
Method Article
Probing RNA Structure with Dimethyl Sulfate Mutational Profiling with Sequencing In Vitro and in Cells
In This Article
Summary
The protocol provides instruction for modifying RNA with dimethyl sulfate for mutational profiling experiments. It includes in vitro and in vivo probing with two alternative library preparation methods.
Abstract
The role of RNA structure in virtually any biological process has become increasingly evident, especially in the past decade. However, classical approaches to solving RNA structure, such as RNA crystallography or cryo-EM, have failed to keep up with the rapidly evolving field and the need for high-throughput solutions. Mutational profiling with sequencing using dimethyl sulfate (DMS) MaPseq is a sequencing-based approach to infer the RNA structure from a base's reactivity with DMS. DMS methylates the N1 nitrogen in adenosines and the N3 in cytosines at their Watson-Crick face when the base is unpaired. Reverse-transcribing the modified RNA with the thermostable group II intron reverse transcriptase (TGIRT-III) leads to the methylated bases being incorporated as mutations into the cDNA. When sequencing the resulting cDNA and mapping it back to a reference transcript, the relative mutation rates for each base are indicative of the base's "status" as paired or unpaired. Even though DMS reactivities have a high signal-to-noise ratio both in vitro and in cells, this method is sensitive to bias in the handling procedures. To reduce this bias, this paper provides a protocol for RNA treatment with DMS in cells and with in vitro transcribed RNA.
Introduction
Since the discovery that RNA has both structural1,2 and catalytic3 properties, the importance of RNA and its regulatory function in a plethora of biological processes have been gradually uncovered. Indeed, the effect of RNA structure on gene regulation has gained increasing attention4. Like proteins, RNA has primary, secondary, and tertiary structures, referring to the sequence of nucleotides, the 2D mapping of base-pairing interactions, and the 3D folding of these base-paired structures, respectively. While determining the tertiary structure is key to understanding the exact mechanisms behind RNA-dependent processes, the secondary structure is also highly informative regarding RNA function and is the basis for further 3D folding5.
However, determining the RNA structure has been intrinsically challenging with conventional approaches. While for proteins, crystallography, nuclear magnetic resonance (NMR), and cryogenic electron microscopy (cryo-EM) have made it possible to determine the diversity of structural motifs, allowing for structure prediction from the sequence alone6, these approaches are not widely applicable to RNAs. Indeed, RNAs are flexible molecules with building blocks (nucleotides) that have much more conformational and rotational freedom in comparison to their amino acid counterparts. Furthermore, the interactions through base-pairing are more dynamic and versatile than those of amino acid residues. As a result, classical approaches have been successful only for relatively small RNAs with well-defined, highly compact structures7.
Another approach to determine the RNA structure is through chemical probing combined with next-generation sequencing (NGS). This strategy generates information about the binding status of each base in an RNA sequence (i.e., its secondary structure). In brief, the bases in an RNA molecule that are not engaging in base-pairing are differentially modified by small chemical compounds. Reverse-transcribing these RNAs with specialized reverse transcriptases (RTs) incorporates the modifications into complementary deoxyribonucleic acid (cDNA) as mutations. These cDNA molecules are then amplified by the polymerase chain reaction (PCR) and sequenced. To obtain information about their "status" as bound or unbound, the mutation frequencies at each base in an RNA of interest are calculated and entered into structure prediction software as constraints8. Based on nearest neighbor rules9 and minimum free energy calculations10, this software generates structure models that best fit the obtained experimental data11,12.
DMS-MaPseq uses DMS, which methylates the N1 nitrogen in adenosines and N3 nitrogen in cytosines at their Watson-Crick face in a highly specific manner13. Using thermostable group II intron reverse transcriptase (TGIRT-III) in reverse transcription creates mutational profiles with unprecedented signal-to-noise ratios, even allowing for the deconvolution of overlapping profiles generated by two or more alternative conformations14,15. Furthermore, DMS can penetrate cell membranes and whole tissues, making probing within physiological contexts possible. However, the generation of good-quality data is challenging, as variations in the handling procedure can impact the results. Therefore, we provide a detailed protocol for both in vitro and in-cell DMS-MaPseq to reduce bias and guide newcomers to the method through the difficulties they may encounter. Especially in light of the recent SARS-CoV2 pandemic, high-quality data on RNA viruses is an important tool for studying gene expression and finding possible therapeutics.
Protocol
NOTE: See the Table of Materials for details related to all the materials, software, reagents, instruments, and cells used in this protocol.
1. Gene-specific in vitro DMS-MaP
- RNA in vitro transcription
- Obtain the sequence of the RNA of interest as double-stranded (ds)DNA (e.g., as DNA fragments, plasmids, or PCR from pre-existing/genomic DNA). If the DNA sequence contains a polymerase promotor, jump to step 3.
- Perform overlap PCR to attach an RNA polymerase promotor upstream of the desired DNA fragment (forward primer for T7 polymerase: 5' TAATACGACTCACTATAGG + first bases of target sequence 3').
- In vitro transcribe the DNA fragment into RNA. Always keep the RNA on ice.
- Digest the DNA using a DNase.
- Isolate the RNA using a column-based approach (step 2.4) or by ethanol precipitation (step 2.5). Elute in an appropriate volume, expecting a yield of ~50 µg.
- Ensure the RNA integrity by running it on an agarose gel; denature the RNA for 2-3 min at 70 °C before running.
NOTE: The buffer and agarose can contain RNases that degrade RNA and might contaminate the RNA sample. Precast agarose gels have previously been used in this lab; the results (especially with RNA) have been ambiguous at times. The best results were obtained with agarose or PAGE gels. - Directly use of store the RNA at −80 °C for several months unless degradation is visible after thawing.
- In vitro DMS modification (at 105 mM DMS)
- Prepare a sufficient amount of refolding buffer (0.4 M sodium cacodylate, pH 7.2, containing 6 mM MgCl2).
NOTE: For each reaction (final volume of 100 µL), add 89 µL of refolding buffer. - For each reaction, transfer 89 µL of refolding buffer to a designated 1.5 mL tube, and prewarm at 37 °C in a thermoshaker placed underneath a chemical hood.
NOTE: DMS is highly toxic and must always be kept underneath a chemical hood until quenched by a reducing agent. - Elute 1-10 pmol of RNA in 10 µL of nuclease-free water (NF H2O); transfer to a PCR tube.
- Incubate in a thermocycler at 95 °C for 1min to denature the RNA.
- Place on an ice block immediately to avoid misfolding.
- Add the RNA sample to the designated tube with refolding buffer at 37 °C, mix well, and incubate for 10-20 min to refold the RNA.
NOTE: Most RNAs will fold in the order of milliseconds to seconds, although exceptions exist16. - Add 1 µL of 100% (10.5 M) DMS to the RNA sample, and incubate for 5 min while shaking at 800-1,400 rotations per minute (rpm).
NOTE: Shaking (or other means of mixing) at this step is crucial as DMS is hydrophobic and may not fully dissolve in the refolding buffer. Deviations in reaction times might affect the reproducibility of the DMS reactivities. To minimize pipetting error, DMS can be dissolved in 100% ethanol prior to adding it to the sample if a final concentration of 1% (105 mM) DMS is maintained. For an untreated control, DMS can be substituted by dimethyl sulfoxide (DMSO) or water. - After 5 min of reaction time, quench with 60 µL of 100% β-mercaptoethanol (BME), mix well, and immediately place the RNA on ice.
NOTE: The RNA can be safely removed from the hood after quenching the reaction with BME to clean it up. However, direct exposure of BME to the surroundings should still be avoided due to its strong smell and irritating properties. - Clean up the RNA by sodium acetate-ethanol precipitation (see step 2.5) or a column-based approach (see step 2.6), and elute in 10 µL of water.
- Quantify the RNA using a spectrophotometer.
- Directly use of store the modified RNA at −80 °C.
NOTE: Long-term storage should be avoided, as RNA is less stable after DMS treatment.
- Prepare a sufficient amount of refolding buffer (0.4 M sodium cacodylate, pH 7.2, containing 6 mM MgCl2).
- Gene-specific RT-PCR of modified RNA
NOTE: See Figure 1 for the RT-PCR setup of the DMS-treated fragments.- Elute 100 ng of modified RNA in 10 µL of nuclease-free (NF) H2O. Transfer to a PCR tube.
- To the tube, add 4 µL of 5x first strand buffer (FSB), 1 µL of dNTP mix (10 mM each), 1 µL of 0.1 M dithiothreitol (DTT) (avoid freeze-thaw cycles), 1 µL of RNase inhibitor, 1 µL of 10 µM reverse primer (single primer or a pool of primers), and 1 µL of TGIRT III.
NOTE: For a pool of primers, do not add 1 µL of 10 µM of each primer directly to the RT; instead, mix the primers first, and add 1 µL from the mix (at 10 µM total primer concentration). - Incubate at 57 °C for 30 min to 1.5 h (typically, 30 min is sufficient to make a 500 nt product) in a thermocycler.
- Add 1 µL of 4 M NaOH, mix by pipetting, and incubate at 95 °C for 3 min to degrade the RNA.
NOTE: This step is crucial as it releases TGIRT from the cDNA by degrading the RNA. If skipped, the downstream PCR might be affected. - Clean up using a column-based approach (see step 2.6) that sufficiently removes the primers, and elute in 10 µL of NF H2O.
- PCR-amplify the cDNA by using 1 µL of the reverse transcription product per 25 µL of the reaction with a PCR kit designed to balance yield and fidelity.
NOTE: The primers should have a melting temperature of ~60 °C. - Run 2 µL of the PCR product on an agarose gel or a precast agarose gel to verify the PCR success.
- Ideally, only one band should show after the PCR. If so, clean up the reaction using a column-based approach. If alternative bands are present, use the remaining PCR reaction to excise the correct band from the gel. Elute in a sufficiently small volume (e.g., 10 µL).
- Quantify the extracted fragments using a spectrophotometer.
- Index the dsDNA fragments for sequencing using an approach suited to the desired sequencing platform.

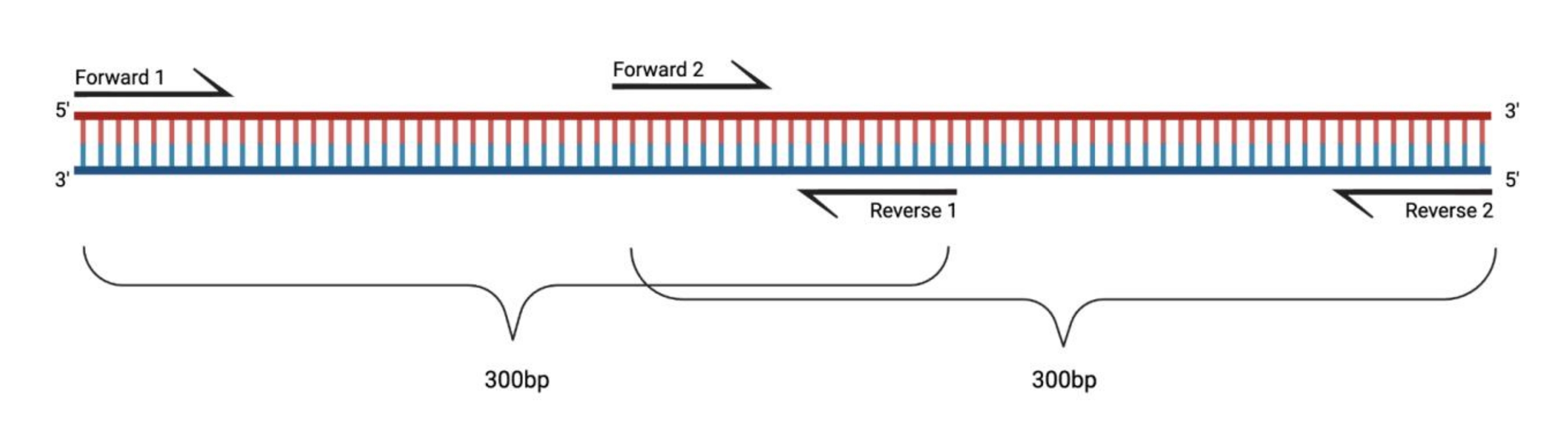
Figure 1: Experimental setup for the RT-PCR of large DMS-treated fragments. When performing reverse transcription on a modified RNA, the modifications on the sequence that the primer anneals to will not be recorded. Thus, when the fragments exceed 400-500 bp in length, fragments overlapping in the primer regions need to be designed, as exemplified here. The length of the fragments depends on the sequencing needs. When using paired-end 150 cycle sequencing, the fragments should not exceed 300 bp. Abbreviations: RT-PCR = reverse transcription polymerase chain reaction; DMS = dimethyl sulfate. Please click here to view a larger version of this figure.
{kind=link}
2. Whole-genome DMS-MaP using virus-infected cells
NOTE: In cells, DMS treatment can also be combined with the gene-specific amplification approach described above. The whole-genome library requires enormous sequencing depth to achieve full coverage on a single gene. However, if viral RNAs make up a significant fraction of the ribodepleted RNA after extraction, whole-genome sequencing would be appropriate. Furthermore, other enrichment methods can be combined with the whole-genome library generation method.
NOTE: Uninfected cells were used for demonstration purposes in the video protocol.
- DMS treatment
- Grow cells infected with the virus until the desired stage of infection.
- Transfer the cell container into a dedicated fume hood that is appropriate for handling both viruses at the required biosafety level and the chemical fumes generated by agents such as DMS.
- Add a 2.5% volume of DMS to the culture medium, and seal the container (typically a 10 cm plate) with parafilm.
NOTE: It is easy to under-modify and over-modify with DMS. When adding DMS directly to cells, it is very important to mix well. Alternatively, prewarm the new medium in a 50 mL conical tube at 37 °C, and add the DMS directly shaking vigorously. Decant the spent medium on the cells, and slowly pipette in the DMS-containing medium. - Transfer to a 37 °C incubator for 5 min.
NOTE: Depending on the amount of time it takes to handle the DMS outside the incubator, it is possible that 5 min will lead to over-modification. Keep the time from adding the DMS to incubating to ≤1 min. If performing the experiment for the first time, it is recommended to do a DMS titration and vary the incubation time (between 3 min and 10 min) to find the optimal modification rate and ensure that the results are robust across a window of concentrations. - Carefully pipette out the DMS-containing medium (into appropriate chemical waste) and gently add 10 mL of stop buffer (PBS with 30% BME [e.g., 3 mL of BME and 7 mL of PBS]).
NOTE: The addition of DMS and BME can lift the cells from the plate if the cells are not strongly adherent. If the cells are lifting, they can be treated as suspension cells-instead of removing the DMS-containing medium, add the stop buffer directly, and scrape out the cells with DMS and BME into a 50 mL conical tube. Pellet the cells by centrifuging for 3 min at 3,000 × g; make sure to get rid of any residual DMS, which can pellet under the cells in large droplets. An extra wash step in 30% BME is recommended if the DMS medium cannot be removed initially. - Scrape the cells, and transfer them to a 15 mL conical tube.
- Pellet by centrifugation at 3,000 × g for 3 min.
- Remove the supernatant and wash 2x with 10 mL of PBS.
- Carefully remove as much residual PBS as possible.
- Dissolve the pellet in an appropriate amount of the RNA isolation reagent (e.g., 3 mL for a T75 culture flask, 1 mL for a 10 cm plate).
NOTE: Insufficient amounts of the reagent might impact the RNA yield.
- RNA extraction and ribosomal RNA (rRNA) depletion
- To 1 mL of homogenized cells in the RNA isolation reagent, add 200 µL of chloroform, vortex for 15-20 s until bright pink, and then incubate for up to 3 min until phase separation is visible.
NOTE: The pink lipid phase should settle at the bottom. If this is not the case, the vortexing time was likely insufficient. - Spin at maximum speed (~ 20,000 × g) for 15 min at 4 °C.
- Transfer the upper aqueous phase to a new tube.
- Clean up the RNA by sodium acetate-ethanol precipitation (see step 2.5) or a column-based approach (see step 2.6), and elute in a sufficient volume of NF H2O.
- Check the RNA integrity on an agarose gel. Look for two bands corresponding to the two ribosomal subunits.
- Deplete the rRNAs using the preferred approach, and elute in an adequate volume (typically 20-50 µL) of NF H2O.
NOTE: For downstream applications, ~500 ng of total RNA is suggested in a volume of 8 µL. Non-ribosomal RNAs typically make up only 5%-10% of the total RNA. - Quantify using a spectrophotometer.
- To 1 mL of homogenized cells in the RNA isolation reagent, add 200 µL of chloroform, vortex for 15-20 s until bright pink, and then incubate for up to 3 min until phase separation is visible.
- Library generation
- Use gene-specific RT-PCR or other approaches to generate libraries15. If using random hexamers for priming, add an incubation step at a low Tm (37-42 °C) to allow for hexamer annealing.
NOTE: Standard library generation kits can also be used by replacing the RT enzyme with TGIRT and changing the RT temperature to 57 °C.
- Use gene-specific RT-PCR or other approaches to generate libraries15. If using random hexamers for priming, add an incubation step at a low Tm (37-42 °C) to allow for hexamer annealing.
- Column-based RNA cleanup using the RNA Clean & Concentrator columns
NOTE: All steps should be conducted at room temperature.- Add NF H2O to the sample tube to bring it to a volume of 50 µL.
- Add 100 µL of binding buffer and 150 µL of 100% ethanol to the sample.
- Mix and transfer to a spin column.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- Add 400 µL of RNA prep buffer.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- Add 700 µL of RNA wash buffer.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- Add 400 µL of RNA wash buffer.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- (Optional) Transfer the column to a new collection tube, and spin at 10,000-16,000 × g for 2 min.
- Transfer the column to a clean RNAse-free tube and add an appropriate amount of NF H2O.
- Spin at 10,000-16,000 × g for 1 min.
- Acid phenol-chloroform RNA extraction.
- Add an equal volume of acid phenol:chloroform:isoamyl alcohol.
- Vortex thoroughly, and centrifuge at 14,000 × g for 5 min.
- If there is no phase separation, add 20 µL of 2 M NaCl, and repeat the centrifugation.
- Transfer the aqueous phase into a new tube.
- Add 500 µL of isopropanol and 2 µL of co-precipitant.
- Mix and incubate at RT for 3 min; then, incubate at −80 °C overnight.
- Pellet the RNA by centrifugation at maximum speed (~ 20,000 × g) for 30 min at 4 °C.
- Wash the pellet with 200 µL of ice-cold 70% ethanol.
- Spin at maximum speed (~ 20,000 × g) for 5 min; discard the flowthrough.
- Resuspend the pellet in the appropriate amount of NF H2O.
- Column-based cDNA cleanup using the Oligo Clean and Concentrator columns
NOTE: All the steps should be conducted at room temperature.- Add NF H2O to the sample tube to bring it to a volume of 50 µL.
- Add 100 µL of binding buffer and 400 µL of 100% ethanol.
- Mix and transfer to a spin column.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- Add 750 µL of DNA wash buffer.
- Spin at 10,000-16,000 × g for 30 s; discard the flowthrough.
- (Optional) Transfer the column to a new collection tube and spin at 10,000-16,000 × g for 2 min.
- Transfer the column to a clean RNAse-free tube, and add an appropriate amount of NF H2O.
- Spin at 10,000-16,000 × g for 1 min.
3. Analysis of the sequencing data
NOTE: To create RNA secondary structure models from the DMS-MaP sequencing data, the resulting .fastq files must be processed by several different steps. These steps can be automatically performed using the
- Trim the adaptor sequences with TrimGalore or Cutadapt.
- Map the reads to the reference sequences (.fasta format) using Bowtie2.
- Count the reads with specialized RNA structure software (e.g., DREEM14, RNA-Framework17, or similar), and create reactivity profiles.
- (Optional) Cluster the reads to find alternative RNA conformations using DREEM14, DRACO17, DANCE-MaP18, or similar.
- Predict the minimum free energy structure based on the reactivity profiles using RNAStructure12, ViennaRNA, or similar.
- Visualize the RNA11 structure using VARNA (https://varna.lri.fr/) or similar.
NOTE: For practicality, software such as DREEM (www.rnadreem.org) and RNA-Framework19 vastly incorporate steps 1–-5 in their pipelines, which streamlines the analysis process. However, any structure prediction should be handled with care (e.g., by verifying the structure's agreement with the data20.
Results
Gene-specific in vitro DMS-MaP
To study the 5'UTR of SARS2, the virus' first 300 bp were ordered as a gBlock sequence, alongside three primers. Those included two primers to propagate the fragment ("FW" & "RV") via PCR, as well as one to attach the T7 promotor ("FW-T7"). These sequences can be seen in Table 1.
| Name | Sequence (5’->3’) |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
Table 1: Primer sequence for DMS-MaP RT-PCR of SARS-CoV2 5'UTR. Here, FW-T7 and RV are needed to generate a DNA template for in vitro transcription, the RV is used in the reverse transcription and the FW-RV primer-pair is used in the subsequent PCR amplification of the cDNA. The primers anneal to the very beginning of the SARS-CoV2 genome (FW) and the sequence right downstream of the region of interest. Abbreviations: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; RT-PCR = reverse transcription polymerase chain reaction; SARS-CoV2 = severe acute respiratory syndrome-coronavirus 2; UTR = untranslated region; RV = reverse primer; FW = forward primer.
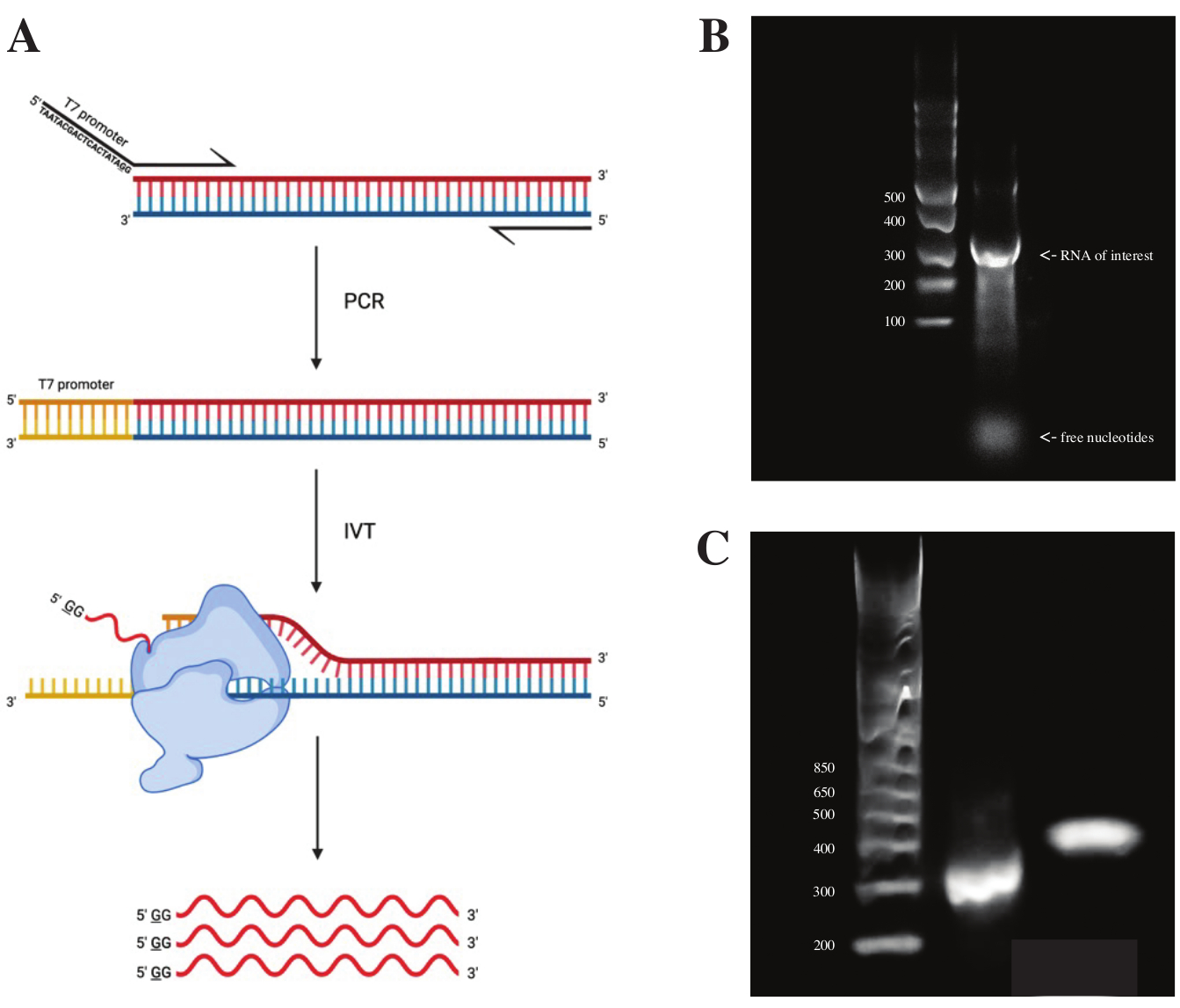
To generate RNA from the gBlock fragment, the sequence of the T7 polymerase promotor was attached using overlap PCR using the PCR premix according to the scheme seen in Figure 2A. From the elongated fragment, RNA was generated using the T7 Transcription Kit. The DNA template was subsequently digested using the DNase and RNA isolated using RNA Clean & Concentrator columns.
Quality control of the in vitro transcription was done by running the RNA product on a 1% agarose gel alongside an ssRNA ladder. As there was only one band visible, in vitro DMS probing and RT-PCR were performed (see Figure 2B).
To verify the success of the PCR reaction, the sample was run on a 2% agarose gel using a dsDNA ladder. After indexing, the band should run ~150 bp higher on the same gel, accounting for the size of the indexing primers.

Figure 2: In vitro transcription of the DNA template. (A)To in vitro transcribe a DNA template that does not yet have an intrinsic RNA polymerase promotor, the template must be attached by overlap PCR first. This is done by using a forward primer, which includes the sequence TAATACGACTCACTATAGG (in the case of the T7 RNA polymerase) upstream of the first bases overlapping with the desired fragment. The underlined base here symbolizes the transcription start site of the polymerase. Once the promoter has attached to the dsDNA fragment, it can be transcribed by the T7 polymerase. Importantly, the polymerase uses the strand opposed to the mentioned promoter sequence as the template (blue), effectively creating RNA identical to the sequence immediately downstream of the indicated promoter sequence (red). (B) A 1% agarose gel with an ssRNA Ladder (lane 1) and the in vitro transcribed RNA product at 300 nt (lane 2). (C) A 2% agarose gel with GeneRuler 1 kb plus Ladder (lane 1), the PCR product after RT-PCR running at 300 bp (lane 2), and the indexed fragment after library preparation running at 470 bp (lane 3). Abbreviations: RT-PCR = reverse transcription polymerase chain reaction; DMS = dimethyl sulfate; nt = nucleotides; dsDNA = double-stranded DNA; ssRNA = single-stranded RNA. Please click here to view a larger version of this figure.
{kind=link}
Whole-genome in vivo DMS-MaP using virus-infected cells
Prior to the DMS treatment, the HCT-8 cells were infected with OC43. When a cytopathic effect (CPE) was observed 4 days post infection (dpi) (as seen in Figure 3A), these cells were treated, and the RNA was extracted and ribodepleted. When running the total RNA on an agarose gel, two bright bands were visible, accounting for the 40S and 60S subunits of the ribosome, which make up approximately 95% of total RNA mass (see Figure 3B). When RNA extraction was unsuccessful or was degraded (e.g., by multiple freeze-thaw cycles), the RNA degradation products were visible on the bottom of the gel (see Figure 3C, second lane). Furthermore, after rRNA depletion, the two bright bands disappeared, leaving a smear in the lane (see Figure 3C, third lane). Finally, after library preparation, the samples had varying size distributions and were shown as a smear on the final PAGE gel. The band was excised between 200 nucleotides (nt) and 500 nt, in agreement with the 150 x 150 paired-end sequencing run planned to analyze these libraries. Most importantly, the adaptor dimers running at ~150 nt were separated out (see Figure 3D).

Figure 3: Checkpoints of in vivo DMS-MaP with virus-infected cells. (A) Light microscopy image of virus-infected HCT-8 cells, 4 days dpi. To obtain the highest possible yield of viral RNA from the total RNA while minimizing the adverse effects due to cell death, DMS should be added when CPE starts or even before that, as seen in the image. (B) A 1% agarose gel with six samples of 1 µg of total RNA. In each lane, two bright bands, accounting for the 40S and 60S subunits, are visible, as ribosomal RNA makes up ~95% of total RNA. Note: In-cell DMS treatment causes some RNA fragmentation and smearing, but the two rRNA bands should still be visible. Mild fragmentation post modification is tolerated because the information containing the methylation mark is generated and reports on the RNA structure during the DMS incubation while the cells are still alive. (C) A 1% Agarose gel of GeneRuler 1 kb plus ladder DNA marker (lane 1) total RNA previously stored at −80 °C for 6 months (lane 2) and ribodepleted RNA (lane 3). When storing RNA for a long time with several freeze-thaw cycles, the RNA starts degrading and possibly should not be used for probing experiments. Furthermore, after ribodepleting the total RNA, the two bright bands, accounting for the 40S and 60S subunits of the ribosome, fade, and a smear of the residual RNAs starts to show. (D) A PAGE gel of GeneRuler 1 kb plus ladder DNA marker (lane 1) and a library sample of whole-genome prepared RNA. The gel should be excised based on the sequencing needs. For a paired-end sequencing run spanning 150 cycles from both sides, the gel should be excised between 300 bp and 500 bp. Adaptor dimers (running at 170 bp) should be separated out. Abbreviations: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; dpi = days post infection; CPE = cytopathic effect. Please click here to view a larger version of this figure.
{kind=link}
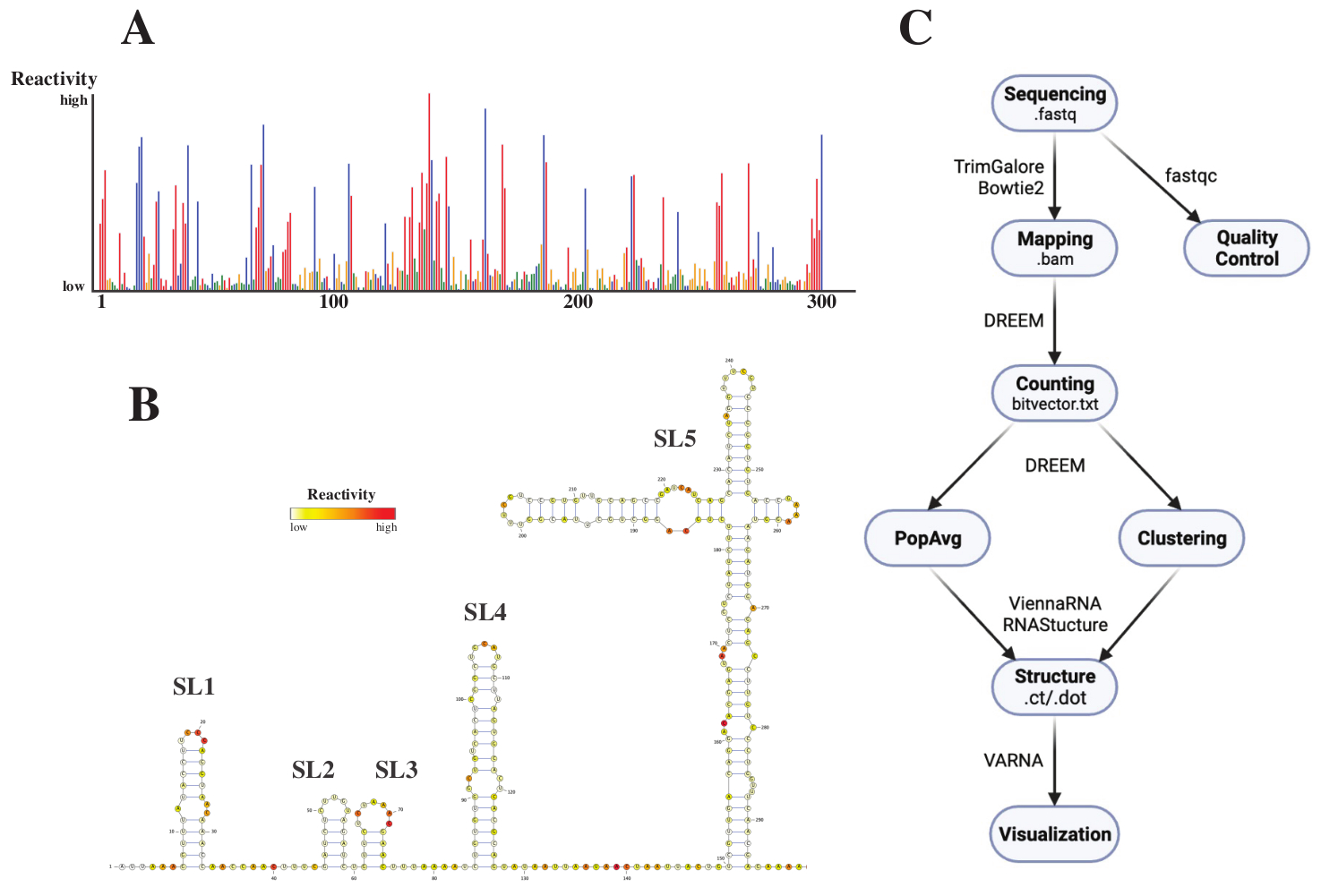
After sequencing, the .fastq files were analyzed by submitting a job to the DREEM webserver (http://rnadreem.org/), together with a .fasta reference file. The output generated by the server includes quality control files generated by fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), as well as other output files containing the population average mutation frequencies. Apart from the diagram showing the mutation frequencies with an interactive .html (see Figure 4A) format and a .csv file with the raw reactivites per base and a struct_constraint.txt file, readable by several RNA structure prediction software, this also includes a bitvector.txt file reporting on the by-read mutations. From these, the population average structures were calculated by submitting the .fasta and struct_constraint.txt files to the RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). This uses the ViennaRNA software to generate structure predictions based on the minimum free energy, which can be viewed online or downloaded in ct or Vienna format. To generate RNA structure models, these downloadable files were submitted to VARNA (https://varna.lri.fr/, see Figure 4B). Lastly, bitvector.txt files can be used by the stable version of DREEM (https://codeocean.com/capsule/6175523/tree/v1) to search for alternative RNA conformations. To obtain good structure models using DREEM, a coverage of 10,000 reads per base should be achieved; for clustering, up to 100,000 reads per base might be required. An overview of the whole workflow can be found in Figure 4C.

Figure 4: Exemplary data obtained from chemical probing experiments of the SARS-CoV2 5'UTR. (A) Reactivity profile of the first 300 bases of the SARS-CoV2 genome colored by base (A: red, C: blue, U: green, G: yellow). The raw reactivities are calculated as the absolute mutation frequency divided by the coverage. Bases with open conformation have high reactivity values; bases engaging in base-pairing have low reactivity values. U and G are not modified by DMS and have low reactivity values, originating from polymerase infidelity. The predictions were made with the DREEM webserver. (B) Structure model of the SARS-CoV2 5'UTR predicted from reactivity values made with VARNA. Bases with high reactivity values are colored in red; bases with low reactivity values are colored in white. (C) Workflow of the DMS-MaP analysis starting with the .fastq files obtained from sequencing. These can be quality-controlled using fastqc; the adaptor sequences are trimmed using TrimGalore and then mapped back to a reference sequence using Bowtie2. From the obtained .bam files, DREEM counts the mutations in each read, creating a mutation map or .bitvector.txt file. These report the mutations of each read in a position-dependent way, based on which the population average reactivity profiles can be created. Alternatively, bitvectors can be clustered using DREEM to search for alternative RNA conformations. Lastly, the obtained structure models are visualized using software (e.g., VARNA). Abbreviations: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; SARS-CoV2 = severe acute respiratory syndrome-coronavirus 2. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The protocol here describes how to probe RNA in vitro and in cells using DMS mutational profiling experiments. Furthermore, it gives instructions on how to prepare libraries for Illumina sequencing to generate gene-specific data and analyze the obtained .fastq files. Additionally, genome-wide library approaches can be used. However, gene-specific RT-PCR produces the highest quality and most robust data. Therefore, if comparing between samples, it is important to ensure that they are prepared with identical sequencing strategies, as the library generation causes some bias. The reproducibility should always be measured by using replicates.
Several precautions
RNA is an unstable molecule that is sensitive to degradation both through elevated temperatures and by RNases. Therefore, special measures — the use of personal protective equipment (PPE), RNAse-free material, and RNAse inhibitors — is recommended. Most importantly, RNA should be kept on ice whenever possible. This especially applies to methylated RNA, which is even more sensitive to high temperatures.
It is important to confirm that the RNA structure of interest is not sensitive to the DMS concentration and buffer conditions. Buffers such as 100 mM Tris, 100 mM MOPS, and 100 mM HEPES at pH 7-7.5 give a high signal but may not be sufficient to maintain the pH during the reaction21. As DMS hydrolyzes in water, which decreases the pH, a strong buffer is critical to maintain a neutral pH during the modification reaction. The addition of bicine has been shown to help maintain the pH as slightly basic21 but results in low DMS modification on Gs and Us, which could be informative but should be analyzed separately due to the production of a much lower signal than As and Cs and is not discussed further in this protocol.
In gene-specific RT-PCR, the modified RNA is reverse-transcribed into the DNA and amplified in fragments by PCR. While the size of the RNA can theoretically be unlimited, these PCR fragments should not exceed a length of 400-500 base pairs (bp) to prevent bias during the reverse transcription reaction. Ideally, the fragments should be within the scope of the sequencing run (i.e., if sequencing is conducted using a 150 x 150 cycle paired-end sequencing program, a single fragment should not exceed 300 bp). When using sequencing programs with fewer cycles, the PCR products can be fragmented using a dsDNase. Furthermore, as sequences within the primer sequences do not hold any structural information, the fragments must overlap when the probed RNA comprises >1 fragment. RT reactions can contain multiple RT primers for different fragments (up to 10 different RT primers). Depending on the sequences, pooling the RT primers can make the reverse transcription less efficient but typically works well. Each PCR reaction should be conducted separately.
When probing RNA with DMS, the experimental conditions play an additional role, as many RNAs are thermodynamically unstable and change their conformation based on environmental factors such as temperature. To avoid irregularities, the experimental conditions should be kept as constant as possible, also with respect to reaction times. The buffer conditions seem to be exchangeable to a certain degree17,20,22,23 when the basic conditions are maintained — the buffering capacity and presence of monovalent (Na) and divalent ions (Mg) — to ensure proper folding of RNA24.
With respect to the library preparation of modified RNAs, several aspects must be taken into consideration. First, as mentioned before, modified RNAs are less stable than their unmodified counterparts, meaning they might require the optimization of the fragmentation times for optimal fragment size distribution. Furthermore, certain RNA library preparation kits, as well as many other RNAseq approaches, use random primers in the reverse transcription kit. This might lead to lower coverage of the reference, especially in the 3' of a gene, and, ultimately, to insufficient coverage depth. If the coverage of a certain region is too low, it might be necessary to remove those bases from the structure prediction. Apart from RT-PCR and whole-genome RNAseq kits, other library preparation approaches can be used. Protocols that include the ligation of 3' and/or 5' adaptors to the RNA are advantageous when using small fragments of RNA or when the loss of probing information in the primer regions must be avoided.
Lastly, the analysis of the chemical probing experiments must always be interpreted carefully. Currently, there is no software that predicts the RNA structure of any RNA from the sequence alone with high accuracy. Although chemical probing constraints greatly improve the accuracy, generating good models for long RNAs (>500 nt) is still challenging. These models should be further tested by other approaches and/or mutagenesis. RNA prediction software optimizes for the maximum number of base pairs, thus significantly penalizing open conformations, which may not accurately represent RNA folding5. Thus, the obtained structure model should be tested by quantifying the prediction agreement with the underlying chemical probing data (e.g., by AUROC) and between replicates (e.g., by mFMI), as exemplified by Lan et al.20.
Ideally, several experiments in different systems to challenge the obtained structure model should be used to strengthen one's hypothesis. These can include the usage of in vitro and in-cell approaches, compensatory mutations, and different cell lines and species. Moreover, raw reactivities are often just as or even more informative than structure predictions, as they record the "ground truth" snapshot of the RNA folding ensemble. As such, raw reactivities are very suitable and informative for comparing structure changes between different conditions. Importantly, the lowest free energy structures calculated using chemical probing constraints with computational prediction should only be used as a starting hypothesis toward a complete structure model.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgements
None
Materials
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
References
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved