
Method Article
Untersuchung der RNA-Struktur mit Dimethylsulfat-Mutationsprofilierung mit Sequenzierung in vitro und in Zellen
In diesem Artikel
Zusammenfassung
Das Protokoll enthält Anweisungen zur Modifizierung von RNA mit Dimethylsulfat für Mutationsprofilierungsexperimente. Es umfasst in vitro und in vivo Sondierung mit zwei alternativen Bibliothekspräparationsmethoden.
Zusammenfassung
Die Rolle der RNA-Struktur in praktisch jedem biologischen Prozess ist vor allem in den letzten zehn Jahren immer deutlicher geworden. Klassische Ansätze zur Lösung der RNA-Struktur, wie RNA-Kristallographie oder Kryo-EM, haben jedoch nicht mit dem sich schnell entwickelnden Feld und dem Bedarf an Hochdurchsatzlösungen Schritt gehalten. Mutationsprofilierung mit Sequenzierung unter Verwendung von Dimethylsulfat (DMS) MaPseq ist ein sequenzierungsbasierter Ansatz, um die RNA-Struktur aus der Reaktivität einer Base mit DMS abzuleiten. DMS methyliert den N1-Stickstoff in Adenosinen und das N3 in Cytosinen an ihrer Watson-Crick-Seite, wenn die Base ungepaart ist. Die umgekehrte Transkription der modifizierten RNA mit der thermostabilen Intron-Reverse-Transkriptase der Gruppe II (TGIRT-III) führt dazu, dass die methylierten Basen als Mutationen in die cDNA eingebaut werden. Bei der Sequenzierung der resultierenden cDNA und der Zuordnung zu einem Referenztranskript sind die relativen Mutationsraten für jede Base ein Hinweis auf den "Status" der Base als gepaart oder ungepaart. Obwohl DMS-Reaktivitäten sowohl in vitro als auch in Zellen ein hohes Signal-Rausch-Verhältnis aufweisen, ist diese Methode empfindlich gegenüber Verzerrungen in den Handhabungsverfahren. Um diese Verzerrung zu reduzieren, bietet dieser Artikel ein Protokoll für die RNA-Behandlung mit DMS in Zellen und mit in vitro transkribierter RNA.
Einleitung
Seit der Entdeckung, dass RNA sowohl strukturelle1,2- als auch katalytische3-Eigenschaften besitzt, wurde die Bedeutung der RNA und ihre regulatorische Funktion in einer Vielzahl biologischer Prozesse nach und nach aufgedeckt. Tatsächlich hat der Einfluss der RNA-Struktur auf die Genregulation zunehmend Aufmerksamkeit erregt4. Wie Proteine hat RNA primäre, sekundäre und tertiäre Strukturen, die sich auf die Sequenz von Nukleotiden, die 2D-Kartierung von Basenpaarungsinteraktionen und die 3D-Faltung dieser Basenpaarstrukturen beziehen. Während die Bestimmung der Tertiärstruktur der Schlüssel zum Verständnis der genauen Mechanismen hinter RNA-abhängigen Prozessen ist, ist die Sekundärstruktur auch sehr informativ in Bezug auf die RNA-Funktion und ist die Grundlage für eine weitere 3D-Faltung5.
Die Bestimmung der RNA-Struktur war jedoch mit herkömmlichen Ansätzen eine intrinsische Herausforderung. Während für Proteine Kristallographie, Kernspinresonanz (NMR) und kryogene Elektronenmikroskopie (Kryo-EM) es ermöglicht haben, die Vielfalt der Strukturmotive zu bestimmen, was eine Strukturvorhersage allein aus der Sequenz ermöglicht6, sind diese Ansätze auf RNAs nicht allgemein anwendbar. Tatsächlich sind RNAs flexible Moleküle mit Bausteinen (Nukleotiden), die im Vergleich zu ihren Aminosäure-Gegenstücken viel mehr Konformations- und Rotationsfreiheit haben. Darüber hinaus sind die Wechselwirkungen durch Basenpaarung dynamischer und vielseitiger als die von Aminosäureresten. Infolgedessen waren klassische Ansätze nur für relativ kleine RNAs mit gut definierten, sehr kompakten Strukturen erfolgreich7.
Ein weiterer Ansatz zur Bestimmung der RNA-Struktur ist die chemische Sondierung in Kombination mit der Next-Generation-Sequenzierung (NGS). Diese Strategie generiert Informationen über den Bindungsstatus jeder Base in einer RNA-Sequenz (d.h. ihre Sekundärstruktur). Kurz gesagt, die Basen in einem RNA-Molekül, die nicht an Basenpaarung beteiligt sind, werden durch kleine chemische Verbindungen differentiell modifiziert. Die umgekehrte Transkription dieser RNAs mit spezialisierten reversen Transkriptasen (RTs) beinhaltet die Modifikationen in komplementäre Desoxyribonukleinsäure (cDNA) als Mutationen. Diese cDNA-Moleküle werden dann durch die Polymerase-Kettenreaktion (PCR) amplifiziert und sequenziert. Um Informationen über ihren "Status" als gebunden oder ungebunden zu erhalten, werden die Mutationshäufigkeiten an jeder Base in einer interessierenden RNA berechnet und als Randbedingungen8 in Strukturvorhersagesoftware eingegeben. Basierend auf den Regeln des nächsten Nachbarn9 und den Berechnungen der minimalen freien Energie 10 generiert diese Software Strukturmodelle, die am besten zu den erhaltenen experimentellen Daten11,12 passen.
DMS-MaPseq verwendet DMS, das den N1-Stickstoff in Adenosinen und den N3-Stickstoff in Cytosinen an ihrer Watson-Crick-Seite auf hochspezifische Weise methyliert13. Die Verwendung der thermostabilen Intron-Reverse-Transkriptase (TGIRT-III) der Gruppe II in umgekehrter Transkription erzeugt Mutationsprofile mit beispiellosen Signal-Rausch-Verhältnissen, die sogar die Dekonvolution von überlappenden Profilen ermöglichen, die von zwei oder mehr alternativen Konformationen erzeugt werden14,15. Darüber hinaus kann DMS Zellmembranen und ganze Gewebe durchdringen, wodurch Sondierungen in physiologischen Kontexten möglich werden. Die Generierung qualitativ hochwertiger Daten ist jedoch eine Herausforderung, da sich Unterschiede im Handhabungsverfahren auf die Ergebnisse auswirken können. Daher stellen wir ein detailliertes Protokoll für DMS-MaPseq in vitro und in der Zelle zur Verfügung, um Verzerrungen zu reduzieren und Neulinge durch die Schwierigkeiten, auf die sie stoßen können, an die Methode heranzuführen. Insbesondere vor dem Hintergrund der jüngsten SARS-CoV2-Pandemie sind qualitativ hochwertige Daten zu RNA-Viren ein wichtiges Instrument, um die Genexpression zu untersuchen und mögliche Therapeutika zu finden.
Protokoll
HINWEIS: In der Materialtabelle finden Sie Details zu allen Materialien, Software, Reagenzien, Instrumenten und Zellen, die in diesem Protokoll verwendet werden.
1. Genspezifisches in vitro DMS-MaP
- RNA in vitro Transkription
- Erhalten Sie die Sequenz der interessierenden RNA als doppelsträngige (ds) DNA (z. B. als DNA-Fragmente, Plasmide oder PCR aus bereits vorhandener/genomischer DNA). Wenn die DNA-Sequenz einen Polymerase-Promotor enthält, fahren Sie mit Schritt 3 fort.
- Führen Sie eine Überlappungs-PCR durch, um einen RNA-Polymerase-Promotor stromaufwärts des gewünschten DNA-Fragments anzuhängen (Vorwärtsprimer für T7-Polymerase: 5' TAATACGACTCACTATAGG + erste Basen der Zielsequenz 3').
- In vitro transkribieren Sie das DNA-Fragment in RNA. Halten Sie die RNA immer auf Eis.
- Verdauen Sie die DNA mit einer DNase.
- Isolieren Sie die RNA mit einem säulenbasierten Ansatz (Schritt 2.4) oder durch Ethanolfällung (Schritt 2.5). Eluieren Sie in einem angemessenen Volumen, wobei eine Ausbeute von ~50 μg erwartet wird.
- Stellen Sie die RNA-Integrität sicher, indem Sie sie auf einem Agarosegel laufen lassen; Denaturierung der RNA für 2-3 min bei 70 °C vor dem Laufen.
HINWEIS: Der Puffer und die Agarose können RNasen enthalten, die RNA abbauen und die RNA-Probe kontaminieren können. Vorgefertigte Agarosegele wurden zuvor in diesem Labor verwendet; die Ergebnisse (insbesondere bei RNA) waren manchmal mehrdeutig. Die besten Ergebnisse wurden mit Agarose- oder PAGE-Gelen erzielt. - Direkte Verwendung von Lagerung der RNA bei −80 °C für mehrere Monate, es sei denn, der Abbau ist nach dem Auftauen sichtbar.
- In vitro DMS-Modifikation (bei 105 mM DMS)
- Bereiten Sie eine ausreichende Menge Rückfaltungspuffer vor (0,4 M Natriumcacodylat, pH 7,2, enthält 6 mMMgCl2).
HINWEIS: Für jede Reaktion (Endvolumen von 100 μL) fügen Sie 89 μL Rückfaltpuffer hinzu. - Für jede Reaktion werden 89 μl Rückfaltpuffer in ein dafür vorgesehenes 1,5-ml-Röhrchen überführt und bei 37 °C in einem Thermoshaker unter einer chemischen Haube vorgewärmt.
HINWEIS: DMS ist hochgiftig und muss immer unter einer chemischen Haube aufbewahrt werden, bis es durch ein Reduktionsmittel abgeschreckt wird. - Elue 1-10 pmol RNA in 10 μL nukleasefreiem Wasser (NFH2O); Transfer in eine PCR-Röhre.
- In einem Thermocycler bei 95 °C für 1min inkubieren, um die RNA zu denaturieren.
- Sofort auf einen Eisblock legen, um ein Fehlfalten zu vermeiden.
- Die RNA-Probe wird mit Rückfaltungspuffer bei 37 °C in das dafür vorgesehene Röhrchen gegeben, gut gemischt und 10-20 min inkubiert, um die RNA zu rekonstruieren.
HINWEIS: Die meisten RNAs falten sich in der Größenordnung von Millisekunden bis Sekunden, obwohl es Ausnahmen gibt16. - 1 μL 100% (10,5 M) DMS in die RNA-Probe geben und 5 min lang inkubieren, während sie bei 800-1.400 Umdrehungen pro Minute (U/min) geschüttelt wird.
HINWEIS: Das Schütteln (oder andere Mischmethoden) bei diesem Schritt ist von entscheidender Bedeutung, da DMS hydrophob ist und sich möglicherweise nicht vollständig im Rückfaltungspuffer auflöst. Abweichungen in den Reaktionszeiten können die Reproduzierbarkeit der DMS-Reaktivitäten beeinträchtigen. Um Pipettierfehler zu minimieren, kann DMS vor der Zugabe in die Probe in 100% Ethanol gelöst werden, wenn eine Endkonzentration von 1% (105 mM) DMS beibehalten wird. Für eine unbehandelte Kontrolle kann DMS durch Dimethylsulfoxid (DMSO) oder Wasser substituiert werden. - Nach 5 Minuten Reaktionszeit mit 60 μL 100% β-Mercaptoethanol (BME) abschrecken, gut mischen und die RNA sofort auf Eis legen.
HINWEIS: Die RNA kann sicher aus der Haube entfernt werden, nachdem die Reaktion mit BME gelöscht wurde, um sie zu reinigen. Eine direkte Exposition von BME gegenüber der Umgebung sollte jedoch aufgrund seines starken Geruchs und seiner irritierenden Eigenschaften vermieden werden. - Die RNA wird durch Natriumacetat-Ethanol-Fällung (siehe Schritt 2.5) oder einen säulenbasierten Ansatz (siehe Schritt 2.6) gereinigt und in 10 μl Wasser eluiert.
- Quantifizieren Sie die RNA mit einem Spektralphotometer.
- Die modifizierte RNA wird direkt bei −80 °C gespeichert.
HINWEIS: Eine Langzeitlagerung sollte vermieden werden, da die RNA nach der DMS-Behandlung weniger stabil ist.
- Bereiten Sie eine ausreichende Menge Rückfaltungspuffer vor (0,4 M Natriumcacodylat, pH 7,2, enthält 6 mMMgCl2).
- Genspezifische RT-PCR modifizierter RNA
HINWEIS: Siehe Abbildung 1 für den RT-PCR-Aufbau der DMS-behandelten Fragmente.- Elue 100 ng modifizierte RNA in 10 μL nukleasefrei (NF)H2O. Transfer in ein PCR-Röhrchen.
- Fügen Sie dem Röhrchen 4 μL 5x Erststrangpuffer (FSB), 1 μL dNTP-Mischung (je 10 mM), 1 μL 0,1 M Dithiothreitol (DTT) (Gefrier-Tau-Zyklen vermeiden), 1 μL RNase-Inhibitor, 1 μL 10 μM Reverse Primer (Einzelprimer oder ein Pool von Primern) und 1 μL TGIRT III hinzu.
HINWEIS: Bei einem Pool von Primern nicht 1 μL von 10 μM jedes Primers direkt in die RT geben; Mischen Sie stattdessen zuerst die Primer und fügen Sie 1 μL aus der Mischung hinzu (bei 10 μM Gesamtprimerkonzentration). - Inkubieren bei 57 °C für 30 min bis 1,5 h (typischerweise reichen 30 min aus, um ein 500-nt-Produkt herzustellen) in einem Thermocycler.
- Fügen Sie 1 μL 4 M NaOH hinzu, mischen Sie durch Pipettieren und inkubieren Sie bei 95 °C für 3 min, um die RNA abzubauen.
HINWEIS: Dieser Schritt ist entscheidend, da TGIRT aus der cDNA freigesetzt wird, indem die RNA abgebaut wird. Wenn sie übersprungen wird, kann die Downstream-PCR betroffen sein. - Bereinigen Sie mit einem säulenbasierten Ansatz (siehe Schritt 2.6), der die Primer ausreichend entfernt, und eluieren Sie in 10 μL NF H2O.
- PCR-Amplifikation der cDNA unter Verwendung von 1 μL des reversen Transkriptionsprodukts pro 25 μL der Reaktion mit einem PCR-Kit, das entwickelt wurde, um Ausbeute und Wiedergabetreue auszugleichen.
HINWEIS: Die Grundierungen sollten eine Schmelztemperatur von ~60 °C haben. - Lassen Sie 2 μL des PCR-Produkts auf ein Agarosegel oder ein vorgefertigtes Agarosegel laufen, um den PCR-Erfolg zu überprüfen.
- Idealerweise sollte nach der PCR nur eine Band auftreten. Wenn ja, bereinigen Sie die Reaktion mit einem spaltenbasierten Ansatz. Wenn alternative Banden vorhanden sind, verwenden Sie die verbleibende PCR-Reaktion, um die richtige Bande aus dem Gel zu entfernen. Eluieren in einem ausreichend kleinen Volumen (z. B. 10 μL).
- Quantifizieren Sie die extrahierten Fragmente mit einem Spektralphotometer.
- Indexieren Sie die dsDNA-Fragmente für die Sequenzierung mit einem Ansatz, der für die gewünschte Sequenzierungsplattform geeignet ist.

Abbildung 1: Versuchsaufbau für die RT-PCR großer DMS-behandelter Fragmente. Bei der reversen Transkription einer modifizierten RNA werden die Modifikationen der Sequenz, zu der der Primer übergeht, nicht aufgezeichnet. Wenn also die Fragmente eine Länge von 400-500 bp überschreiten, müssen Fragmente, die sich in den Primerbereichen überlappen, entworfen werden, wie hier veranschaulicht. Die Länge der Fragmente hängt von den Sequenzierungsanforderungen ab. Bei Verwendung der Paired-End-150-Zyklus-Sequenzierung sollten die Fragmente 300 bp nicht überschreiten. Abkürzungen: RT-PCR = reverse transcription polymerase chain reaction; DMS = Dimethylsulfat. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
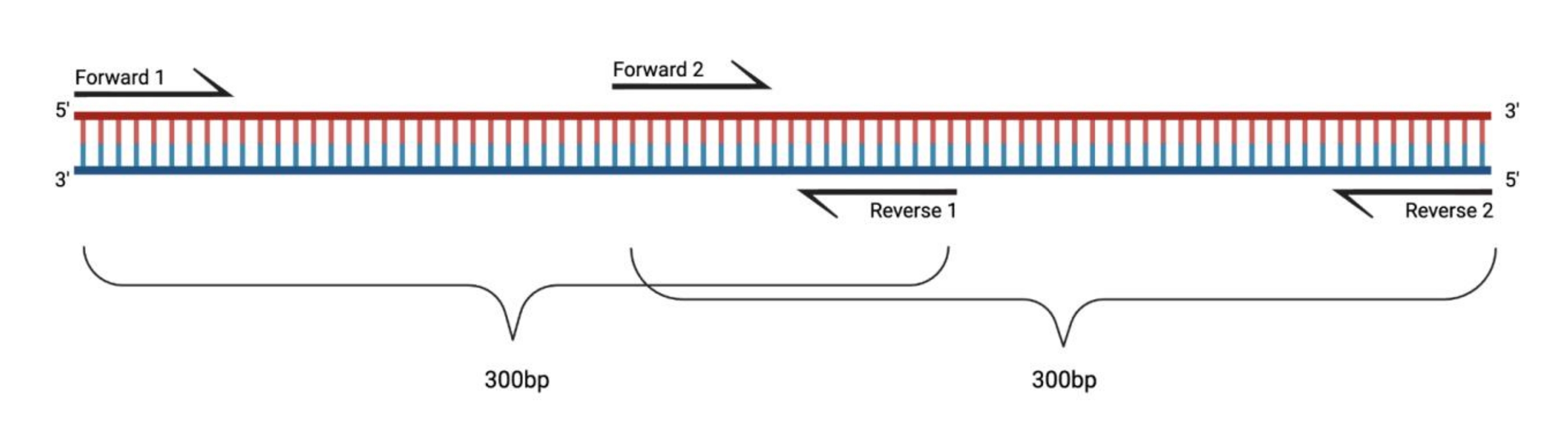{kind=link}
2. DMS-MaP des gesamten Genoms mit virusinfizierten Zellen
HINWEIS: In Zellen kann die DMS-Behandlung auch mit dem oben beschriebenen genspezifischen Amplifikationsansatz kombiniert werden. Die gesamte Genombibliothek erfordert eine enorme Sequenzierungstiefe, um eine vollständige Abdeckung eines einzelnen Gens zu erreichen. Wenn jedoch virale RNAs nach der Extraktion einen signifikanten Anteil der ribodepletierten RNA ausmachen, wäre eine Sequenzierung des gesamten Genoms angemessen. Darüber hinaus können andere Anreicherungsmethoden mit der Methode der Generierung der gesamten Genombibliothek kombiniert werden.
- DMS-Behandlung
- Züchten Sie Zellen, die mit dem Virus infiziert sind, bis zum gewünschten Stadium der Infektion.
- Überführen Sie den Zellbehälter in einen speziellen Abzug, der sowohl für den Umgang mit Viren auf dem erforderlichen Biosicherheitsniveau als auch für die von Wirkstoffen wie DMS erzeugten chemischen Dämpfe geeignet ist.
- Fügen Sie dem Kulturmedium ein Volumen von 2,5% DMS hinzu und verschließen Sie den Behälter (typischerweise eine 10-cm-Platte) mit Parafilm.
HINWEIS: Es ist leicht, mit DMS zu unter- und zu viel zu modifizieren. Wenn DMS direkt zu Zellen hinzugefügt wird, ist es sehr wichtig, gut zu mischen. Alternativ können Sie das neue Medium in einem 50 mL konischen Röhrchen bei 37 °C vorwärmen und das DMS direkt schüttelnd hinzufügen. Dekantieren Sie das verbrauchte Medium auf den Zellen und pipettieren Sie langsam in das DMS-haltige Medium. - Überführung in einen 37 °C Inkubator für 5 min.
HINWEIS: Abhängig von der Zeit, die benötigt wird, um das DMS außerhalb des Inkubators zu handhaben, ist es möglich, dass 5 Minuten zu einer Übermodifikation führen. Halten Sie die Zeit vom Hinzufügen des DMS bis zur Inkubation auf ≤1 min. Wenn Sie das Experiment zum ersten Mal durchführen, wird empfohlen, eine DMS-Titration durchzuführen und die Inkubationszeit (zwischen 3 min und 10 min) zu variieren, um die optimale Modifikationsrate zu finden und sicherzustellen, dass die Ergebnisse über ein Konzentrationsfenster robust sind. - Pipettieren Sie das DMS-haltige Medium vorsichtig aus (in geeignete chemische Abfälle) und fügen Sie vorsichtig 10 mL Stop-Puffer hinzu (PBS mit 30% BME [z. B. 3 ml BME und 7 ml PBS]).
HINWEIS: Die Zugabe von DMS und BME kann die Zellen von der Platte heben, wenn die Zellen nicht stark haften. Wenn sich die Zellen heben, können sie als Suspensionszellen behandelt werden - anstatt das DMS-haltige Medium zu entfernen, fügen Sie den Stopppuffer direkt hinzu und kratzen Sie die Zellen mit DMS und BME in ein 50-ml-konisches Röhrchen ab. Pelletieren Sie die Zellen durch Zentrifugieren für 3 min bei 3.000 × g; Stellen Sie sicher, dass Sie alle DMS-Reste entfernen, die in großen Tröpfchen unter den Zellen pelletieren können. Ein zusätzlicher Waschschritt in 30% BME wird empfohlen, wenn das DMS-Medium zunächst nicht entfernt werden kann. - Schaben Sie die Zellen ab und übertragen Sie sie in ein 15 ml konisches Röhrchen.
- Pellet durch Zentrifugation bei 3.000 × g für 3 min.
- Entfernen Sie den Überstand und waschen Sie ihn 2x mit 10 ml PBS.
- Entfernen Sie vorsichtig so viel Rest-PBS wie möglich.
- Das Pellet wird in einer geeigneten Menge des RNA-Isolierreagenz gelöst (z. B. 3 ml für einen T75-Kulturkolben, 1 ml für eine 10-cm-Platte).
HINWEIS: Unzureichende Mengen des Reagenzes können die RNA-Ausbeute beeinträchtigen.
- RNA-Extraktion und ribosomale RNA (rRNA)-Depletion
- Zu 1 ml homogenisierter Zellen im RNA-Isolierreagenz 200 μL Chloroform, Wirbel für 15-20 s bis hellrosa hinzufügen und dann bis zu 3 min inkubieren, bis eine Phasentrennung sichtbar ist.
HINWEIS: Die rosa Lipidphase sollte sich unten absetzen. Wenn dies nicht der Fall ist, war die Wirbelzeit wahrscheinlich unzureichend. - Schleudern Sie mit maximaler Geschwindigkeit (~ 20.000 × g) für 15 min bei 4 °C.
- Die obere wässrige Phase in ein neues Röhrchen überführen.
- Die RNA wird durch Natriumacetat-Ethanol-Fällung (siehe Schritt 2.5) oder einen säulenbasierten Ansatz (siehe Schritt 2.6) gereinigt und in einem ausreichenden Volumen von NFH2Oeluiert.
- Überprüfen Sie die RNA-Integrität auf einem Agarose-Gel. Suchen Sie nach zwei Banden, die den beiden ribosomalen Untereinheiten entsprechen.
- Die rRNAs werden mit dem bevorzugten Ansatz depletiert und in einem ausreichenden Volumen (typischerweise 20-50 μL) NFH2Oeluiert.
HINWEIS: Für nachgeschaltete Anwendungen werden ~500 ng Gesamt-RNA in einem Volumen von 8 μL empfohlen. Nicht-ribosomale RNAs machen typischerweise nur 5%-10% der gesamten RNA aus. - Quantifizieren Sie mit einem Spektralphotometer.
- Zu 1 ml homogenisierter Zellen im RNA-Isolierreagenz 200 μL Chloroform, Wirbel für 15-20 s bis hellrosa hinzufügen und dann bis zu 3 min inkubieren, bis eine Phasentrennung sichtbar ist.
- Bibliotheksgenerierung
- Verwenden Sie genspezifische RT-PCR oder andere Ansätze, um Bibliotheken zu generieren15. Wenn Sie zufällige Hexamer für das Priming verwenden, fügen Sie einen Inkubationsschritt bei einem niedrigen Tm (37-42 °C) hinzu, um das Hexamerglühen zu ermöglichen.
HINWEIS: Standard-Bibliotheksgenerierungskits können auch verwendet werden, indem das RT-Enzym durch TGIRT ersetzt und die RT-Temperatur auf 57 °C geändert wird.
- Verwenden Sie genspezifische RT-PCR oder andere Ansätze, um Bibliotheken zu generieren15. Wenn Sie zufällige Hexamer für das Priming verwenden, fügen Sie einen Inkubationsschritt bei einem niedrigen Tm (37-42 °C) hinzu, um das Hexamerglühen zu ermöglichen.
- Säulenbasierte RNA-Aufreinigung mit den RNA Clean & Concentrator Säulen
HINWEIS: Alle Schritte sollten bei Raumtemperatur durchgeführt werden.- Geben Sie NF H2O in das Probenröhrchen, um es auf ein Volumen von 50 μL zu bringen.
- Geben Sie 100 μL Bindungspuffer und 150 μL 100% Ethanol in die Probe.
- Mischen und in eine Spin-Spalte übertragen.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- Fügen Sie 400 μL RNA-Prep-Puffer hinzu.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- Fügen Sie 700 μL RNA-Waschpuffer hinzu.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- Fügen Sie 400 μL RNA-Waschpuffer hinzu.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- (Optional) Die Säule in ein neues Sammelröhrchen geben und mit 10.000-16.000 × g für 2 min drehen.
- Die Säule wird in ein sauberes RNAse-freies Röhrchen überführt und eine entsprechende Menge NFH2Ozugegeben.
- Bei 10.000-16.000 × g für 1 min drehen.
- Saure Phenol-Chloroform-RNA-Extraktion.
- Fügen Sie ein gleiches Volumen an saurem Phenol: Chloroform: Isoamylalkohol hinzu.
- Wirbeln gründlich und bei 14.000 × g für 5 min zentrifugieren.
- Wenn es keine Phasentrennung gibt, fügen Sie 20 μL 2 M NaCl hinzu und wiederholen Sie die Zentrifugation.
- Die wässrige Phase in ein neues Röhrchen überführen.
- Fügen Sie 500 μL Isopropanol und 2 μL Co-Präzipitant hinzu.
- Mischen und bei RT für 3 min inkubieren; dann über Nacht bei −80 °C inkubieren.
- Pelletieren Sie die RNA durch Zentrifugation bei maximaler Geschwindigkeit (~ 20.000 × g) für 30 min bei 4 °C.
- Waschen Sie das Pellet mit 200 μL eiskaltem 70% Ethanol.
- Drehen Sie mit maximaler Geschwindigkeit (~ 20.000 × g) für 5 min; Verwerfen Sie den Flowthrough.
- Resuspendieren Sie das Pellet in der entsprechenden Menge NFH2O.
- Säulenbasierte cDNA-Bereinigung mit den Oligo Clean- und Concentrator-Säulen
HINWEIS: Alle Schritte sollten bei Raumtemperatur durchgeführt werden.- Geben Sie NF H2O in das Probenröhrchen, um es auf ein Volumen von 50 μL zu bringen.
- Fügen Sie 100 μL Bindungspuffer und 400 μL 100% Ethanol hinzu.
- Mischen und in eine Spin-Spalte übertragen.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- Fügen Sie 750 μL DNA-Waschpuffer hinzu.
- Spin bei 10.000-16.000 × g für 30 s; Verwerfen Sie den Flowthrough.
- (Optional) Die Säule in ein neues Sammelröhrchen geben und mit 10.000-16.000 × g für 2 min drehen.
- Die Säule wird in ein sauberes RNAse-freies Röhrchen überführt und eine entsprechende Menge NFH2Ohinzugefügt.
- Bei 10.000-16.000 × g für 1 min drehen.
3. Analyse der Sequenzierungsdaten
HINWEIS: Um RNA-Sekundärstrukturmodelle aus den DMS-MaP-Sequenzierungsdaten zu erstellen, müssen die resultierenden .fastq-Dateien in mehreren verschiedenen Schritten verarbeitet werden. Diese Schritte können automatisch mit dem Symbol
- Schneiden Sie die Adaptersequenzen mit TrimGalore oder Cutadapt.
- Ordnen Sie die Lesevorgänge den Referenzsequenzen (.fasta-Format) mit Bowtie2 zu.
- Zählen Sie die Lesevorgänge mit spezieller RNA-Struktursoftware (z. B. DREEM14, RNA-Framework17 oder ähnlich) und erstellen Sie Reaktivitätsprofile.
- (Optional) Clustern Sie die Lesevorgänge, um alternative RNA-Konformationen mit DREEM14, DRACO 17, DANCE-MaP18 oder ähnlichem zu finden.
- Vorhersage der minimalen freien Energiestruktur basierend auf den Reaktivitätsprofilen mit RNAStructure12, ViennaRNA oder ähnlichem.
- Visualisieren Sie die RNA11-Struktur mit VARNA (https://varna.lri.fr/) oder ähnlichem.
HINWEIS: Aus praktischen Gründen integrieren Software wie DREEM (www.rnadreem.org) und RNA-Framework19 die Schritte 1-5 weitgehend in ihre Pipelines, was den Analyseprozess rationalisiert. Jede Strukturvorhersage sollte jedoch mit Vorsicht behandelt werden (z. B. durch Überprüfung der Übereinstimmung der Struktur mit den Daten20.
Ergebnisse
Genspezifisches in vitro DMS-MaP
Um die 5'UTR von SARS2 zu untersuchen, wurden die ersten 300 bp des Virus als gBlock-Sequenz zusammen mit drei Primern bestellt. Dazu gehörten zwei Primer zur Vermehrung des Fragments ("FW" & "RV") über PCR sowie einer zum Anbringen des T7-Promotors ("FW-T7"). Diese Sequenzen sind in Tabelle 1 zu sehen.
| Name | Sequenz (5'->3') |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| WOHNMOBIL | GCAAACTGAGTTGGACGTGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTTTATACCTTCCCAGGTAAC |
Tabelle 1: Primersequenz für DMS-MaP RT-PCR von SARS-CoV2 5'UTR. Hier werden FW-T7 und RV benötigt, um eine DNA-Vorlage für die in vitro Transkription zu erzeugen, das RV wird in der reversen Transkription verwendet und das FW-RV-Primer-Paar wird in der anschließenden PCR-Amplifikation der cDNA verwendet. Die Primer glühen bis zum Anfang des SARS-CoV2-Genoms (FW) und der Sequenz direkt stromabwärts der interessierenden Region. Abkürzungen: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; RT-PCR = Reverse Transkription Polymerase Chain Reaction; SARS-CoV2 = schweres akutes respiratorisches Syndrom-Coronavirus 2; UTR = nicht übersetzte Region; RV = umgekehrte Grundierung; FW = Vorwärtsgrundierung.
Um RNA aus dem gBlock-Fragment zu erzeugen, wurde die Sequenz des T7-Polymerase-Promotors mittels Overlap-PCR unter Verwendung der PCR-Vormischung gemäß dem in Abbildung 2A gezeigten Schema angehängt. Aus dem länglichen Fragment wurde mit dem T7 Transcription Kit RNA erzeugt. Die DNA-Vorlage wurde anschließend mit der DNase und RNA verdaut, die mit RNA Clean & Concentrator-Säulen isoliert wurde.
Die Qualitätskontrolle der In-vitro-Transkription wurde durchgeführt, indem das RNA-Produkt auf einem 1% igen Agarosegel entlang einer ssRNA-Leiter ausgeführt wurde. Da nur eine Bande sichtbar war, wurden in vitro DMS-Sondierung und RT-PCR durchgeführt (siehe Abbildung 2B).
Um den Erfolg der PCR-Reaktion zu überprüfen, wurde die Probe auf einem 2% igen Agarosegel unter Verwendung einer dsDNA-Leiter durchgeführt. Nach der Indizierung sollte das Band ~ 150 bp höher auf dem gleichen Gel laufen, was die Größe der Indexierungsprimer berücksichtigt.

Abbildung 2: In-vitro-Transkription der DNA-Vorlage . (A) Um in vitro eine DNA-Vorlage zu transkribieren, die noch keinen intrinsischen RNA-Polymerase-Promotor hat, muss die Vorlage zuerst durch Überlappungs-PCR angehängt werden. Dies geschieht unter Verwendung eines Vorwärtsprimers, der die Sequenz TAATACGACTCACTATAGG (im Fall der T7-RNA-Polymerase) stromaufwärts der ersten Basen enthält, die mit dem gewünschten Fragment überlappen. Die unterstrichene Basis symbolisiert hier die Transkriptionsstartstelle der Polymerase. Sobald der Promotor an das dsDNA-Fragment gebunden ist, kann es von der T7-Polymerase transkribiert werden. Wichtig ist, dass die Polymerase den der genannten Promotorsequenz entgegengesetzten Strang als Vorlage (blau) verwendet, wodurch effektiv RNA erzeugt wird, die mit der Sequenz unmittelbar nach der angegebenen Promotorsequenz (rot) identisch ist. (B) Ein 1%iges Agarosegel mit einer ssRNA-Leiter (Spur 1) und dem in vitro transkribierten RNA-Produkt bei 300 nt (Spur 2). (C) Ein 2%iges Agarosegel mit GeneRuler 1 kb plus Ladder (Lane 1), dem PCR-Produkt nach RT-PCR mit 300 bp (lane 2) und dem indizierten Fragment nach Bibliotheksvorbereitung mit 470 bp (lane 3). Abkürzungen: RT-PCR = reverse transcription polymerase chain reaction; DMS = Dimethylsulfat; NT = Nukleotide; dsDNA = doppelsträngige DNA; ssRNA = einzelsträngige RNA. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
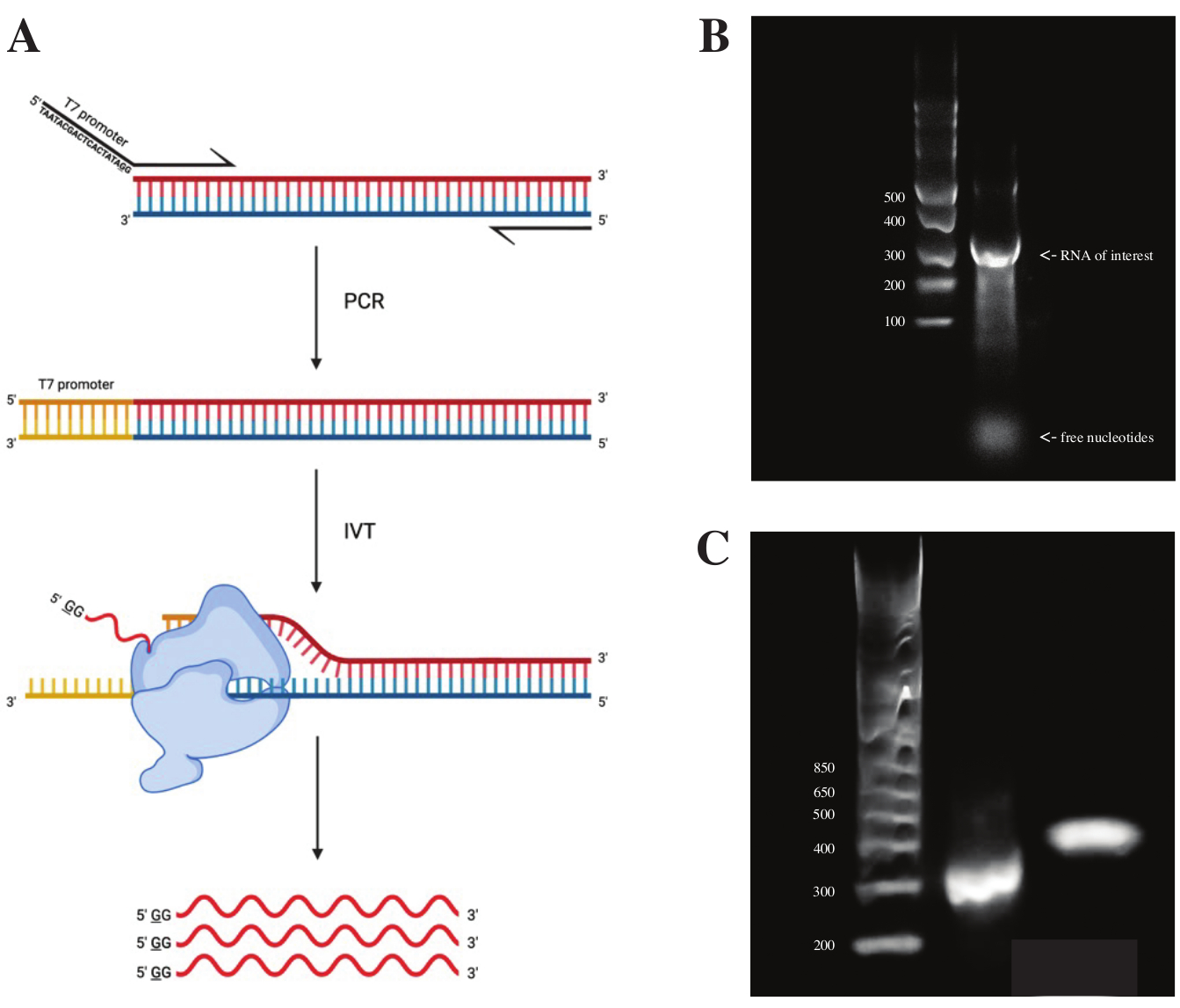{kind=link}
Gesamtgenom in vivo DMS-MaP mit virusinfizierten Zellen
Vor der DMS-Behandlung wurden die HCT-8-Zellen mit OC43 infiziert. Wenn eine zytopathische Wirkung (CPE) 4 Tage nach der Infektion (dpi) beobachtet wurde (wie in Abbildung 3A zu sehen), wurden diese Zellen behandelt, und die RNA wurde extrahiert und ribodepletiert. Beim Ausführen der Gesamt-RNA auf einem Agarosegel waren zwei helle Banden sichtbar, die die 40S- und 60S-Untereinheiten des Ribosoms ausmachen, die etwa 95% der gesamten RNA-Masse ausmachen (siehe Abbildung 3B). Wenn die RNA-Extraktion nicht erfolgreich war oder abgebaut wurde (z. B. durch mehrere Gefrier-Tau-Zyklen), waren die RNA-Abbauprodukte auf der Unterseite des Gels sichtbar (siehe Abbildung 3C, zweite Spur). Darüber hinaus verschwanden die beiden hellen Bänder nach der rRNA-Depletion und hinterließen einen Abstrich in der Spur (siehe Abbildung 3C, dritte Spur). Schließlich hatten die Proben nach der Bibliotheksvorbereitung unterschiedliche Größenverteilungen und wurden als Abstrich auf dem endgültigen PAGE-Gel gezeigt. Die Bande wurde zwischen 200 Nukleotiden (nt) und 500 nt herausgeschnitten, in Übereinstimmung mit dem 150 x 150 Paired-End-Sequenzierungslauf, der zur Analyse dieser Bibliotheken geplant war. Am wichtigsten ist, dass die Adapterdimere, die mit ~150 nt laufen, getrennt wurden (siehe Abbildung 3D).

Abbildung 3: Checkpoints von in vivo DMS-MaP mit virusinfizierten Zellen. (A) Lichtmikroskopische Aufnahme von virusinfizierten HCT-8-Zellen, 4 Tage dpi. Um die höchstmögliche Ausbeute an viraler RNA aus der Gesamt-RNA zu erhalten und gleichzeitig die nachteiligen Auswirkungen aufgrund des Zelltods zu minimieren, sollte DMS hinzugefügt werden, wenn CPE beginnt oder sogar davor, wie im Bild zu sehen ist. (B) Ein 1%iges Agarosegel mit sechs Proben von 1 μg Gesamt-RNA. In jeder Spur sind zwei helle Bänder sichtbar, die die Untereinheiten 40S und 60S ausmachen, da ribosomale RNA ~ 95% der gesamten RNA ausmacht. Hinweis: Die In-Zell-DMS-Behandlung verursacht eine gewisse RNA-Fragmentierung und Verschmierung, aber die beiden rRNA-Banden sollten immer noch sichtbar sein. Eine leichte Fragmentierung nach der Modifikation wird toleriert, da die Information, die die Methylierungsmarkierung enthält, während der DMS-Inkubation erzeugt wird und über die RNA-Struktur berichtet, während die Zellen noch am Leben sind. (C) Ein 1%iges Agarose-Gel aus GeneRuler 1 kb plus Leiter-DNA-Marker (Spur 1) Gesamt-RNA, die zuvor bei −80 °C für 6 Monate gelagert wurde (Spur 2) und ribodepletierte RNA (Spur 3). Bei längerer Lagerung von RNA mit mehreren Gefrier-Tau-Zyklen beginnt die RNA abzubauen und sollte möglicherweise nicht für Sondierungsexperimente verwendet werden. Darüber hinaus verblassen nach der Ribodepletion der Gesamt-RNA die beiden hellen Banden, die die 40S- und 60S-Untereinheiten des Ribosoms ausmachen, und ein Abstrich der verbleibenden RNAs beginnt sich zu zeigen. (D) Ein PAGE-Gel aus GeneRuler 1 kb plus Leiter-DNA-Marker (Spur 1) und eine Bibliotheksprobe der für das gesamte Genom präparierten RNA. Das Gel sollte basierend auf den Sequenzierungsanforderungen herausgeschnitten werden. Für einen Paired-End-Sequenzierlauf, der 150 Zyklen von beiden Seiten umfasst, sollte das Gel zwischen 300 bp und 500 bp herausgeschnitten werden. Adapterdimere (mit 170 bp) sollten getrennt werden. Abkürzungen: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; dpi = Tage nach der Infektion; CPE = zytopathische Wirkung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Nach der Sequenzierung wurden die .fastq-Dateien analysiert, indem ein Job zusammen mit einer .fasta-Referenzdatei an den DREEM-Webserver (http://rnadreem.org/) gesendet wurde. Die vom Server generierte Ausgabe umfasst Qualitätskontrolldateien, die von fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) und TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) generiert wurden, sowie andere Ausgabedateien, die die durchschnittlichen Mutationshäufigkeiten der Bevölkerung enthalten. Neben dem Diagramm, das die Mutationshäufigkeiten mit einem interaktiven .html Format (siehe Abbildung 4A) zeigt, und einer .csv Datei mit den Rohreaktiviten pro Base und einer struct_constraint.txt Datei, die von mehreren RNA-Strukturvorhersagesoftware gelesen werden kann, enthält dies auch eine Bitvektor.txt Datei, die über die By-Read-Mutationen berichtet. Daraus wurden die durchschnittlichen Strukturen der Bevölkerung berechnet, indem die .fasta- und struct_constraint.txt-Dateien an den RNAfold-Webserver gesendet wurden (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Diese nutzt die ViennaRNA-Software, um Strukturvorhersagen auf Basis der minimalen freien Energie zu generieren, die online eingesehen oder im ct- oder Vienna-Format heruntergeladen werden können. Um RNA-Strukturmodelle zu generieren, wurden diese herunterladbaren Dateien an VARNA übermittelt (https://varna.lri.fr/, siehe Abbildung 4B). Schließlich können Bitvektor.txt Dateien von der stabilen Version von DREEM (https://codeocean.com/capsule/6175523/tree/v1) verwendet werden, um nach alternativen RNA-Konformationen zu suchen. Um gute Strukturmodelle mit DREEM zu erhalten, sollte eine Abdeckung von 10.000 Lesevorgängen pro Basis erreicht werden. Für Clustering sind möglicherweise bis zu 100.000 Lesevorgänge pro Basis erforderlich. Eine Übersicht über den gesamten Workflow finden Sie in Abbildung 4C.

Abbildung 4: Beispielhafte Daten aus chemischen Sondierungsexperimenten des SARS-CoV2 5'UTR. (A) Reaktivitätsprofil der ersten 300 Basen des SARS-CoV2-Genoms eingefärbt nach Base (A: rot, C: blau, U: grün, G: gelb). Die rohen Reaktivitäten werden als absolute Mutationshäufigkeit dividiert durch die Abdeckung berechnet. Basen mit offenem Exterieur haben hohe Reaktivitätswerte; Basen, die Basenpaarung betreiben, haben niedrige Reaktivitätswerte. U und G werden durch DMS nicht modifiziert und haben niedrige Reaktivitätswerte, die von Polymerase-Untreue herrühren. Die Vorhersagen wurden mit dem DREEM-Webserver getroffen. (B) Strukturmodell des SARS-CoV2 5'UTR, vorhergesagt aus Reaktivitätswerten, die mit VARNA erstellt wurden. Basen mit hohen Reaktivitätswerten sind rot eingefärbt; Basen mit niedrigen Reaktivitätswerten sind weiß eingefärbt. (C) Workflow der DMS-MaP-Analyse beginnend mit den .fastq-Dateien, die aus der Sequenzierung gewonnen werden. Diese können mit fastqc qualitätskontrolliert werden; Die Adaptersequenzen werden mit TrimGalore getrimmt und dann mit Bowtie2 wieder einer Referenzsequenz zugeordnet. Aus den erhaltenen BAM-Dateien zählt DREEM die Mutationen in jedem Lesevorgang und erstellt eine Mutationszuordnung oder .bitvector.txt Datei. Diese melden die Mutationen jedes Lesevorgangs positionsabhängig, anhand derer die populationsdurchschnittlichen Reaktivitätsprofile erstellt werden können. Alternativ können Bitvektoren mit DREEM geclustert werden, um nach alternativen RNA-Konformationen zu suchen. Schließlich werden die erhaltenen Strukturmodelle mit Software (z.B. VARNA) visualisiert. Abkürzungen: DMS-MaP = Mutational profiling with sequencing using dimethyl sulfate; SARS-CoV2 = schweres akutes respiratorisches Syndrom-Coronavirus 2. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
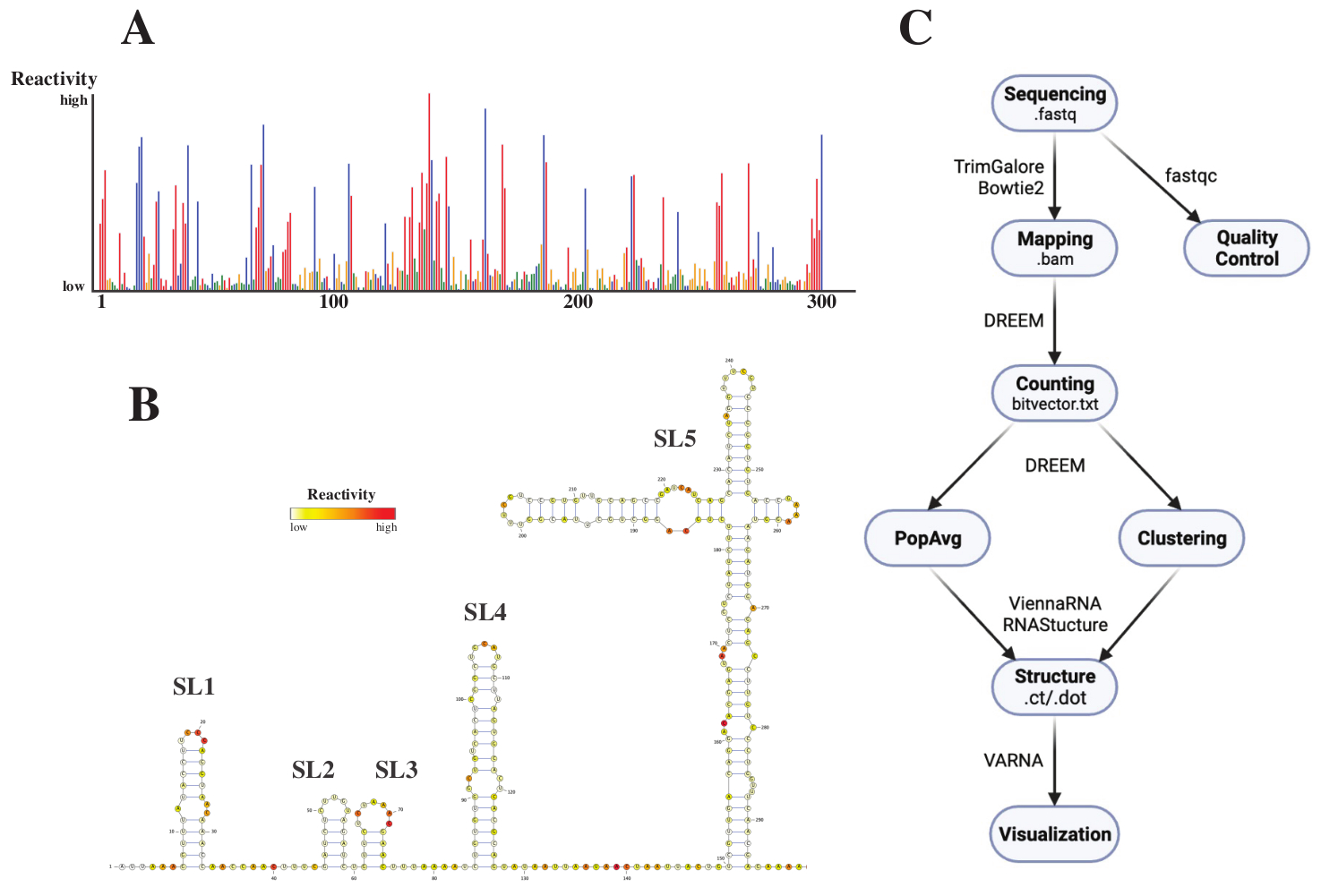{kind=link}
Diskussion
Das Protokoll hier beschreibt, wie RNA in vitro und in Zellen mit DMS-Mutationsprofilierungsexperimenten untersucht werden kann. Darüber hinaus gibt es Anweisungen, wie Bibliotheken für die Illumina-Sequenzierung vorbereitet werden, um genspezifische Daten zu generieren und die erhaltenen .fastq-Dateien zu analysieren. Darüber hinaus können genomweite Bibliotheksansätze verwendet werden. Die genspezifische RT-PCR liefert jedoch die hochwertigsten und robustesten Daten. Daher ist es beim Vergleich zwischen Stichproben wichtig sicherzustellen, dass sie mit identischen Sequenzierungsstrategien vorbereitet werden, da die Bibliotheksgenerierung eine gewisse Verzerrung verursacht. Die Reproduzierbarkeit sollte immer mit Hilfe von Replikaten gemessen werden.
Mehrere Vorsichtsmaßnahmen
RNA ist ein instabiles Molekül, das sowohl durch erhöhte Temperaturen als auch durch RNasen empfindlich auf Abbau reagiert. Daher werden besondere Maßnahmen - die Verwendung von persönlicher Schutzausrüstung (PSA), RNAse-freiem Material und RNASE-Inhibitoren - empfohlen. Am wichtigsten ist, dass RNA wann immer möglich auf Eis gehalten werden sollte. Dies gilt insbesondere für methylierte RNA, die noch empfindlicher auf hohe Temperaturen reagiert.
Es ist wichtig zu bestätigen, dass die interessierende RNA-Struktur nicht empfindlich auf die DMS-Konzentration und die Pufferbedingungen reagiert. Puffer wie 100 mM Tris, 100 mM MOPS und 100 mM HEPES bei pH 7-7,5 geben ein hohes Signal, reichen aber möglicherweise nicht aus, um den pH-Wert während der Reaktion aufrechtzuerhalten21. Da DMS in Wasser hydrolysiert, was den pH-Wert senkt, ist ein starker Puffer entscheidend, um während der Modifizierungsreaktion einen neutralen pH-Wert aufrechtzuerhalten. Es wurde gezeigt, dass die Zugabe von Bic dazu beiträgt, den pH-Wert als leicht basisch21 aufrechtzuerhalten, führt jedoch zu einer niedrigen DMS-Modifikation bei Gs und Us, was informativ sein könnte, aber aufgrund der Erzeugung eines viel niedrigeren Signals als As und Cs separat analysiert werden sollte und in diesem Protokoll nicht weiter diskutiert wird.
Bei der genspezifischen RT-PCR wird die modifizierte RNA reverse-transkribiert in die DNA transkribiert und mittels PCR fragmentiert amplifiziert. Während die Größe der RNA theoretisch unbegrenzt sein kann, sollten diese PCR-Fragmente eine Länge von 400-500 Basenpaaren (bp) nicht überschreiten, um Verzerrungen während der reversen Transkriptionsreaktion zu verhindern. Idealerweise sollten sich die Fragmente im Rahmen des Sequenzierungslaufs befinden (d. h. wenn die Sequenzierung mit einem 150 x 150-Zyklus-Paired-End-Sequenzierungsprogramm durchgeführt wird, sollte ein einzelnes Fragment 300 bp nicht überschreiten). Bei Verwendung von Sequenzierungsprogrammen mit weniger Zyklen können die PCR-Produkte mithilfe einer dsDNase fragmentiert werden. Da Sequenzen innerhalb der Primersequenzen keine Strukturinformationen enthalten, müssen sich die Fragmente überlappen, wenn die untersuchte RNA >1-Fragment umfasst. RT-Reaktionen können mehrere RT-Primer für verschiedene Fragmente enthalten (bis zu 10 verschiedene RT-Primer). Abhängig von den Sequenzen kann das Pooling der RT-Primer die umgekehrte Transkription weniger effizient machen, funktioniert aber in der Regel gut. Jede PCR-Reaktion sollte separat durchgeführt werden.
Bei der Sondierung von RNA mit DMS spielen die experimentellen Bedingungen eine zusätzliche Rolle, da viele RNAs thermodynamisch instabil sind und ihre Konformation aufgrund von Umweltfaktoren wie Temperatur ändern. Um Unregelmäßigkeiten zu vermeiden, sollten die experimentellen Bedingungen auch hinsichtlich der Reaktionszeiten so konstant wie möglich gehalten werden. Die Pufferbedingungen scheinen bis zu einem gewissen Grad austauschbar zu sein 17,20,22,23, wenn die Grundbedingungen - die Pufferkapazität und das Vorhandensein von einwertigen (Na) und zweiwertigen Ionen (Mg) - aufrechterhalten werden, um eine ordnungsgemäße Faltung von RNA 24 zu gewährleisten.
Bei der Bibliothekspräparation modifizierter RNAs müssen mehrere Aspekte berücksichtigt werden. Erstens sind modifizierte RNAs, wie bereits erwähnt, weniger stabil als ihre unmodifizierten Gegenstücke, was bedeutet, dass sie möglicherweise die Optimierung der Fragmentierungszeiten für eine optimale Fragmentgrößenverteilung erfordern. Darüber hinaus verwenden bestimmte RNA-Bibliotheksvorbereitungskits sowie viele andere RNAseq-Ansätze zufällige Primer im reversen Transkriptionskit. Dies könnte zu einer geringeren Abdeckung der Referenz, insbesondere in den 3' eines Gens, und letztendlich zu einer unzureichenden Abdeckungstiefe führen. Wenn die Abdeckung einer bestimmten Region zu gering ist, kann es notwendig sein, diese Basen aus der Strukturvorhersage zu entfernen. Neben RT-PCR und Whole-Genome RNAseq Kits können auch andere Ansätze zur Bibliothekspräparation verwendet werden. Protokolle, die die Ligation von 3'- und/oder 5'-Adaptern an die RNA beinhalten, sind vorteilhaft, wenn kleine RNA-Fragmente verwendet werden oder wenn der Verlust von Sondierungsinformationen in den Primerbereichen vermieden werden muss.
Schließlich muss die Analyse der chemischen Sondierungsexperimente immer sorgfältig interpretiert werden. Derzeit gibt es keine Software, die die RNA-Struktur einer RNA allein aus der Sequenz mit hoher Genauigkeit vorhersagt. Obwohl chemische Sondierungsbeschränkungen die Genauigkeit erheblich verbessern, ist die Erstellung guter Modelle für lange RNAs (>500 nt) immer noch eine Herausforderung. Diese Modelle sollten durch andere Ansätze und/oder Mutagenese weiter getestet werden. Die RNA-Vorhersagesoftware optimiert für die maximale Anzahl von Basenpaaren und bestraft so offene Konformationen, die die RNA-Faltung möglicherweise nicht genau darstellen5. Daher sollte das erhaltene Strukturmodell getestet werden, indem die Vorhersageübereinstimmung mit den zugrunde liegenden chemischen Sondierungsdaten (z. B. durch AUROC) und zwischen Replikaten (z. B. durch mFMI) quantifiziert wird, wie von Lan et al.20 veranschaulicht.
Idealerweise sollten mehrere Experimente in verschiedenen Systemen verwendet werden, um das erhaltene Strukturmodell in Frage zu stellen, um die eigene Hypothese zu stärken. Diese können die Verwendung von In-vitro - und In-Cell-Ansätzen, kompensatorischen Mutationen und verschiedenen Zelllinien und -spezies umfassen. Darüber hinaus sind rohe Reaktivitäten oft genauso informativ oder sogar informativer als Strukturvorhersagen, da sie die "Ground Truth"-Momentaufnahme des RNA-Faltungsensembles aufzeichnen. Daher sind rohe Reaktivitäten sehr geeignet und informativ, um Strukturänderungen zwischen verschiedenen Bedingungen zu vergleichen. Wichtig ist, dass die Strukturen mit der niedrigsten freien Energie, die unter Verwendung chemischer Sondierungsbeschränkungen mit rechnerischer Vorhersage berechnet werden, nur als Ausgangshypothese für ein vollständiges Strukturmodell verwendet werden sollten.
Offenlegungen
Die Autoren haben keine Interessenkonflikte zu erklären.
Danksagungen
Nichts
Materialien
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
Referenzen
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten