
Method Article
Sondeo de la estructura del ARN con perfil mutacional de sulfato de dimetilo con secuenciación in vitro y en células
En este artículo
Resumen
El protocolo proporciona instrucciones para modificar el ARN con sulfato de dimetilo para experimentos de perfiles mutacionales. Incluye sondeo in vitro e in vivo con dos métodos alternativos de preparación de bibliotecas.
Resumen
El papel de la estructura del ARN en prácticamente cualquier proceso biológico se ha vuelto cada vez más evidente, especialmente en la última década. Sin embargo, los enfoques clásicos para resolver la estructura del ARN, como la cristalografía de ARN o la crio-EM, no han logrado mantenerse al día con el campo en rápida evolución y la necesidad de soluciones de alto rendimiento. Perfil mutacional con secuenciación usando sulfato de dimetilo (DMS) MaPseq es un enfoque basado en la secuenciación para inferir la estructura de ARN a partir de la reactividad de una base con DMS. DMS metila el nitrógeno N1 en adenosinas y el N3 en citosinas en su cara de Watson-Crick cuando la base no está apareada. La transcripción inversa del ARN modificado con la transcriptasa inversa del intrón termoestable del grupo II (TGIRT-III) conduce a que las bases metiladas se incorporen como mutaciones en el ADNc. Al secuenciar el ADNc resultante y asignarlo a una transcripción de referencia, las tasas de mutación relativas para cada base son indicativas del "estado" de la base como emparejada o no apareada. Aunque las reactividades DMS tienen una alta relación señal-ruido tanto in vitro como en células, este método es sensible al sesgo en los procedimientos de manipulación. Para reducir este sesgo, este documento proporciona un protocolo para el tratamiento de ARN con DMS en células y con ARN transcrito in vitro .
Introducción
Desde el descubrimiento de que el ARN tiene propiedades estructurales1,2 y catalíticas3, la importancia del ARN y su función reguladora en una plétora de procesos biológicos se han descubierto gradualmente. De hecho, el efecto de la estructura del ARN en la regulación génica ha ganado cada vez más atención4. Al igual que las proteínas, el ARN tiene estructuras primarias, secundarias y terciarias, que se refieren a la secuencia de nucleótidos, el mapeo 2D de las interacciones de emparejamiento de bases y el plegamiento 3D de estas estructuras pareadas de bases, respectivamente. Si bien determinar la estructura terciaria es clave para comprender los mecanismos exactos detrás de los procesos dependientes del ARN, la estructura secundaria también es muy informativa sobre la función del ARN y es la base para un mayor plegamiento 3D5.
Sin embargo, determinar la estructura del ARN ha sido intrínsecamente desafiante con los enfoques convencionales. Mientras que para las proteínas, la cristalografía, la resonancia magnética nuclear (RMN) y la microscopía electrónica criogénica (crio-EM) han permitido determinar la diversidad de motivos estructurales, permitiendo la predicción de la estructura a partir de la secuencia sola6, estos enfoques no son ampliamente aplicables a los ARN. De hecho, los ARN son moléculas flexibles con bloques de construcción (nucleótidos) que tienen mucha más libertad conformacional y rotacional en comparación con sus contrapartes de aminoácidos. Además, las interacciones a través del emparejamiento de bases son más dinámicas y versátiles que las de los residuos de aminoácidos. Como resultado, los enfoques clásicos han tenido éxito solo para ARN relativamente pequeños con estructuras bien definidas y altamente compactas7.
Otro enfoque para determinar la estructura del ARN es a través del sondeo químico combinado con la secuenciación de próxima generación (NGS). Esta estrategia genera información sobre el estado de unión de cada base en una secuencia de ARN (es decir, su estructura secundaria). En resumen, las bases en una molécula de ARN que no participan en el emparejamiento de bases se modifican diferencialmente por pequeños compuestos químicos. La transcripción inversa de estos ARN con transcriptasas inversas especializadas (RT) incorpora las modificaciones en ácido desoxirribonucleico complementario (ADNc) como mutaciones. Estas moléculas de ADNc son amplificadas por la reacción en cadena de la polimerasa (PCR) y secuenciadas. Para obtener información sobre su "estado" como unido o no unido, las frecuencias de mutación en cada base en un ARN de interés se calculan y se introducen en el software de predicción de estructuras como restricciones8. Basado en las reglas del vecino más cercano9 y los cálculos mínimos de energía libre 10, este software genera modelos de estructura que mejor se ajustan a los datos experimentales obtenidos11,12.
DMS-MaPseq utiliza DMS, que metila el nitrógeno N1 en adenosinas y el nitrógeno N3 en citosinas en su cara de Watson-Crick de una manera altamente específica13. El uso de la transcriptasa inversa intrón termoestable del grupo II (TGIRT-III) en la transcripción inversa crea perfiles mutacionales con relaciones señal-ruido sin precedentes, incluso permitiendo la deconvolución de perfiles superpuestos generados por dos o más conformaciones alternativas14,15. Además, el DMS puede penetrar las membranas celulares y los tejidos enteros, lo que hace posible el sondeo dentro de contextos fisiológicos. Sin embargo, la generación de datos de buena calidad es un desafío, ya que las variaciones en el procedimiento de manejo pueden afectar los resultados. Por lo tanto, proporcionamos un protocolo detallado para DMS-MaPseq in vitro y en la célula para reducir el sesgo y guiar a los recién llegados al método a través de las dificultades que pueden encontrar. Especialmente a la luz de la reciente pandemia de SARS-CoV2, los datos de alta calidad sobre los virus de ARN son una herramienta importante para estudiar la expresión génica y encontrar posibles terapias.
Protocolo
NOTA: Consulte la Tabla de materiales para obtener detalles relacionados con todos los materiales, software, reactivos, instrumentos y celdas utilizados en este protocolo.
1. DMS-MaP in vitro específico del gen
- Transcripción in vitro de ARN
- Obtener la secuencia del ARN de interés como ADN bicatenario (ds) (por ejemplo, como fragmentos de ADN, plásmidos o PCR a partir de ADN preexistente/genómico). Si la secuencia de ADN contiene un promotor de la polimerasa, vaya al paso 3.
- Realizar PCR de superposición para unir un promotor de ARN polimerasa aguas arriba del fragmento de ADN deseado (cebador directo para la polimerasa T7: 5' TAATACGACTCACTATAGG + primeras bases de la secuencia diana 3').
- Transcribir in vitro el fragmento de ADN en ARN. Siempre mantenga el ARN en hielo.
- Digiere el ADN usando una DNasa.
- Aislar el ARN utilizando un enfoque basado en columnas (paso 2.4) o por precipitación de etanol (paso 2.5). Eluir en un volumen apropiado, esperando un rendimiento de ~50 μg.
- Asegurar la integridad del ARN ejecutándolo en un gel de agarosa; desnaturalizar el ARN durante 2-3 min a 70 °C antes de correr.
NOTA: El tampón y la agarosa pueden contener RNasas que degradan el ARN y pueden contaminar la muestra de ARN. Los geles prefabricados de agarosa se han utilizado previamente en este laboratorio; los resultados (especialmente con ARN) han sido ambiguos a veces. Los mejores resultados se obtuvieron con los geles de agarosa o PAGE. - Uso directo de almacenar el ARN a −80 °C durante varios meses, a menos que la degradación sea visible después de la descongelación.
- In vitro Modificación de DMS (a 105 mM DMS)
- Preparar una cantidad suficiente de tampón de repliegue (0,4 M de cacodilato de sodio, pH 7,2, que contiene 6 mM de MgCl2).
NOTA: Para cada reacción (volumen final de 100 μL), añadir 89 μL de tampón de replegado. - Para cada reacción, transfiera 89 μL de tampón de repliegue a un tubo designado de 1,5 ml y precaliente a 37 °C en un termoagitador colocado debajo de una campana química.
NOTA: El DMS es altamente tóxico y siempre debe mantenerse debajo de una capucha química hasta que se apague con un agente reductor. - Eluya 1-10 pmol de ARN en 10 μL de agua libre de nucleasa (NFH2O); transferencia a un tubo de PCR.
- Incubar en un termociclador a 95 °C durante 1 min para desnaturalizar el ARN.
- Colóquelo en un bloque de hielo inmediatamente para evitar el plegado incorrecto.
- Agregue la muestra de ARN al tubo designado con tampón de repliegue a 37 ° C, mezcle bien e incube durante 10-20 minutos para renovar el ARN.
NOTA: La mayoría de los ARN se doblarán en el orden de milisegundos a segundos, aunque existen excepciones16. - Agregue 1 μL de DMS al 100% (10,5 M) a la muestra de ARN e incube durante 5 minutos mientras agita a 800-1.400 rotaciones por minuto (rpm).
NOTA: La agitación (u otros medios de mezcla) en este paso es crucial ya que el DMS es hidrófobo y puede no disolverse completamente en el tampón de replegado. Las desviaciones en los tiempos de reacción pueden afectar la reproducibilidad de las reactividades del DMS. Para minimizar el error de pipeteo, el DMS se puede disolver en etanol al 100% antes de agregarlo a la muestra si se mantiene una concentración final de 1% (105 mM) de DMS. Para un control no tratado, el DMS puede ser sustituido por dimetilsulfóxido (DMSO) o agua. - Después de 5 minutos de tiempo de reacción, enfriar con 60 μL de β-mercaptoetanol (BME) al 100%, mezclar bien e inmediatamente colocar el ARN en hielo.
NOTA: El ARN se puede eliminar de forma segura de la campana después de apagar la reacción con BME para limpiarlo. Sin embargo, la exposición directa de BME al entorno debe evitarse debido a su fuerte olor y propiedades irritantes. - Limpiar el ARN mediante precipitación de acetato de sodio-etanol (ver paso 2.5) o un enfoque basado en columnas (ver paso 2.6), y eluya en 10 μL de agua.
- Cuantificar el ARN usando un espectrofotómetro.
- Uso directo de almacenar el ARN modificado a −80 °C.
NOTA: Se debe evitar el almacenamiento a largo plazo, ya que el ARN es menos estable después del tratamiento con DMS.
- Preparar una cantidad suficiente de tampón de repliegue (0,4 M de cacodilato de sodio, pH 7,2, que contiene 6 mM de MgCl2).
- RT-PCR específica del gen del ARN modificado
NOTA: Consulte la Figura 1 para la configuración de RT-PCR de los fragmentos tratados con DMS.- Eluya 100 ng de ARN modificado en 10 μL deH2O. Libre de nucleasa (NF) Transferencia a un tubo de PCR.
- Al tubo, agregue 4 μL de 5x tampón de primera cadena (FSB), 1 μL de mezcla de dNTP (10 mM cada uno), 1 μL de 0.1 M de ditiothreitol (DTT) (evite ciclos de congelación-descongelación), 1 μL de inhibidor de RNasa, 1 μL de cebador inverso de 10 μM (cebador único o un grupo de cebadores) y 1 μL de TGIRT III.
NOTA: Para un grupo de cebadores, no añadir 1 μL de 10 μM de cada cebador directamente al RT; en su lugar, mezcle primero los cebadores y agregue 1 μL de la mezcla (a una concentración total de imprimación de 10 μM). - Incubar a 57 °C durante 30 min a 1,5 h (normalmente, 30 min es suficiente para hacer un producto de 500 nt) en un termociclador.
- Añadir 1 μL de NaOH 4 M, mezclar por pipeteo e incubar a 95 °C durante 3 min para degradar el ARN.
NOTA: Este paso es crucial ya que libera TGIRT del ADNc degradando el ARN. Si se omite, la PCR posterior podría verse afectada. - Limpie utilizando un enfoque basado en columnas (ver paso 2.6) que elimine suficientemente los cebadores y eluya en 10 μL de NFH2O.
- La PCR amplifica el ADNc utilizando 1 μL del producto de transcripción inversa por cada 25 μL de la reacción con un kit de PCR diseñado para equilibrar el rendimiento y la fidelidad.
NOTA: Las imprimaciones deben tener una temperatura de fusión de ~ 60 ° C. - Ejecute 2 μL del producto de PCR en un gel de agarosa o un gel de agarosa prefabricado para verificar el éxito de la PCR.
- Idealmente, solo una banda debe mostrarse después de la PCR. Si es así, limpie la reacción utilizando un enfoque basado en columnas. Si hay bandas alternativas, use la reacción de PCR restante para extirpar la banda correcta del gel. Eluyar en un volumen suficientemente pequeño (por ejemplo, 10 μL).
- Cuantificar los fragmentos extraídos utilizando un espectrofotómetro.
- Indexar los fragmentos de dsDNA para secuenciación utilizando un enfoque adecuado para la plataforma de secuenciación deseada.

Figura 1: Configuración experimental para la RT-PCR de fragmentos grandes tratados con DMS. Al realizar la transcripción inversa en un ARN modificado, no se registrarán las modificaciones en la secuencia a la que se dirige el cebador. Por lo tanto, cuando los fragmentos superan los 400-500 pb de longitud, los fragmentos que se superponen en las regiones de cebador deben diseñarse, como se ejemplifica aquí. La longitud de los fragmentos depende de las necesidades de secuenciación. Cuando se utiliza la secuenciación pareada de 150 ciclos, los fragmentos no deben exceder los 300 pb. Abreviaturas: RT-PCR = reacción en cadena de la polimerasa con transcriptasa inversa; DMS = sulfato de dimetilo. Haga clic aquí para ver una versión más grande de esta figura.
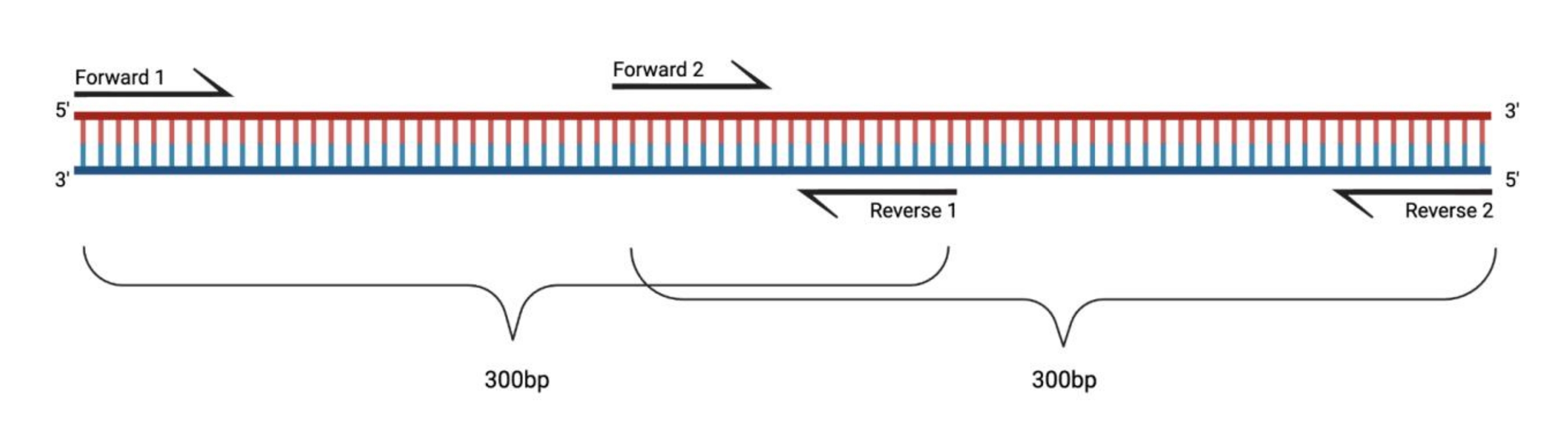{kind=link}
2. DMS-MaP de genoma completo utilizando células infectadas por virus
NOTA: En las células, el tratamiento con DMS también se puede combinar con el enfoque de amplificación específico del gen descrito anteriormente. La biblioteca del genoma completo requiere una enorme profundidad de secuenciación para lograr una cobertura completa en un solo gen. Sin embargo, si los ARN virales constituyen una fracción significativa del ARN ribodepleto después de la extracción, la secuenciación del genoma completo sería apropiada. Además, otros métodos de enriquecimiento se pueden combinar con el método de generación de bibliotecas de genoma completo.
- Tratamiento DMS
- Cultivar células infectadas con el virus hasta la etapa deseada de la infección.
- Transfiera el contenedor de la celda a una campana de humos dedicada que sea apropiada para manejar tanto virus con el nivel de bioseguridad requerido como los humos químicos generados por agentes como DMS.
- Agregue un volumen de 2.5% de DMS al medio de cultivo y selle el recipiente (típicamente una placa de 10 cm) con parafilm.
NOTA: Es fácil sub-modificar y sobre-modificar con DMS. Al agregar DMS directamente a las células, es muy importante mezclar bien. Alternativamente, precaliente el nuevo medio en un tubo cónico de 50 ml a 37 °C y agregue el DMS directamente agitando vigorosamente. Decantar el medio gastado en las células, y pipetear lentamente en el medio que contiene DMS. - Traslado a una incubadora a 37 °C durante 5 min.
NOTA: Dependiendo de la cantidad de tiempo que se tarde en manejar el DMS fuera de la incubadora, es posible que 5 minutos conduzcan a una modificación excesiva. Evite que el tiempo pase de agregar el DMS a la incubación a ≤1 min. Si se realiza el experimento por primera vez, se recomienda realizar una titulación DMS y variar el tiempo de incubación (entre 3 min y 10 min) para encontrar la tasa de modificación óptima y garantizar que los resultados sean robustos en una ventana de concentraciones. - Pipetear cuidadosamente el medio que contiene DMS (en residuos químicos apropiados) y añadir suavemente 10 ml de tampón de parada (PBS con 30% de BME [por ejemplo, 3 ml de BME y 7 ml de PBS]).
NOTA: La adición de DMS y BME puede levantar las células de la placa si las células no son fuertemente adherentes. Si las células se están levantando, se pueden tratar como células de suspensión, en lugar de eliminar el medio que contiene DMS, agregue el tampón de parada directamente y raspe las células con DMS y BME en un tubo cónico de 50 ml. Granular las células centrifugando durante 3 min a 3.000 × g; asegúrese de deshacerse de cualquier DMS residual, que puede granularse debajo de las células en gotas grandes. Se recomienda un paso de lavado adicional en BME al 30% si el medio DMS no se puede eliminar inicialmente. - Raspa las células y transfiérelas a un tubo cónico de 15 ml.
- Pellet por centrifugación a 3.000 × g durante 3 min.
- Retire el sobrenadante y lave 2 veces con 10 ml de PBS.
- Retire con cuidado la mayor cantidad posible de PBS residual.
- Disuelva el pellet en una cantidad adecuada del reactivo de aislamiento de ARN (p. ej., 3 ml para un matraz de cultivo T75, 1 ml para una placa de 10 cm).
NOTA: Cantidades insuficientes del reactivo podrían afectar el rendimiento de ARN.
- Extracción de ARN y agotamiento del ARN ribosómico (ARNr)
- A 1 ml de células homogeneizadas en el reactivo de aislamiento de ARN, agregue 200 μL de cloroformo, vórtice durante 15-20 s hasta que sea de color rosa brillante, y luego incubar hasta 3 minutos hasta que la separación de fases sea visible.
NOTA: La fase lipídica rosa debe asentarse en la parte inferior. Si este no es el caso, el tiempo de vórtice probablemente fue insuficiente. - Girar a velocidad máxima (~ 20.000 × g) durante 15 min a 4 °C.
- Transfiera la fase acuosa superior a un nuevo tubo.
- Limpiar el ARN mediante precipitación de acetato de sodio-etanol (ver paso 2.5) o un enfoque basado en columnas (ver paso 2.6), y eluye en un volumen suficiente de NFH2O.
- Verifique la integridad del ARN en un gel de agarosa. Busque dos bandas correspondientes a las dos subunidades ribosómicas.
- Agotar los ARNr utilizando el enfoque preferido y eluyar en un volumen adecuado (típicamente 20-50 μL) de NFH2O.
NOTA: Para aplicaciones posteriores, se sugiere ~ 500 ng de ARN total en un volumen de 8 μL. Los ARN no ribosómicos generalmente representan solo el 5% -10% del ARN total. - Cuantificar usando un espectrofotómetro.
- A 1 ml de células homogeneizadas en el reactivo de aislamiento de ARN, agregue 200 μL de cloroformo, vórtice durante 15-20 s hasta que sea de color rosa brillante, y luego incubar hasta 3 minutos hasta que la separación de fases sea visible.
- Generación de bibliotecas
- Utilizar RT-PCR específica de genes u otros enfoques para generar bibliotecas15. Si utiliza hexámeros aleatorios para el cebado, agregue un paso de incubación a unbajo T m (37-42 °C) para permitir el recocido del hexámero.
NOTA: Los kits de generación de bibliotecas estándar también se pueden utilizar reemplazando la enzima RT con TGIRT y cambiando la temperatura RT a 57 °C.
- Utilizar RT-PCR específica de genes u otros enfoques para generar bibliotecas15. Si utiliza hexámeros aleatorios para el cebado, agregue un paso de incubación a unbajo T m (37-42 °C) para permitir el recocido del hexámero.
- Limpieza de ARN basada en columnas utilizando las columnas RNA Clean & Concentrator
NOTA: Todos los pasos deben realizarse a temperatura ambiente.- Añadir NFH2Oal tubo de muestra para llevarlo a un volumen de 50 μL.
- Añadir 100 μL de tampón de unión y 150 μL de etanol 100% a la muestra.
- Mezclar y transferir a una columna de centrifugado.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- Agregue 400 μL de tampón de preparación de ARN.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- Añadir 700 μL de tampón de lavado de ARN.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- Añadir 400 μL de tampón de lavado de ARN.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- (Opcional) Transfiera la columna a un nuevo tubo de recolección y gire a 10,000-16,000 × g durante 2 min.
- Transfiera la columna a un tubo limpio libre de ARNasa y agregue una cantidad adecuada de NFH2O.
- Girar a 10.000-16.000 × g durante 1 min.
- Extracción de ARN ácido fenol-cloroformo.
- Añadir un volumen igual de ácido fenol:cloroformo:alcohol isoamílico.
- Vórtice a fondo, y centrifugar a 14.000 × g durante 5 min.
- Si no hay separación de fases, añadir 20 μL de NaCl 2 M y repetir la centrifugación.
- Transfiera la fase acuosa a un nuevo tubo.
- Añadir 500 μL de isopropanol y 2 μL de coprecipitante.
- Mezclar e incubar en RT durante 3 min; luego, incubar a −80 °C durante la noche.
- Granular el ARN por centrifugación a velocidad máxima (~ 20.000 × g) durante 30 min a 4 °C.
- Lave el pellet con 200 μL de etanol helado al 70%.
- Girar a velocidad máxima (~ 20.000 × g) durante 5 min; Descarte el flujo continuo.
- Vuelva a suspender el pellet en la cantidad adecuada de NFH2O.
- Limpieza de ADNc basada en columnas utilizando las columnas Oligo Clean y Concentrator
NOTA: Todos los pasos deben realizarse a temperatura ambiente.- Añadir NFH2Oal tubo de muestra para llevarlo a un volumen de 50 μL.
- Añadir 100 μL de tampón de unión y 400 μL de etanol al 100%.
- Mezclar y transferir a una columna de centrifugado.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- Añadir 750 μL de tampón de lavado de ADN.
- Girar a 10.000-16.000 × g durante 30 s; Descarte el flujo continuo.
- (Opcional) Transfiera la columna a un nuevo tubo de recolección y gire a 10,000-16,000 × g durante 2 min.
- Transfiera la columna a un tubo limpio libre de ARNasa y agregue una cantidad adecuada de NFH2O.
- Girar a 10.000-16.000 × g durante 1 min.
3. Análisis de los datos de secuenciación
NOTA: Para crear modelos de estructura secundaria de ARN a partir de los datos de secuenciación DMS-MaP, los archivos .fastq resultantes deben procesarse mediante varios pasos diferentes. Estos pasos se pueden realizar automáticamente utilizando el comando
- Recorte las secuencias del adaptador con TrimGalore o Cutadapt.
- Asigne las lecturas a las secuencias de referencia (formato .fasta) mediante Bowtie2.
- Cuente las lecturas con un software especializado de estructura de ARN (por ejemplo, DREEM14, RNA-Framework17 o similar) y cree perfiles de reactividad.
- (Opcional) Agrupe las lecturas para encontrar conformaciones alternativas de ARN utilizando DREEM14, DRACO 17, DANCE-MaP18 o similares.
- Predecir la estructura mínima de energía libre basada en los perfiles de reactividad utilizando RNAStructure12, ViennaRNA, o similar.
- Visualice la estructura del ARN11 utilizando VARNA (https://varna.lri.fr/) o similar.
NOTA: Para mayor practicidad, software como DREEM (www.rnadreem.org) y RNA-Framework19 incorporan ampliamente los pasos 1--5 en sus tuberías, lo que agiliza el proceso de análisis. Sin embargo, cualquier predicción de estructura debe manejarse con cuidado (por ejemplo, verificando la concordancia de la estructura con los datos20.
Resultados
DMS-MaP in vitro específico del gen
Para estudiar el 5'UTR del SARS2, los primeros 300 pb del virus se ordenaron como una secuencia gBlock, junto con tres cebadores. Estos incluyeron dos cebadores para propagar el fragmento ("FW" y "RV") a través de PCR, así como uno para conectar el promotor T7 ("FW-T7"). Estas secuencias se pueden ver en la Tabla 1.
| Nombre | Secuencia (5'->3') |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
Tabla 1: Secuencia de cebadores para DMS-MaP RT-PCR de SARS-CoV2 5'UTR. Aquí, FW-T7 y RV son necesarios para generar una plantilla de ADN para la transcripción in vitro , el RV se usa en la transcripción inversa y el par de cebadores FW-RV se usa en la posterior amplificación por PCR del ADNc. Los cebadores se remontan al comienzo del genoma del SARS-CoV2 (FW) y la secuencia justo aguas abajo de la región de interés. Abreviaturas: DMS-MaP = Perfil mutacional con secuenciación utilizando sulfato de dimetilo; RT-PCR = reacción en cadena de la polimerasa con transcriptasa inversa; SARS-CoV2 = síndrome respiratorio agudo grave-coronavirus 2; UTR = región no traducida; RV = cebador inverso; FW = cebador delantero.
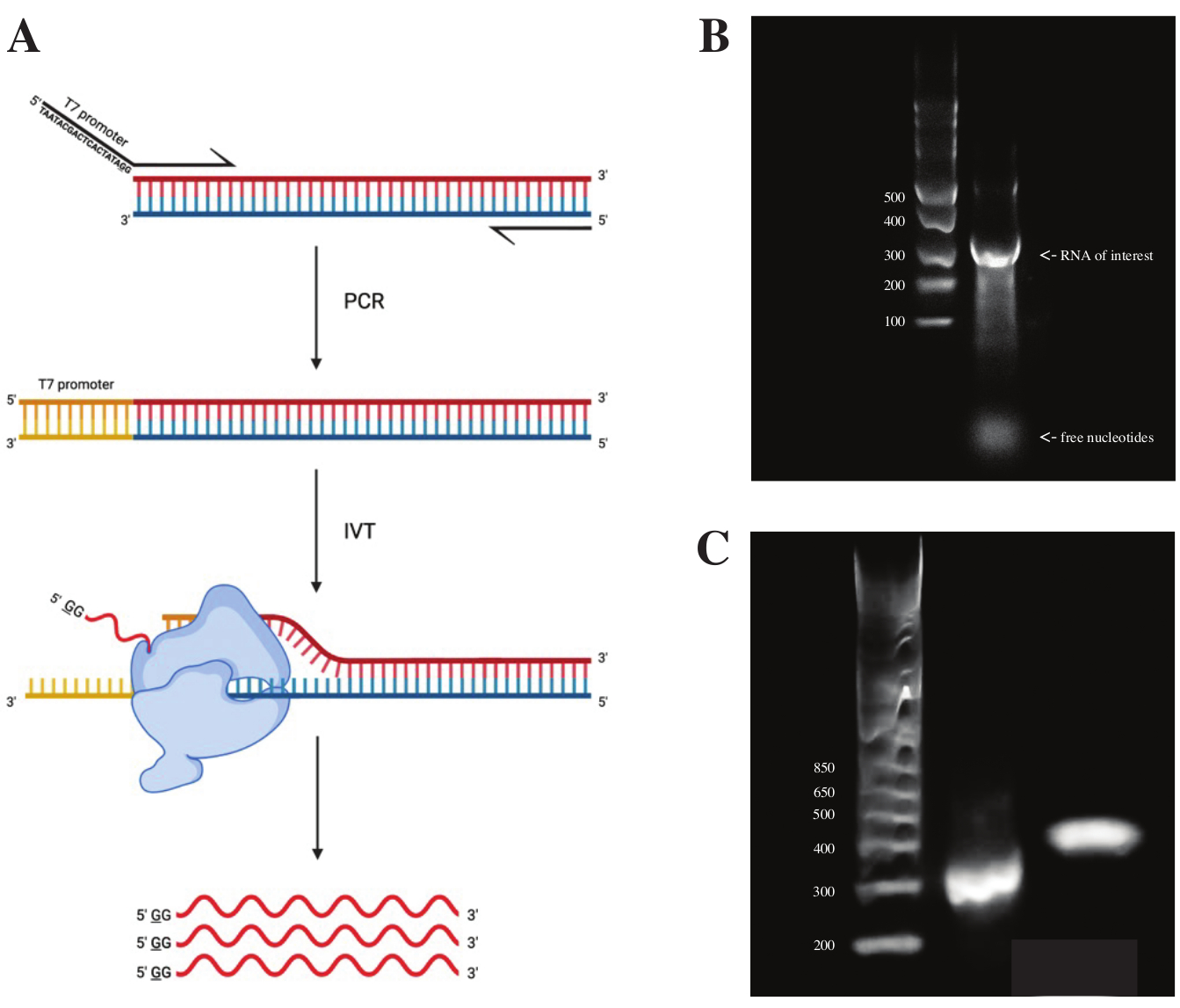
Para generar ARN a partir del fragmento gBlock, la secuencia del promotor de la polimerasa T7 se unió utilizando PCR superpuesta utilizando la premezcla de PCR de acuerdo con el esquema visto en la Figura 2A. A partir del fragmento alargado, el ARN se generó utilizando el kit de transcripción T7. La plantilla de ADN se digirió posteriormente utilizando la DNasa y el ARN aislados utilizando columnas RNA Clean & Concentrator.
El control de calidad de la transcripción in vitro se realizó ejecutando el producto de ARN en un gel de agarosa al 1% junto con una escalera de ARNs. Como solo había una banda visible, se realizó un sondaje DMS in vitro y RT-PCR (ver Figura 2B).
Para verificar el éxito de la reacción de PCR, la muestra se ejecutó en un gel de agarosa al 2% utilizando una escalera de dsDNA. Después de la indexación, la banda debe correr ~ 150 pb más alto en el mismo gel, teniendo en cuenta el tamaño de los cebadores de indexación.

Figura 2: Transcripción in vitro de la plantilla de ADN. (A) Para transcribir in vitro una plantilla de ADN que aún no tiene un promotor intrínseco de ARN polimerasa, la plantilla debe adjuntarse primero por PCR superpuesta. Esto se hace mediante el uso de un cebador directo, que incluye la secuencia TAATACGACTCACTATAGG (en el caso de la ARN polimerasa T7) aguas arriba de las primeras bases que se superponen con el fragmento deseado. La base subrayada aquí simboliza el sitio de inicio de la transcripción de la polimerasa. Una vez que el promotor se ha unido al fragmento de dsDNA, puede ser transcrito por la polimerasa T7. Es importante destacar que la polimerasa utiliza la hebra opuesta a la secuencia promotora mencionada como plantilla (azul), creando efectivamente ARN idéntico a la secuencia inmediatamente posterior de la secuencia promotora indicada (rojo). (B) Un gel de agarosa al 1% con una escalera de ARNs ssn (carril 1) y el producto de ARN transcrito in vitro a 300 nt (carril 2). (C) Un gel de agarosa al 2% con GeneRuler 1 kb más Ladder (carril 1), el producto de PCR después de RT-PCR funcionando a 300 pb (carril 2) y el fragmento indexado después de la preparación de la biblioteca funcionando a 470 pb (carril 3). Abreviaturas: RT-PCR = reacción en cadena de la polimerasa con transcriptasa inversa; DMS = sulfato de dimetilo; nt = nucleótidos; dsDNA = ADN bicatenario; ssRNA = ARN monocatenario. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Genoma completo in vivo DMS-MaP utilizando células infectadas por virus
Antes del tratamiento con DMS, las células HCT-8 estaban infectadas con OC43. Cuando se observó un efecto citopático (CPE) 4 días después de la infección (dpi) (como se ve en la Figura 3A), estas células fueron tratadas, y el ARN fue extraído y ribodepleto. Al ejecutar el ARN total en un gel de agarosa, se observaron dos bandas brillantes, que representan las subunidades 40S y 60S del ribosoma, que constituyen aproximadamente el 95% de la masa total de ARN (ver Figura 3B). Cuando la extracción de ARN no tuvo éxito o se degradó (por ejemplo, por múltiples ciclos de congelación-descongelación), los productos de degradación de ARN fueron visibles en la parte inferior del gel (ver Figura 3C, segundo carril). Además, después del agotamiento del ARNr, las dos bandas brillantes desaparecieron, dejando una mancha en el carril (ver Figura 3C, tercer carril). Finalmente, después de la preparación de la biblioteca, las muestras tenían distribuciones de tamaño variables y se mostraron como un frotis en el gel PAGE final. La banda se extirpó entre 200 nucleótidos (nt) y 500 nt, de acuerdo con la ejecución de secuenciación de extremo pareado de 150 x 150 planificada para analizar estas bibliotecas. Lo más importante es que los dímeros adaptadores que funcionan a ~150 nt se separaron (consulte la Figura 3D).

Figura 3: Puntos de control de DMS-MaP in vivo con células infectadas por virus. (A) Imagen de microscopía óptica de células HCT-8 infectadas por virus, 4 días dpi. Para obtener el mayor rendimiento posible de ARN viral a partir del ARN total y minimizar los efectos adversos debidos a la muerte celular, se debe agregar DMS cuando comienza CPE o incluso antes de eso, como se ve en la imagen. (B) Un gel de agarosa al 1% con seis muestras de 1 μg de ARN total. En cada carril, dos bandas brillantes, que representan las subunidades 40S y 60S, son visibles, ya que el ARN ribosómico constituye ~ 95% del ARN total. Nota: El tratamiento con DMS en la célula causa cierta fragmentación y manchamiento de ARN, pero las dos bandas de ARNr aún deben ser visibles. La fragmentación leve después de la modificación se tolera porque la información que contiene la marca de metilación se genera e informa sobre la estructura del ARN durante la incubación del DMS mientras las células aún están vivas. (C) Un gel de agarosa al 1% de GeneRuler 1 kb más marcador de ADN de escalera (carril 1) ARN total previamente almacenado a -80 °C durante 6 meses (carril 2) y ARN ribodepleto (carril 3). Cuando se almacena ARN durante mucho tiempo con varios ciclos de congelación-descongelación, el ARN comienza a degradarse y posiblemente no debe usarse para experimentos de sondeo. Además, después de ribodepurar el ARN total, las dos bandas brillantes, que representan las subunidades 40S y 60S del ribosoma, se desvanecen y comienza a aparecer un frotis de los ARN residuales. (D) Un gel PAGE de GeneRuler 1 kb más marcador de ADN de escalera (carril 1) y una muestra de biblioteca de ARN preparado para el genoma completo. El gel debe extirparse en función de las necesidades de secuenciación. Para una ejecución de secuenciación de extremo pareado que abarca 150 ciclos desde ambos lados, el gel debe extirparse entre 300 pb y 500 pb. Los dímeros adaptadores (que funcionan a 170 pb) deben separarse. Abreviaturas: DMS-MaP = Perfil mutacional con secuenciación utilizando sulfato de dimetilo; dpi = días después de la infección; CPE = efecto citopático. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
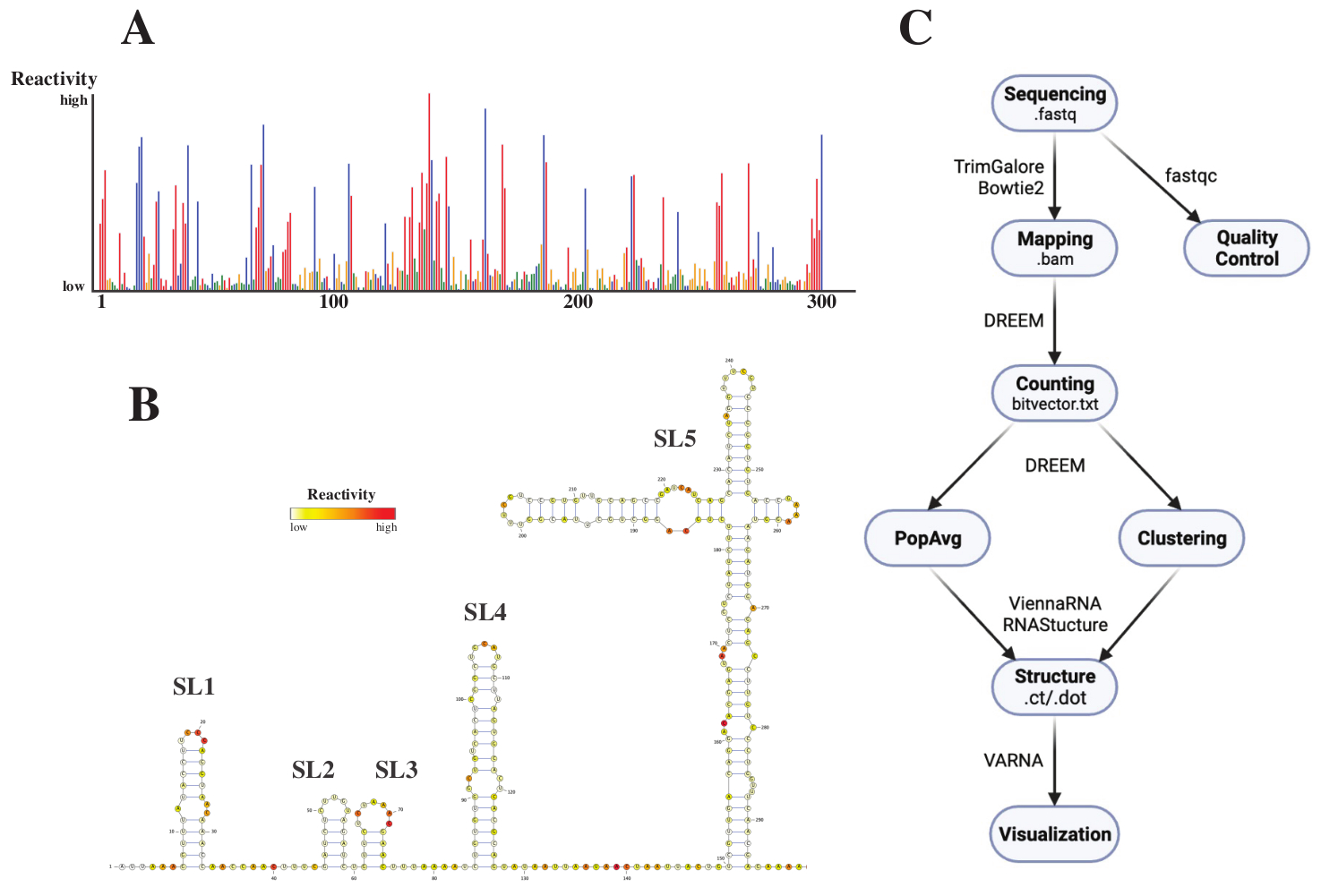
Después de la secuenciación, los archivos .fastq se analizaron enviando un trabajo al servidor web DREEM (http://rnadreem.org/), junto con un archivo de referencia .fasta. La salida generada por el servidor incluye archivos de control de calidad generados por fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) y TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), así como otros archivos de salida que contienen las frecuencias de mutación promedio de la población. Además del diagrama que muestra las frecuencias de mutación con un formato de .html interactivo (ver Figura 4A) y un archivo .csv con las reactivas sin procesar por base y un archivo struct_constraint.txt, legible por varios programas de predicción de estructuras de ARN, esto también incluye un archivo bitvector.txt que informa sobre las mutaciones leídas. A partir de estos, las estructuras promedio de la población se calcularon enviando los archivos .fasta y struct_constraint.txt al servidor web RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Esto utiliza el software ViennaRNA para generar predicciones de estructura basadas en la energía libre mínima, que se pueden ver en línea o descargar en formato ct o Viena. Para generar modelos de estructura de ARN, estos archivos descargables se enviaron a VARNA (https://varna.lri.fr/, consulte la Figura 4B). Por último, los archivos bitvector.txt pueden ser utilizados por la versión estable de DREEM (https://codeocean.com/capsule/6175523/tree/v1) para buscar conformaciones alternativas de ARN. Para obtener buenos modelos de estructura utilizando DREEM, se debe lograr una cobertura de 10,000 lecturas por base; Para la agrupación en clústeres, es posible que se requieran hasta 100.000 lecturas por base. En la figura 4C se puede encontrar una descripción general de todo el flujo de trabajo.

Figura 4: Datos ejemplares obtenidos de experimentos de sondeo químico del SARS-CoV2 5'UTR. (A) Perfil de reactividad de las primeras 300 bases del genoma del SARS-CoV2 coloreado por base (A: rojo, C: azul, U: verde, G: amarillo). Las reactividades brutas se calculan como la frecuencia absoluta de mutación dividida por la cobertura. Las bases con conformación abierta tienen altos valores de reactividad; Las bases que participan en el emparejamiento de bases tienen valores de reactividad bajos. U y G no son modificados por DMS y tienen valores de reactividad bajos, originados por la infidelidad a la polimerasa. Las predicciones se realizaron con el servidor web DREEM. (B) Modelo de estructura del SARS-CoV2 5'UTR predicho a partir de valores de reactividad realizados con VARNA. Las bases con altos valores de reactividad están coloreadas en rojo; Las bases con valores de reactividad bajos están coloreadas en blanco. (C) Flujo de trabajo del análisis DMS-MaP a partir de los archivos .fastq obtenidos de la secuenciación. Estos pueden ser controlados por calidad usando fastqc; las secuencias de adaptadores se recortan con TrimGalore y, a continuación, se asignan de nuevo a una secuencia de referencia mediante Bowtie2. A partir de los archivos .bam obtenidos, DREEM cuenta las mutaciones en cada lectura, creando un mapa de mutación o archivo .bitvector.txt. Estos informan las mutaciones de cada lectura de una manera dependiente de la posición, en función de la cual se pueden crear los perfiles de reactividad promedio de la población. Alternativamente, los vectores de bits se pueden agrupar usando DREEM para buscar conformaciones alternativas de ARN. Por último, los modelos de estructura obtenidos se visualizan utilizando software (por ejemplo, VARNA). Abreviaturas: DMS-MaP = Perfil mutacional con secuenciación utilizando sulfato de dimetilo; SARS-CoV2 = síndrome respiratorio agudo grave-coronavirus 2. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El protocolo aquí describe cómo sondear el ARN in vitro y en células utilizando experimentos de perfil mutacional DMS. Además, da instrucciones sobre cómo preparar bibliotecas para la secuenciación de Illumina para generar datos específicos de genes y analizar los archivos .fastq obtenidos. Además, se pueden utilizar enfoques de biblioteca de todo el genoma. Sin embargo, la RT-PCR específica de genes produce la más alta calidad y los datos más sólidos. Por lo tanto, si se compara entre muestras, es importante asegurarse de que estén preparadas con estrategias de secuenciación idénticas, ya que la generación de la biblioteca causa cierto sesgo. La reproducibilidad siempre debe medirse mediante el uso de réplicas.
Varias precauciones
El ARN es una molécula inestable que es sensible a la degradación tanto a través de temperaturas elevadas como por RNasas. Por lo tanto, se recomiendan medidas especiales: el uso de equipo de protección personal (EPP), material libre de ARNasa e inhibidores de la ARNasa. Lo más importante es que el ARN debe mantenerse en hielo siempre que sea posible. Esto se aplica especialmente al ARN metilado, que es aún más sensible a las altas temperaturas.
Es importante confirmar que la estructura de ARN de interés no es sensible a la concentración de DMS y las condiciones de amortiguación. Los tampones como 100 mM Tris, 100 mM MOPS y 100 mM HEPES a pH 7-7.5 dan una señal alta, pero pueden no ser suficientes para mantener el pH durante la reacción21. A medida que el DMS se hidroliza en agua, lo que disminuye el pH, un tampón fuerte es crítico para mantener un pH neutro durante la reacción de modificación. Se ha demostrado que la adición de bicine ayuda a mantener el pH como ligeramente básico21 , pero da como resultado una baja modificación de DMS en Gs y Us, lo que podría ser informativo, pero debe analizarse por separado debido a la producción de una señal mucho más baja que As y Cs y no se discute más en este protocolo.
En la RT-PCR específica del gen, el ARN modificado se transcribe inversamente en el ADN y se amplifica en fragmentos mediante PCR. Si bien el tamaño del ARN puede ser teóricamente ilimitado, estos fragmentos de PCR no deben exceder una longitud de 400-500 pares de bases (pb) para evitar el sesgo durante la reacción de transcripción inversa. Idealmente, los fragmentos deben estar dentro del alcance de la ejecución de secuenciación (es decir, si la secuenciación se realiza utilizando un programa de secuenciación pareada de 150 x 150 ciclos, un solo fragmento no debe exceder los 300 pb). Cuando se utilizan programas de secuenciación con menos ciclos, los productos de PCR se pueden fragmentar utilizando una dsDNasa. Además, como las secuencias dentro de las secuencias de cebador no contienen ninguna información estructural, los fragmentos deben superponerse cuando el ARN sondeado comprende >1 fragmento. Las reacciones RT pueden contener múltiples cebadores RT para diferentes fragmentos (hasta 10 cebadores RT diferentes). Dependiendo de las secuencias, agrupar los cebadores RT puede hacer que la transcripción inversa sea menos eficiente, pero generalmente funciona bien. Cada reacción de PCR debe realizarse por separado.
Al sondear ARN con DMS, las condiciones experimentales juegan un papel adicional, ya que muchos ARN son termodinámicamente inestables y cambian su conformación en función de factores ambientales como la temperatura. Para evitar irregularidades, las condiciones experimentales deben mantenerse lo más constantes posible, también con respecto a los tiempos de reacción. Las condiciones de amortiguación parecen ser intercambiables hasta cierto punto 17,20,22,23 cuando se mantienen las condiciones básicas —la capacidad amortiguadora y la presencia de iones monovalentes (Na) y divalentes (Mg)— para asegurar un plegamiento adecuado del ARN 24.
Con respecto a la preparación de la biblioteca de ARN modificados, se deben tener en cuenta varios aspectos. Primero, como se mencionó anteriormente, los ARN modificados son menos estables que sus contrapartes no modificadas, lo que significa que podrían requerir la optimización de los tiempos de fragmentación para una distribución óptima del tamaño del fragmento. Además, ciertos kits de preparación de bibliotecas de ARN, así como muchos otros enfoques de RNAseq, utilizan cebadores aleatorios en el kit de transcripción inversa. Esto podría conducir a una menor cobertura de la referencia, especialmente en el 3' de un gen, y, en última instancia, a una profundidad de cobertura insuficiente. Si la cobertura de una determinada región es demasiado baja, podría ser necesario eliminar esas bases de la predicción de la estructura. Además de RT-PCR y kits RNAseq de genoma completo, se pueden utilizar otros enfoques de preparación de bibliotecas. Los protocolos que incluyen la ligadura de adaptadores 3' y/o 5' al ARN son ventajosos cuando se utilizan pequeños fragmentos de ARN o cuando se debe evitar la pérdida de información de sondeo en las regiones del cebador.
Por último, el análisis de los experimentos de sondeo químico siempre debe interpretarse cuidadosamente. Actualmente, no existe un software que prediga la estructura de ARN de cualquier ARN a partir de la secuencia sola con alta precisión. Aunque las restricciones de sondeo químico mejoran en gran medida la precisión, la generación de buenos modelos para ARN largos (>500 nt) sigue siendo un desafío. Estos modelos deben ser probados adicionalmente por otros enfoques y / o mutagénesis. El software de predicción de ARN optimiza el número máximo de pares de bases, penalizando significativamente las conformaciones abiertas, que pueden no representar con precisión el plegamiento del ARN5. Por lo tanto, el modelo de estructura obtenido debe probarse cuantificando el acuerdo de predicción con los datos de sondeo químico subyacentes (por ejemplo, por AUROC) y entre réplicas (por ejemplo, por mFMI), como lo ejemplifican Lan et al.20.
Idealmente, varios experimentos en diferentes sistemas para desafiar el modelo de estructura obtenido deberían usarse para fortalecer la hipótesis. Estos pueden incluir el uso de enfoques in vitro e intracelular, mutaciones compensatorias y diferentes líneas celulares y especies. Además, las reactividades en bruto son a menudo tan o incluso más informativas que las predicciones de estructura, ya que registran la instantánea de la "verdad del terreno" del conjunto de plegamiento de ARN. Como tal, las reactividades en bruto son muy adecuadas e informativas para comparar cambios de estructura entre diferentes condiciones. Es importante destacar que las estructuras de energía libre más bajas calculadas utilizando restricciones de sondeo químico con predicción computacional solo deben usarse como una hipótesis de partida hacia un modelo de estructura completa.
Divulgaciones
Los autores no tienen conflictos de intereses que declarar.
Agradecimientos
Ninguno
Materiales
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
Referencias
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados