt che riporta le mutazioni by-read. Da questi, le strutture medie della popolazione sono state calcolate inviando i file .fasta e struct_constraint.txt al server web RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Questo utilizza il software ViennaRNA per generare previsioni di struttura basate sull'energia libera minima, che può essere visualizzata online o scaricata in formato ct o Vienna. Per generare modelli di struttura dell'RNA, questi file scaricabili sono stati inviati a VARNA (https://varna.lri.fr/, vedere Figura 4B). Infine, i file bitvector.txt possono essere utilizzati dalla versione stabile di DREEM (https://codeocean.com/capsule/6175523/tree/v1) per cercare conformazioni alternative dell'RNA. Per ottenere buoni modelli di struttura utilizzando DREEM, è necessario ottenere una copertura di 10.000 letture per base; Per il clustering, potrebbero essere necessarie fino a 100.000 letture per base. Una panoramica dell'intero flusso di lavoro è disponibile nella Figura 4C.

Figura 4: Dati esemplari ottenuti da esperimenti di sondaggio chimico del SARS-CoV2 5'UTR. (A) Profilo di reattività delle prime 300 basi del genoma SARS-CoV2 colorate per base (A: rosso, C: blu, U: verde, G: giallo). Le reattività grezze sono calcolate come la frequenza assoluta di mutazione divisa per la copertura. Le basi a conformazione aperta hanno alti valori di reattività; Le basi impegnate nell'accoppiamento di basi hanno valori di reattività bassi. u e G non sono modificati dal DMS e hanno bassi valori di reattività, originati dall'infedeltà della polimerasi. Le previsioni sono state fatte con il server web DREEM. (B) Modello di struttura del SARS-CoV2 5'UTR previsto dai valori di reattività effettuati con VARNA. Le basi con alti valori di reattività sono colorate in rosso; Le basi con bassi valori di reattività sono colorate in bianco. (C) Flusso di lavoro dell'analisi DMS-MaP a partire dai file .fastq ottenuti dal sequenziamento. Questi possono essere controllati dalla qualità utilizzando fastqc; le sequenze dell'adattatore vengono rifilate utilizzando TrimGalore e quindi mappate a una sequenza di riferimento utilizzando Bowtie2. Dai file .bam ottenuti, DREEM conta le mutazioni in ogni lettura, creando una mappa delle mutazioni o un file .bitvector.txt. Questi riportano le mutazioni di ciascuna lettura in modo dipendente dalla posizione, in base al quale è possibile creare i profili di reattività media della popolazione. In alternativa, i bitvector possono essere raggruppati usando DREEM per cercare conformazioni alternative dell'RNA. Infine, i modelli di struttura ottenuti vengono visualizzati utilizzando software (ad esempio, VARNA). Abbreviazioni: DMS-MaP = Profilo mutazionale con sequenziamento mediante dimetilsolfato; SARS-CoV2 = sindrome respiratoria acuta grave-coronavirus 2. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Discussione
Il protocollo qui descrive come sondare l'RNA in vitro e nelle cellule utilizzando esperimenti di profilazione mutazionale DMS. Inoltre, fornisce istruzioni su come preparare le librerie per il sequenziamento Illumina per generare dati gene-specifici e analizzare i file .fastq ottenuti. Inoltre, è possibile utilizzare approcci di libreria a livello di genoma. Tuttavia, la RT-PCR gene-specifica produce dati di altissima qualità e più robusti. Pertanto, quando si confrontano tra campioni, è importante assicurarsi che siano preparati con strategie di sequenziamento identiche, poiché la generazione della libreria causa alcune distorsioni. La riproducibilità deve sempre essere misurata utilizzando repliche.
Diverse precauzioni
L'RNA è una molecola instabile che è sensibile alla degradazione sia attraverso temperature elevate che da parte delle RNasi. Pertanto, si raccomandano misure speciali - l'uso di dispositivi di protezione individuale (DPI), materiale privo di RNAsi e inibitori della RNAsi. Ancora più importante, l'RNA dovrebbe essere tenuto sul ghiaccio quando possibile. Questo vale soprattutto per l'RNA metilato, che è ancora più sensibile alle alte temperature.
È importante confermare che la struttura dell'RNA di interesse non è sensibile alla concentrazione di DMS e alle condizioni tampone. Tamponi come 100 mM Tris, 100 mM MOPS e 100 mM HEPES a pH 7-7,5 danno un segnale elevato ma potrebbero non essere sufficienti per mantenere il pH durante la reazione21. Poiché il DMS si idrolizza in acqua, che diminuisce il pH, un tampone forte è fondamentale per mantenere un pH neutro durante la reazione di modifica. L'aggiunta di bicina ha dimostrato di aiutare a mantenere il pH leggermente basico21 , ma si traduce in una bassa modifica del DMS su Gs e Us, che potrebbe essere informativa ma dovrebbe essere analizzata separatamente a causa della produzione di un segnale molto più basso di As e Cs e non è discusso ulteriormente in questo protocollo.
Nella RT-PCR gene-specifica, l'RNA modificato viene trascritto inversamente nel DNA e amplificato in frammenti mediante PCR. Mentre la dimensione dell'RNA può teoricamente essere illimitata, questi frammenti PCR non dovrebbero superare una lunghezza di 400-500 coppie di basi (bp) per prevenire la distorsione durante la reazione di trascrizione inversa. Idealmente, i frammenti dovrebbero rientrare nell'ambito del sequenziamento (cioè, se il sequenziamento viene condotto utilizzando un programma di sequenziamento paired-end a ciclo 150 x 150, un singolo frammento non dovrebbe superare i 300 bp). Quando si utilizzano programmi di sequenziamento con meno cicli, i prodotti PCR possono essere frammentati utilizzando una dsDNasi. Inoltre, poiché le sequenze all'interno delle sequenze di primer non contengono alcuna informazione strutturale, i frammenti devono sovrapporsi quando l'RNA sondato comprende >1 frammento. Le reazioni RT possono contenere più primer RT per frammenti diversi (fino a 10 diversi primer RT). A seconda delle sequenze, il raggruppamento dei primer RT può rendere la trascrizione inversa meno efficiente, ma in genere funziona bene. Ogni reazione PCR deve essere condotta separatamente.
Quando si sonda l'RNA con DMS, le condizioni sperimentali svolgono un ruolo aggiuntivo, poiché molti RNA sono termodinamicamente instabili e cambiano la loro conformazione in base a fattori ambientali come la temperatura. Per evitare irregolarità, le condizioni sperimentali dovrebbero essere mantenute il più possibile costanti, anche per quanto riguarda i tempi di reazione. Le condizioni tampone sembrano essere scambiabili in una certa misura 17,20,22,23 quando vengono mantenute le condizioni di base — la capacità tampone e la presenza di ioni monovalenti (Na) e bivalenti (Mg) — per garantire il corretto ripiegamento dell'RNA 24.
Per quanto riguarda la preparazione in libreria di RNA modificati, diversi aspetti devono essere presi in considerazione. In primo luogo, come accennato in precedenza, gli RNA modificati sono meno stabili delle loro controparti non modificate, il che significa che potrebbero richiedere l'ottimizzazione dei tempi di frammentazione per una distribuzione ottimale delle dimensioni dei frammenti. Inoltre, alcuni kit di preparazione della libreria di RNA, così come molti altri approcci RNAseq, utilizzano primer casuali nel kit di trascrizione inversa. Ciò potrebbe portare a una minore copertura del riferimento, specialmente nei 3' di un gene e, in definitiva, a una profondità di copertura insufficiente. Se la copertura di una determinata regione è troppo bassa, potrebbe essere necessario rimuovere tali basi dalla previsione della struttura. Oltre ai kit RT-PCR e RNAseq dell'intero genoma, possono essere utilizzati altri approcci di preparazione della libreria. I protocolli che includono la legatura di adattatori 3' e/o 5' all'RNA sono vantaggiosi quando si utilizzano piccoli frammenti di RNA o quando si deve evitare la perdita di informazioni di sondaggio nelle regioni di primer.
Infine, l'analisi degli esperimenti di sondaggio chimico deve sempre essere interpretata con attenzione. Attualmente, non esiste un software che preveda la struttura dell'RNA di qualsiasi RNA dalla sola sequenza con elevata precisione. Sebbene i vincoli del sondaggio chimico migliorino notevolmente l'accuratezza, generare buoni modelli per RNA lunghi (>500 nt) è ancora impegnativo. Questi modelli dovrebbero essere ulteriormente testati con altri approcci e/o mutagenesi. Il software di predizione dell'RNA ottimizza per il numero massimo di coppie di basi, penalizzando così significativamente le conformazioni aperte, che potrebbero non rappresentare accuratamente il ripiegamento dell'RNA5. Pertanto, il modello di struttura ottenuto dovrebbe essere testato quantificando l'accordo di previsione con i dati di sondaggio chimico sottostanti (ad esempio, da AUROC) e tra le repliche (ad esempio, da mFMI), come esemplificato da Lan et al.20.
Idealmente, diversi esperimenti in diversi sistemi per sfidare il modello di struttura ottenuto dovrebbero essere utilizzati per rafforzare la propria ipotesi. Questi possono includere l'uso di approcci in vitro e in-cellula , mutazioni compensative e diverse linee cellulari e specie. Inoltre, le reattività grezze sono spesso altrettanto o addirittura più informative delle previsioni di struttura, poiché registrano l'istantanea della "verità sul terreno" dell'insieme di ripiegamento dell'RNA. In quanto tali, le reattività grezze sono molto adatte e informative per confrontare i cambiamenti di struttura tra diverse condizioni. È importante sottolineare che le strutture a più bassa energia libera calcolate utilizzando vincoli di sondaggio chimico con previsione computazionale dovrebbero essere utilizzate solo come ipotesi di partenza verso un modello di struttura completo.
Divulgazioni
Gli autori non hanno conflitti di interesse da dichiarare.
Riconoscimenti
Nessuno
Materiali
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
Riferimenti
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati