
Method Article
Sondando a estrutura do RNA com perfil mutacional de sulfato de dimetilo com sequenciamento in vitro e em células
Neste Artigo
Resumo
O protocolo fornece instruções para modificar o RNA com sulfato de dimetilo para experimentos de perfil mutacional. Inclui sondagem in vitro e in vivo com dois métodos alternativos de preparação de bibliotecas.
Resumo
O papel da estrutura do RNA em praticamente qualquer processo biológico tornou-se cada vez mais evidente, especialmente na última década. No entanto, as abordagens clássicas para resolver a estrutura de RNA, como a cristalografia de RNA ou a crio-EM, não conseguiram acompanhar o campo em rápida evolução e a necessidade de soluções de alto rendimento. O perfil mutacional com sequenciamento usando sulfato de dimetilo (DMS) MaPseq é uma abordagem baseada em sequenciamento para inferir a estrutura de RNA a partir da reatividade de uma base com DMS. DMS metila o nitrogênio N1 em adenosinas e o N3 em citosinas em sua face Watson-Crick quando a base é desemparelhada. A transcrição reversa do RNA modificado com a transcriptase reversa do íntron do grupo II termoestável (TGIRT-III) leva à incorporação das bases metiladas como mutações no cDNA. Ao sequenciar o cDNA resultante e mapeá-lo de volta a um transcrito de referência, as taxas de mutação relativas para cada base são indicativas do "status" da base como emparelhada ou desemparelhada. Embora as reatividades do DMS tenham uma alta relação sinal-ruído tanto in vitro quanto nas células, esse método é sensível ao viés nos procedimentos de manuseio. Para reduzir esse viés, este trabalho fornece um protocolo para o tratamento de RNA com DMS em células e com RNA transcrito in vitro .
Introdução
Desde a descoberta de que o RNA tem propriedades estruturais1,2 e catalíticas3, a importância do RNA e sua função reguladora em uma infinidade de processos biológicos foram gradualmente descobertas. De fato, o efeito da estrutura do RNA na regulação gênica tem ganhado cada vez mais atenção4. Como as proteínas, o RNA tem estruturas primárias, secundárias e terciárias, referindo-se à sequência de nucleotídeos, ao mapeamento 2D das interações de emparelhamento de bases e ao dobramento 3D dessas estruturas emparelhadas de bases, respectivamente. Embora a determinação da estrutura terciária seja fundamental para a compreensão dos mecanismos exatos por trás dos processos dependentes de RNA, a estrutura secundária também é altamente informativa sobre a função do RNA e é a base para novas dobras 3D5.
No entanto, determinar a estrutura do RNA tem sido intrinsecamente desafiador com abordagens convencionais. Enquanto para proteínas, cristalografia, ressonância magnética nuclear (RMN) e microscopia eletrônica criogênica (crio-EM) tornaram possível determinar a diversidade de motivos estruturais, permitindo a predição da estrutura a partir da sequência isolada6, essas abordagens não são amplamente aplicáveis aos RNAs. De fato, os RNAs são moléculas flexíveis com blocos de construção (nucleotídeos) que têm muito mais liberdade conformacional e rotacional em comparação com suas contrapartes de aminoácidos. Além disso, as interações através do emparelhamento de bases são mais dinâmicas e versáteis do que as dos resíduos de aminoácidos. Como resultado, as abordagens clássicas têm sido bem-sucedidas apenas para RNAs relativamente pequenos com estruturas bem definidas e altamente compactas7.
Outra abordagem para determinar a estrutura do RNA é através de sondagem química combinada com sequenciamento de próxima geração (NGS). Essa estratégia gera informações sobre o status de ligação de cada base em uma sequência de RNA (ou seja, sua estrutura secundária). Em resumo, as bases em uma molécula de RNA que não estão envolvidas no emparelhamento de bases são diferencialmente modificadas por pequenos compostos químicos. A transcrição reversa desses RNAs com transcriptases reversas (RTs) especializadas incorpora as modificações no ácido desoxirribonucleico complementar (cDNA) como mutações. Essas moléculas de cDNA são então amplificadas pela reação em cadeia da polimerase (PCR) e sequenciadas. Para obter informações sobre seu "status" como ligado ou não, as frequências de mutação em cada base em um RNA de interesse são calculadas e inseridas em software de previsão de estrutura como restrições8. Com base nas regras do vizinho mais próximo9 e nos cálculos mínimos de energia livre 10, este software gera modelos de estrutura que melhor se ajustam aos dados experimentais obtidos11,12.
O DMS-MaPseq utiliza o DMS, que metila o nitrogênio N1 em adenosinas e o nitrogênio N3 em citosinas em sua face Watson-Crick de maneira altamente específica13. O uso da transcriptase reversa de íntron do grupo II termoestável (TGIRT-III) na transcrição reversa cria perfis mutacionais com relações sinal-ruído sem precedentes, permitindo até mesmo a deconvolução de perfis sobrepostos gerados por duas ou mais conformações alternativas14,15. Além disso, o DMS pode penetrar nas membranas celulares e tecidos inteiros, tornando possível a sondagem dentro de contextos fisiológicos. No entanto, a geração de dados de boa qualidade é um desafio, pois variações no procedimento de manuseio podem afetar os resultados. Portanto, fornecemos um protocolo detalhado para DMS-MaPseq in vitro e in-cell para reduzir o viés e orientar os recém-chegados ao método através das dificuldades que possam encontrar. Especialmente à luz da recente pandemia de SARS-CoV2, dados de alta qualidade sobre vírus de RNA são uma ferramenta importante para estudar a expressão gênica e encontrar possíveis terapêuticas.
Protocolo
NOTA: Consulte a Tabela de Materiais para obter detalhes relacionados a todos os materiais, software, reagentes, instrumentos e células usados neste protocolo.
1. DMS-MaP in vitro específico do gene
- Transcrição in vitro de RNA
- Obter a sequência do RNA de interesse como DNA de fita dupla (ds) (por exemplo, como fragmentos de DNA, plasmídeos ou PCR a partir de DNA pré-existente/genômico). Se a sequência de DNA contiver um promotor de polimerase, pule para a etapa 3.
- Executar a sobreposição de PCR para anexar um promotor de RNA polimerase a montante do fragmento de DNA desejado (primer para frente para a polimerase T7: 5' TAATACGACTCACTATAGG + primeiras bases da sequência alvo 3').
- Transcrever in vitro o fragmento de DNA em RNA. Mantenha sempre o RNA no gelo.
- Digerir o DNA usando uma DNase.
- Isolar o ARN utilizando uma abordagem baseada em colunas (passo 2.4) ou por precipitação de etanol (passo 2.5). Elute em um volume apropriado, esperando um rendimento de ~50 μg.
- Garantir a integridade do RNA, executando-o em um gel de agarose; desnaturar o ARN durante 2-3 min a 70 °C antes de correr.
NOTA: O tampão e a agarose podem conter RNases que degradam o RNA e podem contaminar a amostra de RNA. Géis de agarose pré-moldados já foram utilizados neste laboratório; os resultados (especialmente com RNA) têm sido ambíguos às vezes. Os melhores resultados foram obtidos com agarose ou géis PAGE. - Utilização directa do ARN a -80 °C durante vários meses, a menos que a degradação seja visível após o descongelamento.
- In vitro Modificação do DMS (a 105 mM DMS)
- Preparar uma quantidade suficiente de tampão de redobramento (cacodilato de sódio 0,4 M, pH 7,2, contendo 6 mM MgCl2).
NOTA: Para cada reacção (volume final de 100 μL), adicionar 89 μL de tampão de redobramento. - Para cada reação, transfira 89 μL de tampão redobrável para um tubo designado de 1,5 mL e pré-aqueça a 37 °C em um termoshaker colocado sob um exaustor químico.
NOTA: O DMS é altamente tóxico e deve sempre ser mantido sob um exaustor químico até ser apagado por um agente redutor. - Eluto 1-10 pmol de RNA em 10 μL de água livre de nuclease (NF H2O); transferir para um tubo de PCR.
- Incubar num termociclador a 95 °C durante 1min para desnaturar o ARN.
- Coloque em um bloco de gelo imediatamente para evitar dobrar incorretamente.
- Adicione a amostra de RNA ao tubo designado com tampão de redobramento a 37 °C, misture bem e incube por 10-20 min para reaquecer o RNA.
NOTA: A maioria dos RNAs se dobrará na ordem de milissegundos a segundos, embora existam exceções16. - Adicione 1 μL de 100% (10,5 M) de DMS à amostra de RNA e incube por 5 minutos enquanto agita a 800-1.400 rotações por minuto (rpm).
NOTA: Tremer (ou outros meios de mistura) nesta etapa é crucial, pois o DMS é hidrofóbico e pode não se dissolver totalmente no buffer de redobramento. Desvios nos tempos de reação podem afetar a reprodutibilidade das reatividades do DMS. Para minimizar o erro de pipetagem, o DMS pode ser dissolvido em etanol a 100% antes de adicioná-lo à amostra se uma concentração final de 1% (105 mM) de DMS for mantida. Para um controle não tratado, o DMS pode ser substituído por dimetilsulfóxido (DMSO) ou água. - Após 5 min de tempo de reação, tempere com 60 μL de β-mercaptoetanol (BME) a 100%, misture bem e coloque imediatamente o RNA no gelo.
NOTA: O RNA pode ser removido com segurança do exaustor após a extinção da reação com BME para limpá-lo. No entanto, a exposição direta da BME ao ambiente ainda deve ser evitada devido ao seu cheiro forte e propriedades irritantes. - Limpar o ARN por precipitação de acetato-etanol de sódio (ver passo 2.5) ou uma abordagem baseada em colunas (ver passo 2.6) e eluir em 10 μL de água.
- Quantifique o RNA usando um espectrofotômetro.
- Uso direto do armazenamento do RNA modificado a -80 °C.
NOTA: O armazenamento a longo prazo deve ser evitado, uma vez que o ARN é menos estável após o tratamento com DMS.
- Preparar uma quantidade suficiente de tampão de redobramento (cacodilato de sódio 0,4 M, pH 7,2, contendo 6 mM MgCl2).
- RT-PCR gene-específico do RNA modificado
NOTA: Consulte a Figura 1 para obter a configuração RT-PCR dos fragmentos tratados com DMS.- Elute 100 ng de RNA modificado em 10 μL de nuclease-free (NF) H2O. Transferência para um tubo de PCR.
- Ao tubo, adicione 4 μL de 5x primeiro tampão de fita (FSB), 1 μL de mistura de dNTP (10 mM cada), 1 μL de 0,1 M de ditiotreitol (DTT) (evite ciclos de congelamento-descongelamento), 1 μL de inibidor de RNase, 1 μL de primer reverso de 10 μM (primer único ou um conjunto de primers) e 1 μL de TGIRT III.
NOTA: Para um conjunto de iniciadores, não adicione 1 μL de 10 μM de cada primer diretamente ao RT; em vez disso, misture os primers primeiro e adicione 1 μL da mistura (na concentração total do primer de 10 μM). - Incubar a 57 °C durante 30 min a 1,5 h (normalmente, 30 min é suficiente para fazer um produto de 500 nt) num termociclador.
- Adicionar 1 μL de 4 M de NaOH, misturar por pipetagem e incubar a 95 °C durante 3 min para degradar o ARN.
NOTA: Esta etapa é crucial, pois libera TGIRT do cDNA degradando o RNA. Se ignorada, a PCR a jusante pode ser afetada. - Limpar utilizando uma abordagem baseada em colunas (ver passo 2.6) que remova suficientemente os primers e eluir em 10 μL de NF H2O.
- Amplificar o cDNA por PCR usando 1 μL do produto de transcrição reversa por 25 μL da reação com um kit de PCR projetado para equilibrar o rendimento e a fidelidade.
NOTA: Os primers devem ter uma temperatura de fusão de ~60 °C. - Execute 2 μL do produto de PCR em um gel de agarose ou um gel de agarose pré-moldado para verificar o sucesso da PCR.
- Idealmente, apenas uma banda deve aparecer após o PCR. Em caso afirmativo, limpe a reação usando uma abordagem baseada em colunas. Se houver bandas alternativas, use a reação de PCR restante para extirpar a banda correta do gel. Eluir em um volume suficientemente pequeno (por exemplo, 10 μL).
- Quantifique os fragmentos extraídos usando um espectrofotômetro.
- Indexe os fragmentos de dsDNA para sequenciamento usando uma abordagem adequada à plataforma de sequenciamento desejada.

Figura 1: Configuração experimental para a RT-PCR de grandes fragmentos tratados com DMS. Ao realizar a transcrição reversa em um RNA modificado, as modificações na sequência para a qual o primer recozi não serão registradas. Assim, quando os fragmentos excedem 400-500 pb de comprimento, fragmentos sobrepostos nas regiões do primer precisam ser projetados, como exemplificado aqui. O comprimento dos fragmentos depende das necessidades de sequenciamento. Ao usar o sequenciamento de 150 ciclos de extremidade emparelhada, os fragmentos não devem exceder 300 pb. Abreviaturas: RT-PCR = reação em cadeia da polimerase de transcrição reversa; DMS = sulfato de dimetilo. Por favor, clique aqui para ver uma versão maior desta figura.
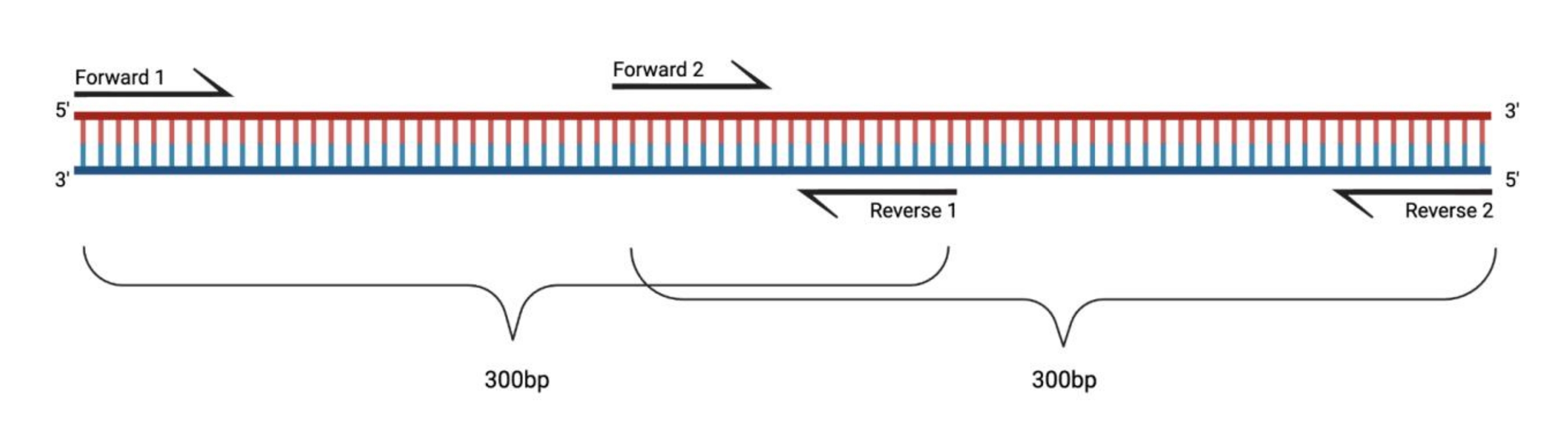{kind=link}
2. DMS-MaP de genoma completo usando células infectadas por vírus
NOTA: Nas células, o tratamento com DMS também pode ser combinado com a abordagem de amplificação específica do gene descrita acima. A biblioteca de todo o genoma requer uma enorme profundidade de sequenciamento para alcançar a cobertura total em um único gene. No entanto, se os RNAs virais constituírem uma fração significativa do RNA ribodepletado após a extração, o sequenciamento do genoma completo seria apropriado. Além disso, outros métodos de enriquecimento podem ser combinados com o método de geração de biblioteca de genoma inteiro.
- Tratamento DMS
- Crescer células infectadas com o vírus até o estágio desejado da infecção.
- Transfira o recipiente de células para um exaustor dedicado que seja apropriado para lidar com vírus no nível de biossegurança exigido e os vapores químicos gerados por agentes como o DMS.
- Adicione um volume de DMS de 2,5% ao meio de cultura e sele o recipiente (normalmente uma placa de 10 cm) com parafilme.
NOTA: É fácil sub-modificar e sobre-modificar com DMS. Ao adicionar DMS diretamente às células, é muito importante misturar bem. Alternativamente, pré-aqueça o novo meio em um tubo cônico de 50 mL a 37 °C e adicione o DMS agitando vigorosamente vigorosamente. Decantar o meio gasto nas células e pipetar lentamente no meio contendo DMS. - Transferir para uma incubadora de 37 °C durante 5 min.
NOTA: Dependendo da quantidade de tempo que leva para lidar com o DMS fora da incubadora, é possível que 5 minutos levem a modificações excessivas. Evite o tempo de adicionar o DMS à incubação para ≤1 min. Se realizar o experimento pela primeira vez, recomenda-se fazer uma titulação DMS e variar o tempo de incubação (entre 3 min e 10 min) para encontrar a taxa de modificação ideal e garantir que os resultados sejam robustos em uma janela de concentrações. - Pipete cuidadosamente o meio contendo DMS (em resíduos químicos apropriados) e adicione suavemente 10 mL de tampão de parada (PBS com 30% de BME [por exemplo, 3 mL de BME e 7 mL de PBS]).
NOTA: A adição de DMS e BME pode levantar as células da placa se as células não forem fortemente aderentes. Se as células estiverem levantando, elas podem ser tratadas como células de suspensão - em vez de remover o meio contendo DMS, adicione o tampão de parada diretamente e raspe as células com DMS e BME em um tubo cônico de 50 mL. Pellet as células por centrifugação por 3 min a 3.000 × g; Certifique-se de se livrar de qualquer DMS residual, que pode pellet sob as células em gotículas grandes. Recomenda-se uma etapa de lavagem extra em 30% BME se o meio DMS não puder ser removido inicialmente. - Raspe as células e transfira-as para um tubo cônico de 15 mL.
- Pellet por centrifugação a 3.000 × g durante 3 min.
- Retire o sobrenadante e lave 2x com 10 mL de PBS.
- Remova cuidadosamente o máximo de PBS residual possível.
- Dissolva o pellet em uma quantidade apropriada do reagente de isolamento de RNA (por exemplo, 3 mL para um frasco de cultura T75, 1 mL para uma placa de 10 cm).
NOTA: Quantidades insuficientes do reagente podem afetar o rendimento de RNA.
- Extração de RNA e depleção de RNA ribossômico (rRNA)
- Para 1 mL de células homogeneizadas no reagente de isolamento de RNA, adicione 200 μL de clorofórmio, vórtice por 15-20 s até ficar rosa brilhante e, em seguida, incube por até 3 min até que a separação de fases seja visível.
NOTA: A fase lipídica rosa deve se estabelecer na parte inferior. Se este não for o caso, o tempo de vórtice provavelmente foi insuficiente. - Gire à velocidade máxima (~ 20.000 × g) durante 15 min a 4 °C.
- Transfira a fase aquosa superior para um novo tubo.
- Limpar o ARN por precipitação de acetato-etanol de sódio (ver passo 2.5) ou uma abordagem baseada em colunas (ver passo 2.6) e eluir num volume suficiente de NF H2O.
- Verifique a integridade do RNA em um gel de agarose. Procure duas bandas correspondentes às duas subunidades ribossomais.
- Esgotar os rRNAs usando a abordagem preferida e eluir em um volume adequado (tipicamente 20-50 μL) de NF H2O.
NOTA: Para aplicações a jusante, ~500 ng de RNA total é sugerido em um volume de 8 μL. RNAs não ribossômicos normalmente compõem apenas 5%-10% do RNA total. - Quantificar usando um espectrofotômetro.
- Para 1 mL de células homogeneizadas no reagente de isolamento de RNA, adicione 200 μL de clorofórmio, vórtice por 15-20 s até ficar rosa brilhante e, em seguida, incube por até 3 min até que a separação de fases seja visível.
- Geração de bibliotecas
- Use RT-PCR específico para genes ou outras abordagens para gerar bibliotecas15. Se estiver a utilizar hexâmeros aleatórios para a escorvamento, adicione um passo de incubação a um baixo Tm (37-42 °C) para permitir o recozimento do hexâmero.
NOTA: Os kits de geração de bibliotecas padrão também podem ser usados substituindo a enzima RT por TGIRT e alterando a temperatura RT para 57 °C.
- Use RT-PCR específico para genes ou outras abordagens para gerar bibliotecas15. Se estiver a utilizar hexâmeros aleatórios para a escorvamento, adicione um passo de incubação a um baixo Tm (37-42 °C) para permitir o recozimento do hexâmero.
- Limpeza de RNA baseada em coluna usando as colunas RNA Clean & Concentrator
NOTA: Todas as etapas devem ser realizadas à temperatura ambiente.- Adicionar NF H2O ao tubo de amostra para o levar a um volume de 50 μL.
- Adicionar 100 μL de tampão de ligação e 150 μL de etanol a 100% à amostra.
- Misture e transfira para uma coluna de rotação.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- Adicione 400 μL de tampão de preparação de RNA.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- Adicione 700 μL de tampão de lavagem de RNA.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- Adicione 400 μL de tampão de lavagem de RNA.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- (Opcional) Transfira a coluna para um novo tubo de coleta e gire a 10.000-16.000 × g por 2 min.
- Transfira a coluna para um tubo limpo e isento de RNAse e adicione uma quantidade apropriada de NF H2O.
- Gire a 10.000-16.000 × g por 1 min.
- Extração ácida de RNA fenol-clorofórmio.
- Adicionar um volume igual de fenol ácido:clorofórmio:álcool isoamílico.
- Vórtice completamente e centrífuga a 14.000 × g por 5 min.
- Se não houver separação de fase, adicione 20 μL de NaCl 2 M e repita a centrifugação.
- Transfira a fase aquosa para um novo tubo.
- Adicionar 500 μL de isopropanol e 2 μL de coprecipitante.
- Misturar e incubar no RT por 3 min; em seguida, incubar a -80 °C durante a noite.
- Pellet o ARN por centrifugação à velocidade máxima (~ 20.000 × g) durante 30 min a 4 °C.
- Lave o pellet com 200 μL de etanol gelado a 70%.
- Gire na velocidade máxima (~ 20.000 × g) por 5 min; descarte o fluxo.
- Ressuspender o pellet na quantidade adequada de NF H2O.
- Limpeza de cDNA baseada em colunas usando as colunas Oligo Clean e Concentrator
NOTA: Todas as etapas devem ser realizadas à temperatura ambiente.- Adicionar NF H2O ao tubo de amostra para o levar a um volume de 50 μL.
- Adicionar 100 μL de tampão de ligação e 400 μL de etanol a 100%.
- Misture e transfira para uma coluna de rotação.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- Adicionar 750 μL de tampão de lavagem de ADN.
- Gire a 10.000-16.000 × g por 30 s; descarte o fluxo.
- (Opcional) Transfira a coluna para um novo tubo de coleta e gire a 10.000-16.000 × g por 2 min.
- Transfira a coluna para um tubo limpo sem RNAse e adicione uma quantidade apropriada de NF H2O.
- Gire a 10.000-16.000 × g por 1 min.
3. Análise dos dados de sequenciamento
Observação : para criar modelos de estrutura secundária de RNA a partir dos dados de sequenciamento DMS-MaP, os arquivos .fastq resultantes devem ser processados por várias etapas diferentes. Essas etapas podem ser executadas automaticamente usando o
- Corte as sequências do adaptador com TrimGalore ou Cutadapt.
- Mapeie as leituras para as sequências de referência (formato .fasta) usando Bowtie2.
- Conte as leituras com um software especializado em estrutura de RNA (por exemplo, DREEM14, RNA-Framework17 ou similar) e crie perfis de reatividade.
- (Opcional) Agrupe as leituras para encontrar conformações alternativas de RNA usando DREEM14, DRACO 17, DANCE-MaP18 ou similar.
- Preveja a estrutura mínima de energia livre com base nos perfis de reatividade usando RNAStructura12, ViennaRNA ou similar.
- Visualize a estrutura do RNA11 usando VARNA (https://varna.lri.fr/) ou similar.
NOTA: Para fins práticos, softwares como o DREEM (www.rnadreem.org) e o RNA-Framework19 incorporam amplamente as etapas de 1 a 5 em seus pipelines, o que simplifica o processo de análise. No entanto, qualquer previsão de estrutura deve ser tratada com cuidado (por exemplo, verificando a concordância da estrutura com os dados20.
Resultados
DMS-MaP in vitro específico para genes
Para estudar os 5'UTR do SARS2, os primeiros 300 pb do vírus foram ordenados como uma sequência de gBlock, juntamente com três primers. Estes incluíram dois primers para propagar o fragmento ("FW" e "RV") via PCR, bem como um para anexar o promotor T7 ("FW-T7"). Essas sequências podem ser observadas na Tabela 1.
| Nome | Sequência (5'->3') |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
Tabela 1: Sequência de primer para DMS-MaP RT-PCR de SARS-CoV2 5'UTR. Aqui, FW-T7 e RV são necessários para gerar um molde de DNA para transcrição in vitro , o VD é usado na transcrição reversa e o par de iniciadores FW-RV é usado na amplificação subsequente por PCR do cDNA. Os primers recoziram até o início do genoma do SARS-CoV2 (FW) e a sequência a jusante da região de interesse. Abreviaturas: DMS-MaP = Perfil mutacional com sequenciamento usando sulfato de dimetila; RT-PCR = reação em cadeia da polimerase de transcrição reversa; SARS-CoV2 = síndrome respiratória aguda grave-coronavírus 2; UTR = região não traduzida; VD = primer reverso; FW = primer para a frente.
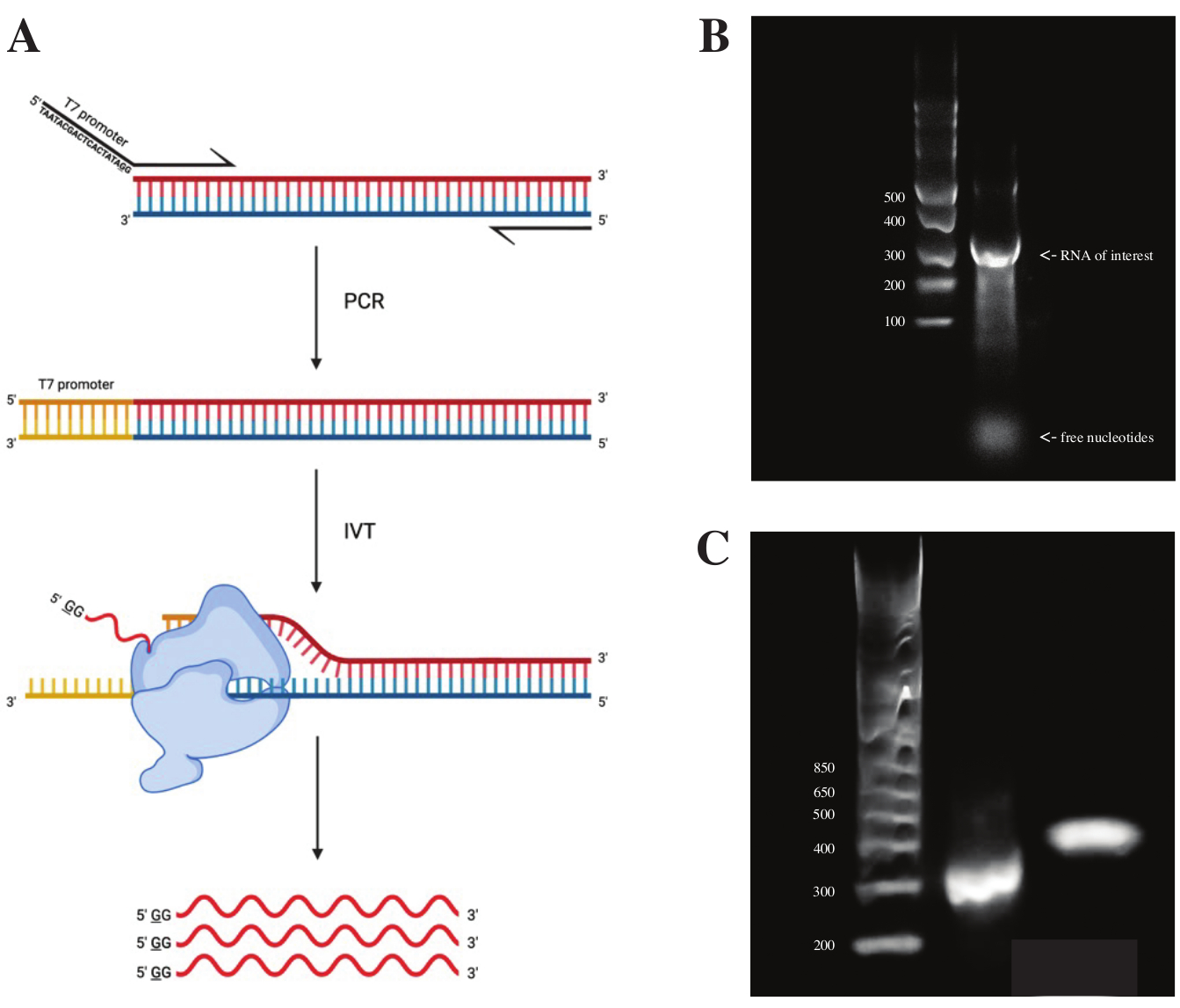
Para gerar RNA a partir do fragmento gBlock, a sequência do promotor de polimerase T7 foi fixada por PCR de sobreposição utilizando a pré-mistura de PCR de acordo com o esquema observado na Figura 2A. A partir do fragmento alongado, o RNA foi gerado utilizando o T7 Transcription Kit. O molde de DNA foi posteriormente digerido usando a DNase e o RNA isolados usando colunas de RNA Clean & Concentrator.
O controle de qualidade da transcrição in vitro foi feito executando o produto de RNA em um gel de agarose a 1% ao lado de uma escada de ssRNA. Como havia apenas uma banda visível, foram realizadas sondagens in vitro de DMS e RT-PCR (ver Figura 2B).
Para verificar o sucesso da reação de PCR, a amostra foi executada em gel de agarose a 2% usando uma escada de dsDNA. Após a indexação, a banda deve correr ~150 pb mais alta no mesmo gel, contabilizando o tamanho dos primers de indexação.

Figura 2: Transcrição in vitro do molde de DNA. (A) Para transcrever in vitro um molde de DNA que ainda não possui um promotor intrínseco de RNA polimerase, o molde deve ser anexado por sobreposição de PCR primeiro. Isso é feito usando um primer avançado, que inclui a sequência TAATACGACTCACTATAGG (no caso da RNA polimerase T7) a montante das primeiras bases sobrepostas ao fragmento desejado. A base sublinhada aqui simboliza o local de início da transcrição da polimerase. Uma vez que o promotor tenha se ligado ao fragmento de dsDNA, ele pode ser transcrito pela polimerase T7. É importante ressaltar que a polimerase usa a fita oposta à sequência promotora mencionada como molde (azul), criando efetivamente RNA idêntico à sequência imediatamente a jusante da sequência promotora indicada (vermelho). (B) Um gel de agarose a 1% com uma escada de ssRNA (faixa 1) e o produto de RNA transcrito in vitro a 300 nt (faixa 2). (C) Um gel de agarose a 2% com GeneRuler 1 kb mais Ladder (faixa 1), o produto de PCR após RT-PCR funcionando a 300 pb (pista 2) e o fragmento indexado após a preparação da biblioteca funcionando a 470 pb (pista 3). Abreviaturas: RT-PCR = reação em cadeia da polimerase de transcrição reversa; DMS = sulfato de dimetilo; nt = nucleotídeos; dsDNA = DNA de fita dupla; ssRNA = RNA de fita simples. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Genoma completo in vivo DMS-MaP usando células infectadas por vírus
Antes do tratamento com DMS, as células HCT-8 estavam infectadas com OC43. Quando um efeito citopático (ECP) foi observado 4 dias após a infecção (dpi) (como visto na Figura 3A), essas células foram tratadas, e o RNA foi extraído e ribodepleto. Ao executar o RNA total em um gel de agarose, duas bandas brilhantes foram visíveis, representando as subunidades 40S e 60S do ribossomo, que compõem aproximadamente 95% da massa total de RNA (ver Figura 3B). Quando a extração de RNA não foi bem-sucedida ou foi degradada (por exemplo, por múltiplos ciclos de congelamento-descongelamento), os produtos de degradação de RNA foram visíveis no fundo do gel (ver Figura 3C, segunda faixa). Além disso, após o esgotamento do rRNA, as duas bandas brilhantes desapareceram, deixando uma mancha na pista (ver Figura 3C, terceira faixa). Finalmente, após o preparo da biblioteca, as amostras apresentaram distribuições de tamanho variáveis e foram mostradas como um esfregaço no gel PAGE final. A banda foi excisada entre 200 nucleotídeos (nt) e 500 nt, de acordo com a execução de sequenciamento final pareado de 150 x 150 planejada para analisar essas bibliotecas. Mais importante ainda, os dímeros do adaptador que funcionavam a ~150 nt foram separados (ver Figura 3D).

Figura 3: Pontos de verificação de DMS-MaP in vivo com células infectadas por vírus. (A) Imagem de microscopia de luz de células HCT-8 infectadas pelo vírus, 4 dias dpi. Para obter o maior rendimento possível de RNA viral a partir do RNA total, minimizando os efeitos adversos devido à morte celular, o DMS deve ser adicionado quando a ECP começa ou mesmo antes disso, como visto na imagem. (B) Um gel de agarose a 1% com seis amostras de 1 μg de ARN total. Em cada pista, duas bandas brilhantes, responsáveis pelas subunidades 40S e 60S, são visíveis, já que o RNA ribossômico compõe ~95% do RNA total. Nota: O tratamento com DMS celular causa alguma fragmentação e mancha de RNA, mas as duas bandas de rRNA ainda devem estar visíveis. A fragmentação leve pós-modificação é tolerada porque a informação contendo a marca de metilação é gerada e relata a estrutura do RNA durante a incubação do DMS enquanto as células ainda estão vivas. (C) Um gel de Agarose a 1% de GeneRuler 1 kb mais marcador de DNA de escada (faixa 1) RNA total previamente armazenado a -80 °C por 6 meses (pista 2) e RNA ribodepletado (faixa 3). Ao armazenar RNA por um longo tempo com vários ciclos de congelamento-descongelamento, o RNA começa a se degradar e, possivelmente, não deve ser usado para experimentos de sondagem. Além disso, após o ribodepleto do RNA total, as duas bandas brilhantes, responsáveis pelas subunidades 40S e 60S do ribossomo, desaparecem e um esfregaço dos RNAs residuais começa a aparecer. (D) Um gel PAGE de GeneRuler 1 kb mais marcador de DNA de escada (faixa 1) e uma amostra de biblioteca de RNA preparado para todo o genoma. O gel deve ser extirpado com base nas necessidades de sequenciamento. Para uma sequência de sequenciamento emparelhada que abranja 150 ciclos de ambos os lados, o gel deve ser excisado entre 300 pb e 500 pb. Os dímeros adaptadores (funcionando a 170 pb) devem ser separados. Abreviaturas: DMS-MaP = Perfil mutacional com sequenciamento usando sulfato de dimetila; dpi = dias pós-infecção; ECP = efeito citopático. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
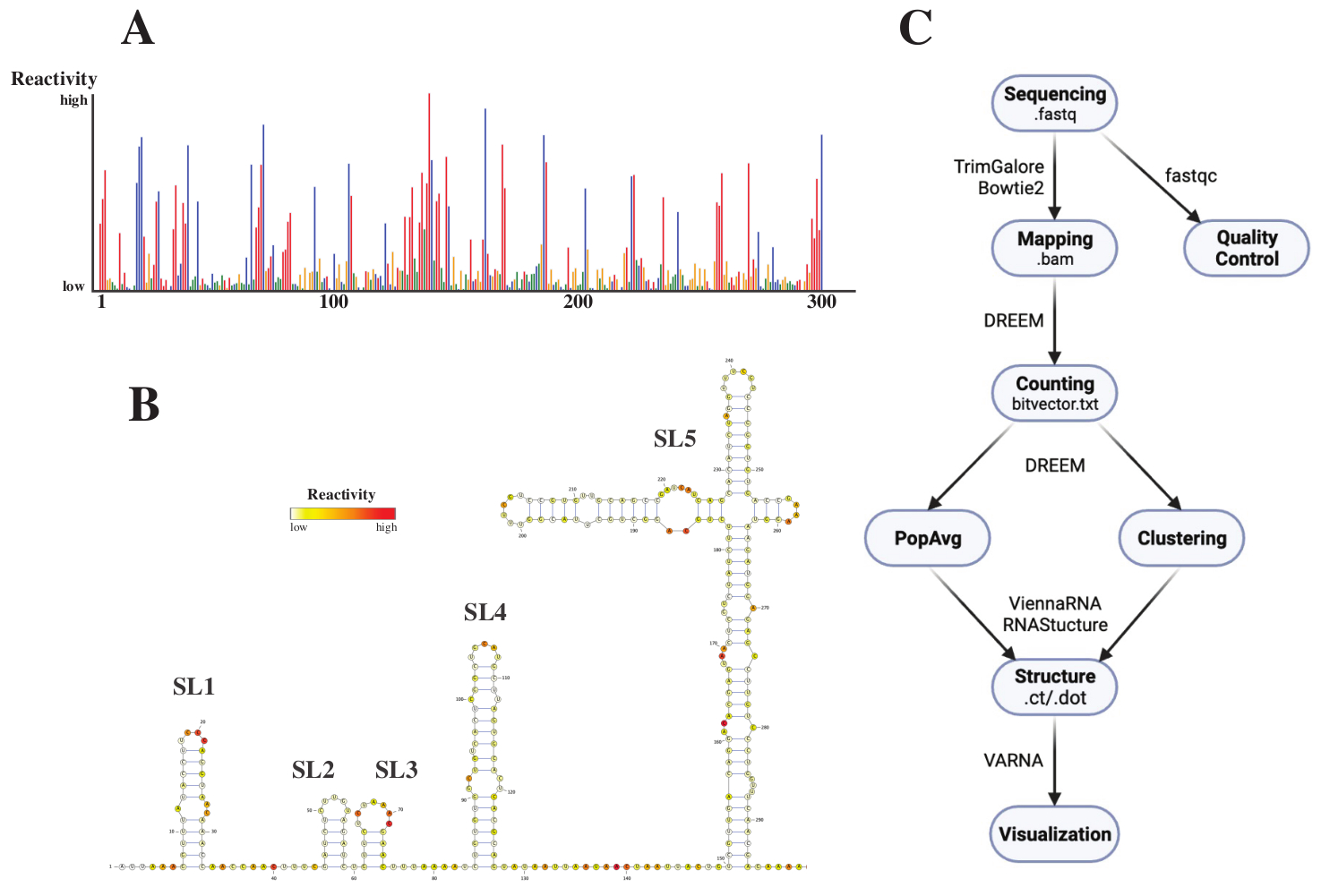
Após o sequenciamento, os arquivos .fastq foram analisados submetendo-se um trabalho ao servidor web DREEM (http://rnadreem.org/), juntamente com um arquivo de referência .fasta. A saída gerada pelo servidor inclui arquivos de controle de qualidade gerados pelo fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) e TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), bem como outros arquivos de saída contendo as frequências médias de mutação da população. Além do diagrama mostrando as frequências de mutação com um formato de .html interativo (veja a Figura 4A) e um arquivo de .csv com as reativites brutas por base e um arquivo de struct_constraint.txt, legível por vários softwares de previsão de estrutura de RNA, isso também inclui um arquivo de vetor de bits.txt relatando as mutações por leitura. A partir destes, as estruturas médias da população foram calculadas submetendo-se os arquivos .fasta e struct_constraint.txt ao servidor web RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Isso usa o software ViennaRNA para gerar previsões de estrutura com base na energia livre mínima, que pode ser visualizada on-line ou baixada no formato ct ou Viena. Para gerar modelos de estrutura de RNA, esses arquivos para download foram submetidos ao VARNA (https://varna.lri.fr/, consulte a Figura 4B). Por fim, os arquivos bitvector.txt podem ser usados pela versão estável do DREEM (https://codeocean.com/capsule/6175523/tree/v1) para procurar conformações alternativas de RNA. Para obter bons modelos de estrutura usando o DREEM, uma cobertura de 10.000 leituras por base deve ser alcançada; para clustering, podem ser necessárias até 100.000 leituras por base. Uma visão geral de todo o fluxo de trabalho pode ser encontrada na Figura 4C.

Figura 4: Dados exemplares obtidos de experimentos de sondagem química do SARS-CoV2 5'UTR. (A) Perfil de reatividade das primeiras 300 bases do genoma do SARS-CoV2 colorido por base (A: vermelho, C: azul, U: verde, G: amarelo). As reatividades brutas são calculadas como a frequência absoluta de mutação dividida pela cobertura. Bases com conformação aberta apresentam altos valores de reatividade; as bases envolvidas no emparelhamento de bases têm baixos valores de reatividade. você e G não são modificados pelo DMS e apresentam baixos valores de reatividade, originados da infidelidade à polimerase. As previsões foram feitas com o servidor web do DREEM. (B) Modelo estrutural do SARS-CoV2 5'UTR previsto a partir de valores de reatividade obtidos com VARNA. Bases com altos valores de reatividade são coloridas em vermelho; bases com baixos valores de reatividade são coloridas em branco. (C) Fluxo de trabalho da análise DMS-MaP começando com os arquivos .fastq obtidos do sequenciamento. Estes podem ser controlados pela qualidade usando fastqc; as sequências do adaptador são aparadas usando TrimGalore e, em seguida, mapeadas de volta para uma sequência de referência usando Bowtie2. A partir dos arquivos .bam obtidos, o DREEM conta as mutações em cada leitura, criando um mapa de mutação ou um arquivo .txt .bitvector. Estes relatam as mutações de cada leitura de uma forma dependente da posição, com base na qual os perfis de reatividade média da população podem ser criados. Alternativamente, os vetores de bits podem ser agrupados usando o DREEM para procurar conformações alternativas de RNA. Por fim, os modelos de estrutura obtidos são visualizados usando software (por exemplo, VARNA). Abreviaturas: DMS-MaP = Perfil mutacional com sequenciamento usando sulfato de dimetila; SARS-CoV2 = síndrome respiratória aguda grave-coronavírus 2. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
O protocolo aqui descreve como investigar o RNA in vitro e em células usando experimentos de perfil mutacional DMS. Além disso, ele fornece instruções sobre como preparar bibliotecas para o sequenciamento Illumina para gerar dados específicos de genes e analisar os arquivos .fastq obtidos. Além disso, abordagens de biblioteca de todo o genoma podem ser usadas. No entanto, o RT-PCR específico do gene produz a mais alta qualidade e os dados mais robustos. Portanto, ao comparar entre as amostras, é importante garantir que elas sejam preparadas com estratégias de sequenciamento idênticas, pois a geração da biblioteca causa algum viés. A reprodutibilidade deve ser sempre medida usando replicações.
Várias precauções
O RNA é uma molécula instável que é sensível à degradação tanto através de temperaturas elevadas quanto por RNases. Portanto, medidas especiais – o uso de equipamentos de proteção individual (EPI), material livre de RNAse e inibidores de RNAse – são recomendadas. Mais importante ainda, o RNA deve ser mantido no gelo sempre que possível. Isso se aplica especialmente ao RNA metilado, que é ainda mais sensível a altas temperaturas.
É importante confirmar que a estrutura de interesse do RNA não é sensível à concentração de DMS e às condições de tampão. Buffers como 100 mM Tris, 100 mM MOPS e 100 mM HEPES em pH 7-7,5 dão um sinal alto, mas podem não ser suficientes para manter o pH durante a reação21. Como o DMS hidrolisa na água, o que diminui o pH, um tampão forte é fundamental para manter um pH neutro durante a reação de modificação. A adição de bicina demonstrou ajudar a manter o pH como ligeiramente básico21 , mas resulta em baixa modificação do DMS em Gs e Us, o que poderia ser informativo, mas deve ser analisado separadamente devido à produção de um sinal muito menor do que As e Cs e não é discutido mais adiante neste protocolo.
Na RT-PCR específica do gene, o RNA modificado é transcrito reversamente no DNA e amplificado em fragmentos por PCR. Embora o tamanho do RNA possa teoricamente ser ilimitado, esses fragmentos de PCR não devem exceder um comprimento de 400-500 pares de bases (pb) para evitar viés durante a reação de transcrição reversa. Idealmente, os fragmentos devem estar dentro do escopo da execução de sequenciamento (ou seja, se o sequenciamento for conduzido usando um programa de sequenciamento final emparelhado de 150 x 150 ciclos, um único fragmento não deve exceder 300 pb). Ao usar programas de sequenciamento com menos ciclos, os produtos de PCR podem ser fragmentados usando um dsDNase. Além disso, como as sequências dentro das sequências de iniciadores não contêm nenhuma informação estrutural, os fragmentos devem se sobrepor quando o RNA sondado compreende o fragmento >1. As reações RT podem conter vários primers RT para diferentes fragmentos (até 10 primers RT diferentes). Dependendo das sequências, o agrupamento dos primers RT pode tornar a transcrição reversa menos eficiente, mas normalmente funciona bem. Cada reação de PCR deve ser conduzida separadamente.
Ao sondar o RNA com DMS, as condições experimentais desempenham um papel adicional, pois muitos RNAs são termodinamicamente instáveis e alteram sua conformação com base em fatores ambientais, como a temperatura. Para evitar irregularidades, as condições experimentais devem ser mantidas o mais constantes possível, também no que diz respeito aos tempos de reação. As condições tampão parecem ser trocáveis até certo grau 17,20,22,23 quando as condições básicas são mantidas — a capacidade de tamponamento e a presença de íons monovalentes (Na) e divalentes (Mg) — para garantir o dobramento adequado do RNA 24.
Com relação à preparação da biblioteca de RNAs modificados, vários aspectos devem ser levados em consideração. Primeiro, como mencionado anteriormente, os RNAs modificados são menos estáveis do que suas contrapartes não modificadas, o que significa que eles podem exigir a otimização dos tempos de fragmentação para uma distribuição ideal do tamanho do fragmento. Além disso, certos kits de preparação de bibliotecas de RNA, bem como muitas outras abordagens de RNAseq, usam primers aleatórios no kit de transcrição reversa. Isso pode levar a uma menor cobertura da referência, especialmente nos 3' de um gene, e, em última análise, a uma profundidade de cobertura insuficiente. Se a cobertura de uma determinada região for muito baixa, pode ser necessário remover essas bases da previsão da estrutura. Além dos kits RT-PCR e RNAseq de genoma completo, outras abordagens de preparação de bibliotecas podem ser usadas. Protocolos que incluem a ligadura de adaptadores de 3' e/ou 5' ao RNA são vantajosos quando se utilizam pequenos fragmentos de RNA ou quando a perda de informações de sondagem nas regiões do primer deve ser evitada.
Por fim, a análise dos experimentos de sondagem química deve sempre ser interpretada com cuidado. Atualmente, não há software que preveja a estrutura de RNA de qualquer RNA a partir da sequência sozinha com alta precisão. Embora as restrições de sondagem química melhorem muito a precisão, a geração de bons modelos para RNAs longos (>500 nt) ainda é um desafio. Estes modelos devem ser testados por outras abordagens e/ou mutagénese. O software de predição de RNA otimiza para o número máximo de pares de bases, penalizando significativamente as conformações abertas, que podem não representar com precisão o dobramento do RNA5. Assim, o modelo de estrutura obtido deve ser testado quantificando a concordância de predição com os dados de sondagem química subjacentes (por exemplo, por AUROC) e entre replicações (por exemplo, por mFMI), como exemplificado por Lan et al.20.
Idealmente, vários experimentos em diferentes sistemas para desafiar o modelo de estrutura obtido devem ser usados para fortalecer a hipótese. Estes podem incluir o uso de abordagens in vitro e in-cell, mutações compensatórias e diferentes linhagens celulares e espécies. Além disso, as reatividades brutas são muitas vezes tão ou mais informativas do que as previsões de estrutura, pois registram o instantâneo da "verdade do solo" do conjunto de dobragem de RNA. Como tal, as reatividades brutas são muito adequadas e informativas para comparar mudanças de estrutura entre diferentes condições. É importante ressaltar que as estruturas de energia livre mais baixas calculadas usando restrições de sondagem química com previsão computacional só devem ser usadas como uma hipótese inicial em direção a um modelo de estrutura completa.
Divulgações
Os autores não têm conflitos de interesse a declarar.
Agradecimentos
Nenhum
Materiais
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
Referências
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados