
Method Article
בדיקת מבנה RNA עם פרופיל מוטציה של דימתיל סולפט עם ריצוף במבחנה ובתאים
In This Article
Summary
הפרוטוקול מספק הוראות לשינוי RNA עם דימתיל סולפט לניסויים בפרופיל מוטציות. הוא כולל בדיקה במבחנה ו - in vivo עם שתי שיטות הכנה חלופיות לספרייה.
Abstract
תפקידו של מבנה הרנ"א כמעט בכל תהליך ביולוגי נעשה ברור יותר ויותר, במיוחד בעשור האחרון. עם זאת, גישות קלאסיות לפתרון מבנה RNA, כגון קריסטלוגרפיה של RNA או cryo-EM, לא הצליחו לעמוד בקצב של התחום המתפתח במהירות והצורך בפתרונות בעלי תפוקה גבוהה. פרופיל מוטציה עם ריצוף באמצעות דימתיל סולפט (DMS) MaPseq היא גישה מבוססת ריצוף כדי להסיק את מבנה הרנ"א מתגובתיות הבסיס עם DMS. DMS מתילאט את החנקן N1 באדנוסינים ואת N3 בציטוזינים על פני ווטסון-קריק שלהם כאשר הבסיס אינו מזווג. תמלול לאחור של הרנ"א המתוקן עם הטרנסקריפטאז ההפוך של הקבוצה התרמוספטיבית II (TGIRT-III) מוביל לכך שהבסיסים שעברו מתילציה משולבים כמוטציות ב-cDNA. כאשר מריצפים את ה-cDNA המתקבל וממפים אותו בחזרה לתעתיק ייחוס, שיעורי המוטציה היחסיים עבור כל בסיס מעידים על "מצבו" של הבסיס כמזווג או לא מזווג. למרות שלתגובתיות DMS יש יחס אות לרעש גבוה הן במבחנה והן בתאים , שיטה זו רגישה להטיה בהליכי הטיפול. כדי להפחית את ההטיה הזו, מאמר זה מספק פרוטוקול לטיפול ב-RNA עם DMS בתאים ועם רנ"א מתועתק במבחנה .
Introduction
מאז הגילוי שלרנ"א יש תכונותמבניות של 1,2 וגם תכונותקטליטיות 3, נחשפו בהדרגה חשיבותו של הרנ"א ותפקודו הרגולטורי במגוון רחב של תהליכים ביולוגיים. ואכן, ההשפעה של מבנה הרנ"א על ויסות גנים זכתה לתשומת לב גוברת4. בדומה לחלבונים, לרנ"א יש מבנים ראשוניים, משניים ושלישוניים, המתייחסים לרצף הנוקלאוטידים, למיפוי הדו-ממדי של אינטראקציות זיווג-בסיס, ולקיפול התלת-ממדי של מבנים אלה המזווגים בבסיס, בהתאמה. בעוד שקביעת המבנה השלישוני היא המפתח להבנת המנגנונים המדויקים העומדים מאחורי תהליכים תלויי RNA, המבנה המשני הוא גם אינפורמטיבי מאוד לגבי תפקוד הרנ"א והוא הבסיס לקיפול תלת-ממדי נוסף5.
עם זאת, קביעת מבנה הרנ"א הייתה מאתגרת במהותה בגישות קונבנציונליות. בעוד שעבור חלבונים, קריסטלוגרפיה, תהודה מגנטית גרעינית (NMR) ומיקרוסקופיית אלקטרונים קריוגנית (cryo-EM) אפשרו לקבוע את מגוון המוטיבים המבניים, מה שמאפשר חיזוי מבנה מהרצף בלבד6, גישות אלה אינן ישימות באופן נרחב על רנ"א. ואכן, רנ"א הן מולקולות גמישות עם אבני בניין (נוקלאוטידים) שיש להן הרבה יותר חופש קונפורמציה וסיבובי בהשוואה למקבילותיהן בחומצות אמינו. יתר על כן, האינטראקציות באמצעות זיווג בסיסים הן דינמיות ורב-תכליתיות יותר מאלו של שאריות חומצות אמינו. כתוצאה מכך, גישות קלאסיות הצליחו רק עבור רנ"א קטנים יחסית עם מבנים מוגדרים היטב וקומפקטיים מאוד7.
גישה נוספת לקביעת מבנה הרנ"א היא באמצעות בדיקה כימית בשילוב עם ריצוף מהדור הבא (NGS). אסטרטגיה זו מייצרת מידע על מצב הקשירה של כל בסיס ברצף RNA (כלומר, המבנה המשני שלו). בקצרה, הבסיסים במולקולת RNA שאינם עוסקים בזיווג בסיסים משתנים באופן דיפרנציאלי על ידי תרכובות כימיות קטנות. תמלול לאחור של רנ"א אלה באמצעות תעתיק הפוך מיוחד (RTs) משלב את השינויים בחומצה דאוקסיריבונוקלאית משלימה (cDNA) כמוטציות. מולקולות cDNA אלה מוגברות לאחר מכן על ידי תגובת שרשרת פולימראז (PCR) ומרוצפות. כדי לקבל מידע על "מצבם" כמאוגדים או לא מאוגדים, תדרי המוטציות בכל בסיס ברנ"א של עניין מחושבים ומוכנסים לתוכנת חיזוי מבנה כאילוצים8. בהתבסס על כללי השכן הקרוב ביותר9 וחישובי אנרגיה חופשית מינימליים 10, תוכנה זו מייצרת מודלים מבניים המתאימים ביותר לנתונים הניסיוניים שהתקבלו11,12.
DMS-MaPseq משתמש ב-DMS, אשר מתילציה של חנקן N1 באדנוסינים וחנקן N3 בציטוזינים על פני ווטסון-קריק שלהם באופן ספציפי מאוד13. שימוש בטרנסקריפטאז הפוך (TGIRT-III) מקבוצה תרמוספטיבית II (TGIRT-III) בתעתיק הפוך יוצר פרופילים מוטציוניים עם יחסי אות לרעש חסרי תקדים, ואף מאפשר פירוק של פרופילים חופפים הנוצרים על ידי שני קונפורמציות חלופיות או יותר14,15. יתר על כן, DMS יכול לחדור לקרומי תאים ולרקמות שלמות, מה שהופך את הבדיקה בהקשרים פיזיולוגיים לאפשרית. עם זאת, יצירת נתונים באיכות טובה היא מאתגרת, מכיוון ששינויים בהליך הטיפול יכולים להשפיע על התוצאות. לכן, אנו מספקים פרוטוקול מפורט הן למבחנה והן ל-DMS-MaPseq בתוך התא כדי להפחית את ההטיה ולהנחות מצטרפים חדשים לשיטה דרך הקשיים שהם עלולים להיתקל בהם. במיוחד לאור מגיפת SARS-CoV2 האחרונה, נתונים באיכות גבוהה על נגיפי RNA הם כלי חשוב לחקר ביטוי גנים ומציאת טיפולים אפשריים.
Protocol
הערה: עיין בטבלת החומרים לקבלת פרטים הקשורים לכל החומרים, התוכנות, הריאגנטים, המכשירים והתאים המשמשים בפרוטוקול זה.
1. DMS-MaP ספציפי לגן במבחנה
- שעתוק RNA במבחנה
- קבל את רצף הרנ"א המעניין כדנ"א דו-גדילי (ds)DNA (למשל, כמקטעי דנ"א, פלסמידים או PCR מדנ"א קיים/גנומי). אם רצף הדנ"א מכיל מקדם פולימראז, קפצו לשלב 3.
- בצע חפיפה PCR כדי לחבר מקדם RNA פולימראז במעלה הזרם של מקטע ה- DNA הרצוי (פריימר קדמי עבור T7 פולימראז: 5' TAATACGACTCACTATAGG + בסיסים ראשונים של רצף מטרה 3).
- במבחנה מתמללים את מקטע הדנ"א לרנ"א. תמיד לשמור את הרנ"א על הקרח.
- לעכל את הדנ"א באמצעות DNase.
- לבודד את הרנ"א באמצעות גישה מבוססת עמודות (שלב 2.4) או על ידי משקעי אתנול (שלב 2.5). אלוטה בנפח מתאים, מצפה לתפוקה של ~ 50 מיקרוגרם.
- להבטיח את שלמות הרנ"א על ידי הרצתו על ג'ל אגרוז; לנטרל את הרנ"א למשך 2-3 דקות ב-70 מעלות צלזיוס לפני הריצה.
הערה: החיץ והאגרוז יכולים להכיל RNases שמפרקים RNA ועלולים לזהם את דגימת ה-RNA. בעבר נעשה שימוש בג'לים של אגרוז בקאסט במעבדה זו; התוצאות (במיוחד עם RNA) היו מעורפלות לפעמים. התוצאות הטובות ביותר הושגו עם agarose או PAGE ג'לים. - שימוש ישיר לאחסן את הרנ"א בטמפרטורה של −80 מעלות צלזיוס למשך מספר חודשים, אלא אם כן ניתן לראות השפלה לאחר ההפשרה.
- במבחנה שינוי DMS (ב-105 mM DMS)
- הכינו כמות מספקת של חיץ מתקפל מחדש (0.4 M נתרן קקודיט, pH 7.2, המכיל 6 mM MgCl2).
הערה: עבור כל תגובה (נפח סופי של 100 μL), הוסף 89 μL של מאגר refolding. - עבור כל תגובה, העבירו 89 μL של חיץ מתקפל מחדש לצינור ייעודי של 1.5 מ"ל, וחימום מוקדם בטמפרטורה של 37 מעלות צלזיוס בתרמושייקר הממוקם מתחת למכסה המנוע הכימי.
הערה: DMS הוא רעיל ביותר ויש לשמור אותו תמיד מתחת למכסה המנוע הכימי עד למרווה על ידי חומר מחזרים. - אלוטה 1-10 pmol של RNA ב 10 μL של מים ללא נוקלאז (NF H2O); העברה לצינור PCR.
- דגירה בתרמוציקלר בטמפרטורה של 95 מעלות צלזיוס למשך דקה אחת כדי לנטרל את הרנ"א.
- יש להניח מיד על גוש קרח כדי למנוע קיפול שגוי.
- מוסיפים את דגימת הרנ"א לצינור המיועד עם חיץ מתקפל מחדש בטמפרטורה של 37 מעלות צלזיוס, מערבבים היטב ודוגרים במשך 10-20 דקות כדי להחזיר את הרנ"א.
הערה: רוב הרנ"א יתקפלו בסדר גודל של אלפיות השנייה לשניות, אם כי קיימים יוצאים מן הכלל16. - הוסיפו לדגימת הרנ"א 1 μL של 100% (10.5 M) DMS, ודגרו במשך 5 דקות תוך כדי רעידות במהירות של 800-1,400 סיבובים לדקה (סל"ד).
הערה: ניעור (או אמצעי ערבוב אחרים) בשלב זה הוא חיוני מכיוון ש-DMS הוא הידרופובי וייתכן שלא יתמוסס במלואו במאגר המתקפל. סטיות בזמני התגובה עשויות להשפיע על יכולת השחזור של תגובתיות ה-DMS. כדי למזער את שגיאת הפיפטינג, ניתן להמיס DMS ב-100% אתנול לפני הוספתו לדגימה אם נשמר ריכוז סופי של 1% (105 mM) DMS. עבור בקרה לא מטופלת, DMS יכול להיות מוחלף על ידי דימתיל סולפוקסיד (DMSO) או מים. - לאחר 5 דקות של זמן תגובה, להרוות עם 60 μL של 100% β-mercaptoethanol (BME), לערבב היטב, ומיד להניח את הרנ"א על קרח.
הערה: ניתן להסיר את הרנ"א בבטחה ממכסה המנוע לאחר המרווה את התגובה באמצעות BME כדי לנקות אותו. עם זאת, חשיפה ישירה של BME לסביבה עדיין יש להימנע בשל ריח חזק תכונות מגרה. - נקו את הרנ"א על ידי משקעי נתרן אצטט-אתנול (ראו שלב 2.5) או גישה מבוססת עמודות (ראו שלב 2.6), והתחמקו מ-10 מיקרון מים.
- לכמת את הרנ"א באמצעות ספקטרופוטומטר.
- שימוש ישיר לאחסן את הרנ"א שהשתנה ב -80 מעלות צלזיוס.
הערה: יש להימנע מאחסון לטווח ארוך, מכיוון שהרנ"א פחות יציב לאחר טיפול DMS.
- הכינו כמות מספקת של חיץ מתקפל מחדש (0.4 M נתרן קקודיט, pH 7.2, המכיל 6 mM MgCl2).
- RT-PCR ספציפי לגן של RNA מהונדס
הערה: ראו איור 1 עבור הגדרת RT-PCR של שברים שטופלו ב-DMS.- Elute 100 ng של RNA שונה ב 10 μL של נטול נוקלאז (NF) H2O. העברה לצינור PCR.
- לצינור, הוסף 4 μL של 5x חיץ גדיל ראשון (FSB), 1 μL של תערובת dNTP (10 mM כל אחד), 1 μL של 0.1 M dithiothreitol (DTT) (להימנע ממחזורי הפשרה בהקפאה), 1 μL של מעכב RNase, 1 μL של 10 μM פריימר הפוך (פריימר יחיד או מאגר של פריימרים), ו 1 μL של TGIRT III.
הערה: עבור מאגר פריימרים, אין להוסיף 1 μL של 10 μM מכל פריימר ישירות ל-RT; במקום זאת, ערבבו תחילה את הפריימרים, והוסיפו 1 μL מהתערובת (בריכוז פריימר כולל של 10 μM). - דגירה ב-57°C למשך 30 דקות עד 1.5 שעות (בדרך כלל, 30 דקות מספיקות כדי לייצר מוצר של 500 nt) בתרמו-ציקלר.
- מוסיפים 1 μL של 4 M NaOH, מערבבים על ידי פיפטציה, ודוגרים ב 95 מעלות צלזיוס במשך 3 דקות כדי לפרק את הרנ"א.
הערה: שלב זה הוא חיוני מכיוון שהוא משחרר TGIRT מה-cDNA על ידי השפלת הרנ"א. אם מדלגים עליו, ה-PCR במורד הזרם עשוי להיות מושפע. - נקה באמצעות גישה מבוססת עמודות (ראה שלב 2.6) שמסירה במידה מספקת את הפריימרים, והתחמק מ-10 μL של NF H2O.
- PCR מגביר את ה-cDNA באמצעות 1 μL של תוצר השעתוק ההפוך לכל 25 μL של התגובה עם ערכת PCR שנועדה לאזן בין תפוקה ונאמנות.
הערה: הפריימרים צריכים להיות בעלי טמפרטורת התכה של ~ 60 מעלות צלזיוס. - הפעל 2 μL של מוצר ה- PCR על ג'ל אגרוז או ג'ל אגרוז טרומי precast כדי לאמת את הצלחת ה- PCR.
- באופן אידיאלי, רק להקה אחת צריכה להופיע אחרי ה-PCR. אם כן, נקה את התגובה באמצעות גישה מבוססת טורים. אם קיימות רצועות חלופיות, השתמש בתגובת ה-PCR הנותרת כדי להוציא את הרצועה הנכונה מהג'ל. אלוטה בנפח קטן מספיק (לדוגמה, 10 μL).
- כמת את השברים שחולצו באמצעות ספקטרופוטומטר.
- אינדקס מקטעי dsDNA לריצוף באמצעות גישה המתאימה לפלטפורמת הריצוף הרצויה.

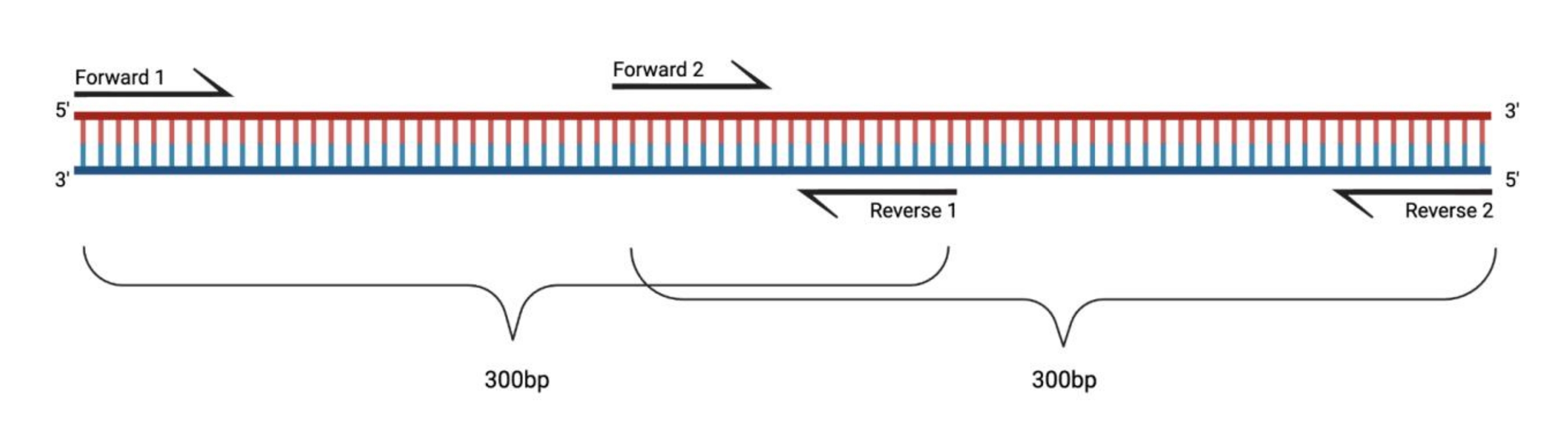
איור 1: מערך ניסיוני עבור RT-PCR של שברים גדולים שטופלו ב-DMS. בעת ביצוע שעתוק הפוך על רנ"א שונה, השינויים ברצף שאליו לא יתועדו הפריימרים. לכן, כאשר השברים עולים על 400-500 bp באורך, שברים חופפים באזורי פריימר צריך להיות מתוכנן, כפי שמודגם כאן. אורך השברים תלוי בצרכי הרצף. בעת שימוש ברצף מחזור 150 זוגי, השברים לא יעלו על 300 bp. קיצורים: RT-PCR = תגובת שרשרת פולימראז שעתוק לאחור; DMS = דימתיל סולפט. אנא לחץ כאן כדי להציג גרסה גדולה יותר של נתון זה.
{kind=link}
2. גנום שלם DMS-MaP באמצעות תאים נגועים בנגיף
הערה: בתאים, ניתן לשלב טיפול ב-DMS גם עם גישת ההגברה הספציפית לגן שתוארה לעיל. ספריית הגנום כולו דורשת עומק ריצוף עצום כדי להשיג כיסוי מלא על גן יחיד. עם זאת, אם רנ"א נגיפי מהווה חלק משמעותי מהרנ"א הריבודפל לאחר המיצוי, ריצוף גנום שלם יהיה מתאים. יתר על כן, ניתן לשלב שיטות העשרה אחרות עם שיטת יצירת ספריית הגנום כולו.
- טיפול DMS
- לגדל תאים נגועים בנגיף עד לשלב הרצוי של ההדבקה.
- העבירו את מיכל התא למכסה אדים ייעודי המתאים לטיפול הן בנגיפים ברמת הבטיחות הביולוגית הנדרשת והן באדים הכימיים הנוצרים על ידי חומרים כגון DMS.
- הוסף נפח של 2.5% של DMS למדיום התרבית, ואטם את המיכל (בדרך כלל צלחת של 10 ס"מ) באמצעות פרפילם.
הערה: קל לשנות פחות ולשנות יתר על המידה באמצעות DMS. כאשר מוסיפים DMS ישירות לתאים, חשוב מאוד לערבב היטב. לחלופין, יש לחמם את המדיום החדש בצינור חרוטי של 50 מ"ל בטמפרטורה של 37 מעלות צלזיוס, ולהוסיף את ה-DMS ישירות לרעוד במרץ. דקנט את המדיום המושקע על התאים, ולאט לאט פיפטה במדיום המכיל DMS. - מעבירים לחממה של 37 מעלות צלזיוס למשך 5 דקות.
הערה: בהתאם למשך הזמן שלוקח לטפל ב- DMS מחוץ לחממה, ייתכן ש- 5 דקות יובילו לשינוי יתר. שמור על הזמן מהוספת ה- DMS לדגירה ל- ≤1 דקות. אם מבצעים את הניסוי בפעם הראשונה, מומלץ לבצע טיטרציה של DMS ולשנות את זמן הדגירה (בין 3 דקות ל-10 דקות) כדי למצוא את קצב השינוי האופטימלי ולוודא שהתוצאות חזקות על פני חלון ריכוזים. - פזרו בזהירות את המדיום המכיל DMS (לפסולת כימית מתאימה) והוסיפו בעדינות 10 מ"ל של מאגר עצירה (PBS עם 30% BME [למשל, 3 מ"ל של BME ו-7 מ"ל של PBS]).
הערה: תוספת של DMS ו-BME יכולה להרים את התאים מהצלחת אם התאים אינם דבקים חזק. אם התאים מתרוממים, ניתן להתייחס אליהם כאל תאי תרחיף - במקום להסיר את המדיום המכיל DMS, להוסיף את חיץ העצירה ישירות, ולגרד את התאים עם DMS ו- BME לתוך צינור חרוטי של 50 מ"ל. גלולה את התאים על ידי צנטריפוגה במשך 3 דקות ב 3,000 × גרם; הקפידו להיפטר מכל שאריות DMS, שיכולות לגלוש מתחת לתאים בטיפות גדולות. שלב שטיפה נוסף ב-30% BME מומלץ אם לא ניתן להסיר את מדיום ה-DMS בתחילה. - לגרד את התאים, ולהעביר אותם צינור חרוטי 15 מ"ל.
- גלולה על ידי צנטריפוגה ב 3,000 × גרם במשך 3 דקות.
- יש להסיר את הסופר-נטנט ולשטוף 2x עם 10 מ"ל של PBS.
- הסר בזהירות כמה שיותר PBS שיורי.
- ממיסים את הכדור בכמות מתאימה של מגיב בידוד RNA (למשל, 3 מ"ל עבור בקבוק תרבית T75, 1 מ"ל עבור צלחת 10 ס"מ).
הערה: כמויות לא מספיקות של המגיב עלולות להשפיע על תפוקת הרנ"א.
- מיצוי RNA ודלדול RNA ריבוזומלי (rRNA)
- ל-1 מ"ל של תאים הומוגניים בריאגנט לבידוד RNA, הוסיפו 200 μL של כלורופורם, מערבולת במשך 15-20 שניות עד לוורוד בהיר, ואז דגרו עד 3 דקות עד להפרדת הפאזה.
הערה: שלב השומנים הוורודים אמור להתיישב בתחתית. אם זה לא המקרה, סביר להניח שזמן המערבולת לא הספיק. - סובב במהירות מרבית (~ 20,000 × גרם) למשך 15 דקות ב-4 מעלות צלזיוס.
- מעבירים את השלב המיימי העליון לצינור חדש.
- נקו את הרנ"א על ידי משקעי נתרן אצטט-אתנול (ראו שלב 2.5) או גישה מבוססת עמודות (ראו שלב 2.6), והתחמקו בנפח מספיק של NF H2O.
- בדוק את שלמות ה- RNA על ג'ל אגרוז. חפשו שתי רצועות המתאימות לשתי יחידות המשנה הריבוזומליות.
- דלדל את ה-rRNAs בגישה המועדפת, והתחמק בנפח מתאים (בדרך כלל 20-50 μL) של NF H2O.
הערה: עבור יישומים במורד הזרם, ~500 ננוגרם של RNA כולל מוצע בנפח של 8 μL. רנ"א שאינם ריבוזומליים מהווים בדרך כלל רק 5%-10% מסך הרנ"א. - כימות באמצעות ספקטרופוטומטר.
- ל-1 מ"ל של תאים הומוגניים בריאגנט לבידוד RNA, הוסיפו 200 μL של כלורופורם, מערבולת במשך 15-20 שניות עד לוורוד בהיר, ואז דגרו עד 3 דקות עד להפרדת הפאזה.
- יצירת ספרייה
- השתמש ב-RT-PCR ספציפי לגן או בגישות אחרות כדי ליצור ספריות15. אם אתם משתמשים בהקסמרים אקראיים לצורך הקדמה, הוסיפו שלב דגירה ב-Tm נמוך (37-42 מעלות צלזיוס) כדי לאפשר חישול הקסמר.
הערה: ניתן להשתמש בערכות ייצור ספריות סטנדרטיות גם על-ידי החלפת האנזים RT ב-TGIRT ושינוי טמפרטורת ה-RT ל-57 °C.
- השתמש ב-RT-PCR ספציפי לגן או בגישות אחרות כדי ליצור ספריות15. אם אתם משתמשים בהקסמרים אקראיים לצורך הקדמה, הוסיפו שלב דגירה ב-Tm נמוך (37-42 מעלות צלזיוס) כדי לאפשר חישול הקסמר.
- ניקוי RNA מבוסס עמודות באמצעות עמודות RNA Clean ו-Concentrator
הערה: כל השלבים צריכים להתבצע בטמפרטורת החדר.- הוסף את NF H2O לצינור הדגימה כדי להביא אותו לנפח של 50 μL.
- הוסף 100 μL של מאגר קשירה ו 150 μL של 100% אתנול לדגימה.
- מערבבים ומעבירים לטור ספין.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- הוסף 400 μL של מאגר הכנה RNA.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- הוסף 700 μL של מאגר שטיפת RNA.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- הוסף 400 μL של מאגר שטיפת RNA.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- (אופציונלי) מעבירים את העמודה לצינור איסוף חדש, ומסתובבים ב-10,000-16,000 × גרם למשך 2 דקות.
- העבירו את העמודה לצינור נקי ללא RNAse והוסיפו כמות מתאימה של NF H2O.
- סובבו ב-10,000-16,000 × גרם למשך דקה.
- מיצוי RNA פנול-כלורופורם חומצה.
- הוסף נפח שווה של פנול חומצה:כלורופורם:אלכוהול איזואמיל.
- מערבולת ביסודיות, וצנטריפוגה ב 14,000 × גרם במשך 5 דקות.
- אם אין הפרדת פאזה, הוסף 20 μL של 2 M NaCl, וחזור על הצנטריפוגה.
- מעבירים את הפאזה המימית לצינור חדש.
- הוסף 500 μL של איזופרופנול ו -2 μL של משקע משותף.
- מערבבים ומדגרים ב-RT למשך 3 דקות; לאחר מכן, לדגור ב -80 מעלות צלזיוס למשך הלילה.
- גלולה את הרנ"א על ידי צנטריפוגה במהירות מקסימלית (~ 20,000 × גרם) במשך 30 דקות ב 4 מעלות צלזיוס.
- לשטוף את הכדור עם 200 μL של קרח קר 70% אתנול.
- סובב במהירות מרבית (~ 20,000 × גרם) למשך 5 דקות; להשליך את הזרימה.
- השהה את הכדור בכמות המתאימה של NF H2O.
- ניקוי cDNA מבוסס עמודות באמצעות העמודות Oligo Clean ו-Concentrator
הערה: כל השלבים צריכים להתבצע בטמפרטורת החדר.- הוסף את NF H2O לצינור הדגימה כדי להביא אותו לנפח של 50 μL.
- הוסף 100 μL של מאגר מחייב ו 400 μL של 100% אתנול.
- מערבבים ומעבירים לטור ספין.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- הוסף 750 μL של מאגר שטיפת DNA.
- ספין ב 10,000-16,000 × גרם במשך 30 שניות; להשליך את הזרימה.
- (אופציונלי) מעבירים את העמודה לצינור איסוף חדש ומסתובבים ב-10,000-16,000 × גרם למשך 2 דקות.
- העבירו את העמודה לצינור נקי ללא RNAse, והוסיפו כמות מתאימה של NF H2O.
- סובבו ב-10,000-16,000 × גרם למשך דקה.
3. ניתוח נתוני הרצף
הערה: כדי ליצור מודלים של מבנה משני של RNA מנתוני הריצוף של DMS-MaP, קבצי ה- .fastq המתקבלים חייבים להיות מעובדים במספר שלבים שונים. ניתן לבצע שלבים אלה באופן אוטומטי באמצעות
- חתוך את רצפי המתאם באמצעות TrimGalore או Cutadapt.
- מפה את הקריאות לרצפי הייחוס (תבנית .fasta) באמצעות Bowtie2.
- ספרו את הקריאות באמצעות תוכנה מיוחדת למבנה RNA (לדוגמה, DREEM14, RNA-Framework17 או דומה), וצרו פרופילי תגובתיות.
- (אופציונלי) אשכול הקריאות כדי למצוא קונפורמציות RNA חלופיות באמצעות DREEM 14, DRACO17, DANCE-MaP18, או דומה.
- לחזות את מבנה האנרגיה החופשית המינימלי בהתבסס על פרופילי התגובה באמצעות RNAStructure12, ViennaRNA או דומה.
- דמיינו את מבנה ה-RNA11 באמצעות VARNA (https://varna.lri.fr/) או דומה.
הערה: עבור מעשיות, תוכנות כגון DREEM (www.rnadreem.org) ו- RNA-Framework19 משלבות במידה רבה שלבים 1--5 בצינורות שלהן, מה שמייעל את תהליך הניתוח. עם זאת, כל חיזוי מבנה צריך להיות מטופל בזהירות (למשל, על ידי אימות הסכמת המבנה עם הנתונים20).
תוצאות
DMS-MaP ספציפי לגנים במבחנה
כדי לחקור את ה-5'UTR של SARS2, הוזמנו 300 ה-bp הראשונים של הנגיף כרצף gBlock, לצד שלושה פריימרים. אלה כללו שני פריימרים להפצת השבר ("FW" ו-"RV") באמצעות PCR, וכן אחד לחיבור מקדם T7 ("FW-T7"). ניתן לראות רצפים אלה בטבלה 1.
| שם | רצף (5'->3') |
| פ.וו. | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| קרוואן | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
טבלה 1: רצף פריימר עבור DMS-MaP RT-PCR של SARS-CoV2 5'UTR. כאן, FW-T7 ו- RV נדרשים ליצירת תבנית DNA לתעתיק חוץ גופי , ה- RV משמש בתעתיק ההפוך וזוג הפריימרים של FW-RV משמש בהגברת ה- PCR הבאה של ה- cDNA. הפריימרים מתקרבים לתחילת הגנום של SARS-CoV2 (FW) והרצף ממש במורד הזרם של אזור העניין. קיצורים: DMS-MaP = פרופיל מוטציה עם ריצוף באמצעות דימתיל סולפט; RT-PCR = תגובת שרשרת פולימראז שעתוק לאחור; SARS-CoV2 = תסמונת נשימה חריפה חמורה-קורונה 2; UTR = אזור לא מתורגם; RV = פריימר הפוך; FW = פריימר קדימה.
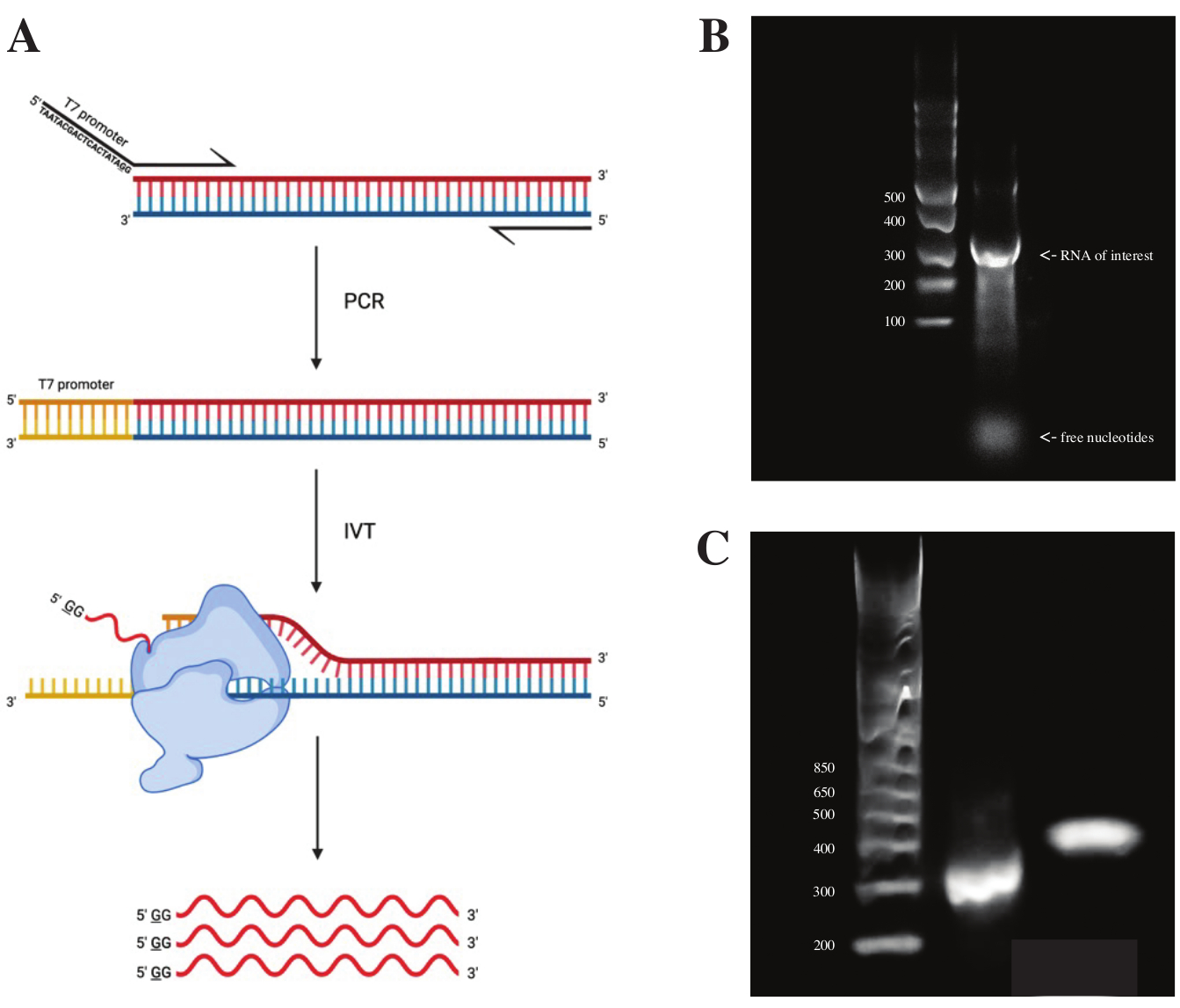
כדי ליצור רנ"א מקטע gBlock, הרצף של מקדם הפולימראז T7 חובר באמצעות PCR חופף באמצעות פרמיקס PCR לפי הסכימה שנראתה באיור 2A. מהקטע המוארך, רנ"א נוצר באמצעות ערכת השעתוק T7. תבנית הדנ"א עוכלכה לאחר מכן באמצעות ה-DNase וה-RNA שבודדו באמצעות עמודות RNA Clean ו-Concentrator.
בקרת האיכות של השעתוק במבחנה נעשתה על ידי הרצת מוצר ה-RNA על ג'ל אגרוז 1% לצד סולם ssRNA. מכיוון שהייתה רק רצועה אחת גלויה, בוצעו בדיקות DMS במבחנה ו-RT-PCR (ראו איור 2B).
כדי לאמת את הצלחת תגובת ה-PCR, הדגימה הופעלה על ג'ל אגרוז של 2% באמצעות סולם dsDNA. לאחר יצירת האינדקס, הרצועה צריכה לרוץ ~ 150 bp גבוה יותר על אותו ג'ל, בהתחשב בגודל של פריימרים אינדקס.

איור 2: שעתוק חוץ גופי של תבנית הדנ"א. (א) כדי לתמלל במבחנה תבנית דנ"א שעדיין אין לה מקדם RNA פולימראז פנימי, יש לחבר את התבנית על ידי חפיפה PCR תחילה. זה נעשה באמצעות פריימר קדימה, הכולל את הרצף TAATACGACTCACTATAGG (במקרה של T7 RNA פולימראז) במעלה הזרם של הבסיסים הראשונים חופפים עם השבר הרצוי. הבסיס המסומן בקו תחתון כאן מסמל את אתר התחלת השעתוק של הפולימראז. לאחר שהמקדם התחבר למקטע dsDNA, ניתן לתמלל אותו על ידי פולימראז T7. חשוב לציין שהפולימראז משתמש בגדיל המתנגד לרצף המקדם שהוזכר כתבנית (כחול), ולמעשה יוצר RNA זהה לרצף מיד במורד הזרם של רצף המקדם המצוין (אדום). (B) ג'ל אגרוז 1% עם סולם ssRNA (נתיב 1) ותוצר RNA מתועתק במבחנה ב-300 nt (נתיב 2). (C) ג'ל אגרוז של 2% עם GeneRuler 1 קילו פלוס סולם (נתיב 1), מוצר ה-PCR לאחר RT-PCR הפועל ב-300 כ"ס (נתיב 2), והמקטע האינדקס לאחר הכנת הספרייה בריצה של 470 כ"ס (נתיב 3). קיצורים: RT-PCR = תגובת שרשרת פולימראז שעתוק לאחור; DMS = דימתיל סולפט; nt = נוקלאוטידים; dsDNA = דנ"א דו-גדילי; ssRNA = RNA חד-גדילי. אנא לחץ כאן כדי להציג גרסה גדולה יותר של נתון זה.
{kind=link}
גנום שלם in vivo DMS-MaP באמצעות תאים נגועים בנגיף
לפני הטיפול ב-DMS, תאי ה-HCT-8 היו נגועים ב-OC43. כאשר נצפתה השפעה ציטופתית (CPE) 4 ימים לאחר ההדבקה (dpi) (כפי שניתן לראות באיור 3A), התאים האלה טופלו, והרנ"א הופק וריבודפל. בעת הרצת הרנ"א הכולל על ג'ל אגרוז, נראו שני פסים בהירים, המהווים את יחידות המשנה 40S ו-60S של הריבוזום, המהוות כ-95% ממסת הרנ"א הכוללת (ראו איור 3B). כאשר מיצוי הרנ"א לא הצליח או נפגע (למשל, על-ידי מחזורי הפשרה מרובים של הקפאה), תוצרי פירוק הרנ"א נראו בתחתית הג'ל (ראו איור 3C, נתיב שני). יתר על כן, לאחר הידלדלות ה-rRNA, שני הפסים הבהירים נעלמו, והותירו כתם בנתיב (ראו איור 3C, נתיב שלישי). לבסוף, לאחר הכנת הספרייה, הדגימות היו בחלוקות בגדלים שונים והוצגו ככתם על ג'ל PAGE הסופי. הרצועה נכרתה בין 200 נוקלאוטידים (nt) ל-500 nt, בהסכמה עם ריצת הריצוף הזוגית בגודל 150 x 150 שתוכננה לנתח ספריות אלה. והכי חשוב, דימרים של מתאמים שפועלים ב~150 nt הופרדו החוצה (ראו איור 3D).

איור 3: מחסומים של in vivo DMS-MaP עם תאים נגועים בנגיף. (A) תמונת מיקרוסקופיה קלה של תאי HCT-8 נגועים בנגיף, 4 ימים dpi. כדי להשיג את התפוקה הגבוהה ביותר האפשרית של RNA נגיפי מסך הרנ"א תוך מזעור ההשפעות השליליות עקב מוות תאי, יש להוסיף DMS כאשר CPE מתחיל או אפילו לפני כן, כפי שניתן לראות בתמונה. (B) ג'ל אגרוז 1% עם שש דגימות של 1 מיקרוגרם של RNA כולל. בכל נתיב נראים שני פסים בהירים, המהווים את יחידות המשנה 40S ו- 60S, מכיוון ש- RNA ריבוזומלי מהווה ~ 95% מסך ה- RNA. הערה: טיפול ב-DMS בתוך התא גורם לפיצול ולמריחה מסוימת של RNA, אך שני רצועות ה-rRNA עדיין צריכות להיות גלויות. פיצול קל לאחר שינוי נסבל מכיוון שהמידע המכיל את סימן המתילציה נוצר ומדווח על מבנה הרנ"א במהלך דגירה של DMS בזמן שהתאים עדיין בחיים. (C) ג'ל אגרוז 1% של GeneRuler 1 kb בתוספת סמן DNA סולם (נתיב 1) סך RNA שאוחסן בעבר בטמפרטורה של 80 מעלות צלזיוס במשך 6 חודשים (נתיב 2) ורנ"א ריבודפל (נתיב 3). כאשר מאחסנים RNA במשך זמן רב עם מספר מחזורי הפשרה בהקפאה, הרנ"א מתחיל להתדרדר ואולי אין להשתמש בו לניסויים ניסיוניים. יתר על כן, לאחר ריבודפל הרנ"א הכולל, שני הפסים הבהירים, המהווים את יחידות המשנה 40S ו-60S של הריבוזום, דהייה וכתם של שאריות ה-RNAs מתחילים להראות. (D) ג'ל PAGE של GeneRuler 1 kb בתוספת סמן DNA סולם (נתיב 1) ודגימת ספרייה של RNA מוכן לגנום שלם. יש לכרות את הג'ל בהתאם לצרכי הרצף. עבור ריצת ריצוף זוגית, המתפרשת על פני 150 מחזורים משני הצדדים, יש לכרות את הג'ל בין 300 כ"ס ל-500 כ"ס. יש להפריד בין דימרים של מתאמים (הפועלים ב-170 כ"ס). קיצורים: DMS-MaP = פרופיל מוטציה עם ריצוף באמצעות דימתיל סולפט; dpi = ימים לאחר ההדבקה; CPE = אפקט ציטופתי. אנא לחץ כאן כדי להציג גרסה גדולה יותר של נתון זה.
{kind=link}
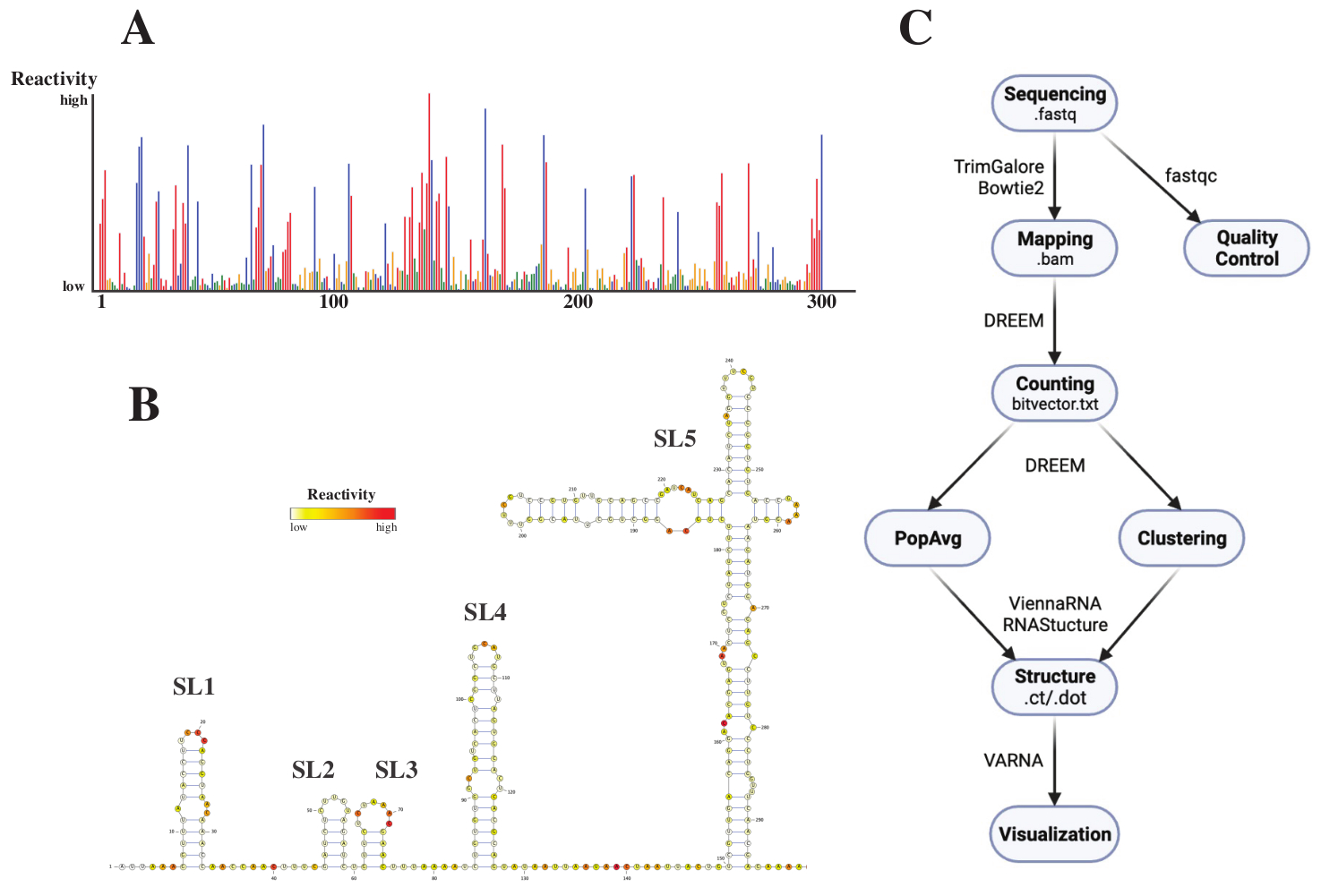
לאחר הרצף, קבצי ה- .fastq נותחו על-ידי שליחת עבודה לשרת האינטרנט DREEM (http://rnadreem.org/), יחד עם קובץ הפניה מסוג .fasta. הפלט שנוצר על ידי השרת כולל קבצי בקרת איכות שנוצרו על ידי fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) ו- TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), כמו גם קבצי פלט אחרים המכילים את תדרי המוטציה הממוצעת של האוכלוסייה. מלבד הדיאגרמה המציגה את תדרי המוטציות בתבנית אינטראקטיבית .html (ראו איור 4A) וקובץ .csv עם ההפעלות הגולמיות לכל בסיס וקובץ struct_constraint.txt, הניתן לקריאה על ידי מספר תוכנות חיזוי מבנה RNA, זה כולל גם קובץ bitvector.txt המדווח על המוטציות שנקראו. מתוך אלה, המבנים הממוצעים של האוכלוסייה חושבו על ידי שליחת קבצי .fasta ו- struct_constraint.txt לשרת האינטרנט RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). זה משתמש בתוכנת ViennaRNA כדי ליצור תחזיות מבנה המבוססות על האנרגיה החופשית המינימלית, אשר ניתן לצפות באינטרנט או להוריד בפורמט ct או וינה. כדי ליצור מודלים של מבנה רנ"א, הקבצים האלה להורדה נשלחו ל-VARNA (https://varna.lri.fr/, ראו איור 4B). לבסוף, קבצים .txt bitvector יכולים לשמש את הגרסה היציבה של DREEM (https://codeocean.com/capsule/6175523/tree/v1) כדי לחפש קונפורמציות RNA חלופיות. כדי להשיג מודלים של מבנה טוב באמצעות DREEM, יש להשיג כיסוי של 10,000 קריאות לכל בסיס; עבור קיבוץ באשכולות, ייתכן שיידרשו עד 100,000 קריאות לכל בסיס. סקירה כללית של זרימת העבודה כולה ניתן למצוא באיור 4C.

איור 4: נתונים לדוגמה שהתקבלו מניסויי בדיקה כימיים של SARS-CoV2 5'UTR. (A) פרופיל תגובתיות של 300 הבסיסים הראשונים של הגנום של SARS-CoV2 הצבועים לפי בסיס (A: אדום, C: כחול, U: ירוק, G: צהוב). התגובות הגולמיות מחושבות כתדירות המוטציה המוחלטת חלקי הכיסוי. לבסיסים עם קונפורמציה פתוחה יש ערכי תגובתיות גבוהים; לבסיסים העוסקים בזיווג בסיסים יש ערכי תגובתיות נמוכים. אתה ו-G אינם משתנים על-ידי DMS ויש להם ערכי תגובתיות נמוכים, שמקורם בבגידה בפולימראז. התחזיות נעשו עם שרת האינטרנט DREEM. (B) מודל המבנה של SARS-CoV2 5'UTR שנחזה מערכי תגובתיות שנעשו עם VARNA. בסיסים עם ערכי תגובתיות גבוהים צבועים באדום; בסיסים עם ערכי תגובתיות נמוכים צבועים בלבן. (C) זרימת עבודה של ניתוח DMS-MaP החל מקבצי .fastq המתקבלים מרצף. אלה יכולים להיות מבוקרי איכות באמצעות fastqc; רצפי המתאם נחתכים באמצעות TrimGalore ולאחר מכן ממופים בחזרה לרצף ייחוס באמצעות Bowtie2. מקבצי ה- .bam המתקבלים, DREEM סופר את המוטציות בכל קריאה, ויוצר מפת מוטציות או קובץ .bitvector.txt. אלה מדווחים על המוטציות של כל קריאה באופן תלוי מיקום, שעל בסיסו ניתן ליצור פרופילי תגובתיות ממוצעים של האוכלוסייה. לחלופין, ניתן לקבץ סיביות באמצעות DREEM כדי לחפש קונפורמציות RNA חלופיות. לבסוף, מודלי המבנה המתקבלים מוצגים באופן חזותי באמצעות תוכנה (למשל, VARNA). קיצורים: DMS-MaP = פרופיל מוטציה עם ריצוף באמצעות דימתיל סולפט; SARS-CoV2 = תסמונת נשימה חריפה חמורה-וירוס קורונה 2. אנא לחץ כאן כדי להציג גרסה גדולה יותר של נתון זה.
{kind=link}
Discussion
הפרוטוקול כאן מתאר כיצד לחקור RNA במבחנה ובתאים באמצעות ניסויי פרופיל מוטציה של DMS. יתר על כן, הוא נותן הוראות כיצד להכין ספריות לריצוף Illumina כדי ליצור נתונים ספציפיים לגנים ולנתח את קבצי ה- .fastq המתקבלים. בנוסף, ניתן להשתמש בגישות ספרייה כלל-גנומית. עם זאת, RT-PCR ספציפי לגן מפיק את הנתונים האיכותיים והחזקים ביותר. לכן, אם משווים בין דוגמאות, חשוב לוודא שהם מוכנים עם אסטרטגיות רצף זהות, שכן יצירת הספרייה גורמת להטיה מסוימת. תמיד יש למדוד את יכולת השכפול באמצעות משכפלים.
מספר אמצעי זהירות
רנ"א היא מולקולה לא יציבה הרגישה להתפרקות הן באמצעות טמפרטורות גבוהות והן על ידי RNases. לכן, מומלץ להשתמש באמצעים מיוחדים - שימוש בציוד מגן אישי (PPE), חומר נטול RNAse ומעכבי RNAse. והכי חשוב, RNA צריך להישמר על קרח במידת האפשר. זה חל במיוחד על RNA מתילציה, אשר רגיש עוד יותר לטמפרטורות גבוהות.
חשוב לוודא שמבנה הרנ"א אינו רגיש לריכוז ה-DMS ולתנאי החיץ. מאגרים כגון 100 mM Tris, 100 mM MOPS ו- 100 mM HEPES ב- pH 7-7.5 נותנים אות גבוה אך עשויים שלא להספיק כדי לשמור על ה- pH במהלך התגובה21. כאשר DMS עובר הידרוליזה במים, מה שמפחית את ה-pH, חיץ חזק הוא קריטי לשמירה על pH נייטרלי במהלך תגובת השינוי. התוספת של bicine הוכחה כמסייעת לשמור על ה- pH כבסיסי מעט21 אך גורמת לשינוי DMS נמוך ב- Gs ו- Us, אשר יכול להיות אינפורמטיבי אך יש לנתח אותו בנפרד בשל ייצור אות נמוך בהרבה מאשר As ו- Cs ואינו נדון בהמשך פרוטוקול זה.
ב-RT-PCR ספציפי לגנים, הרנ"א המהונדס מתועתק לאחור לתוך הדנ"א ומוגבר בשברים על ידי PCR. בעוד שגודל הרנ"א יכול תיאורטית להיות בלתי מוגבל, מקטעי PCR אלה לא יעלו על אורך של 400-500 זוגות בסיסים (bp) כדי למנוע הטיה במהלך תגובת השעתוק ההפוכה. באופן אידיאלי, השברים צריכים להיות בטווח של הרצת הרצף (כלומר, אם הרצף מתבצע באמצעות תוכנית ריצוף של 150 x 150 מחזור, מקטע יחיד לא יעלה על 300 bp). בעת שימוש בתוכניות ריצוף עם פחות מחזורים, ניתן לפצל את מוצרי ה-PCR באמצעות dsDNase. יתר על כן, מכיוון שרצפים בתוך רצפי הפריימר אינם מכילים מידע מבני כלשהו, השברים חייבים לחפוף כאשר הרנ"א הנחקר כולל מקטע >1. תגובות RT יכולות להכיל פריימרים מרובים של RT עבור מקטעים שונים (עד 10 פריימרים שונים של RT). בהתאם לרצפים, איגום פריימרים RT יכול להפוך את השעתוק ההפוך לפחות יעיל, אך בדרך כלל עובד היטב. כל תגובת PCR צריכה להתבצע בנפרד.
כאשר חוקרים RNA עם DMS, תנאי הניסוי ממלאים תפקיד נוסף, שכן רנ"א רבים אינם יציבים מבחינה תרמודינמית ומשנים את הקונפורמציה שלהם בהתבסס על גורמים סביבתיים כגון טמפרטורה. כדי למנוע אי סדרים, יש לשמור על תנאי הניסוי קבועים ככל האפשר, גם ביחס לזמני התגובה. נראה כי תנאי החיץ ניתנים להחלפה במידה מסוימת 17,20,22,23 כאשר התנאים הבסיסיים נשמרים - יכולת האגירה והנוכחות של יונים חד-ערכיים (Na) ודיוולנטיים (Mg) - כדי להבטיח קיפול תקין של RNA 24.
לגבי הכנת הספרייה של רנ"א שונה, יש לקחת בחשבון מספר היבטים. ראשית, כפי שצוין קודם לכן, רנ"א שעברו שינוי הם פחות יציבים ממקביליהם שלא שונו, כלומר הם עשויים לדרוש אופטימיזציה של זמני הפיצול להתפלגות אופטימלית של גודל השבר. יתר על כן, ערכות הכנה מסוימות של ספריית RNA, כמו גם גישות RNAseq רבות אחרות, משתמשות בפריימרים אקראיים בערכת השעתוק ההפוכה. זה עלול להוביל לכיסוי נמוך יותר של הייחוס, במיוחד ב-3' של גן, ובסופו של דבר, לעומק כיסוי לא מספיק. אם הכיסוי של אזור מסוים נמוך מדי, ייתכן שיהיה צורך להסיר בסיסים אלה מחיזוי המבנה. מלבד ערכות RT-PCR ו-RNAseq של גנום שלם, ניתן להשתמש בגישות אחרות להכנת ספרייה. פרוטוקולים הכוללים קשירת מתאמי 3' ו/או 5' ל-RNA הם יתרון כאשר משתמשים בשברי RNA קטנים או כאשר יש להימנע מאובדן מידע בדיקה באזורי הפריימר.
לבסוף, יש לפרש תמיד בזהירות את ניתוח הניסויים הכימיים. נכון לעכשיו, אין תוכנה המנבאת את מבנה הרנ"א של כל רנ"א מהרצף לבדו בדיוק גבוה. למרות שאילוצים של בדיקה כימית משפרים מאוד את הדיוק, יצירת מודלים טובים עבור רנ"א ארוך (>500 nt) עדיין מאתגרת. מודלים אלה צריכים להיבדק עוד יותר על ידי גישות אחרות ו/או מוטגנזה. תוכנת חיזוי RNA מבצעת אופטימיזציה למספר המרבי של זוגות בסיסים, ובכך מענישה באופן משמעותי קונפורמציות פתוחות, שאולי אינן מייצגות במדויק קיפול RNA5. לפיכך, יש לבחון את מודל המבנה המתקבל על ידי כימות הסכם החיזוי עם נתוני הבדיקה הכימיים הבסיסיים (למשל, על ידי AUROC) ובין שכפולים (למשל, על ידי mFMI), כפי שהודגם על ידי Lan et al.20.
באופן אידיאלי, יש להשתמש במספר ניסויים במערכות שונות כדי לאתגר את מודל המבנה המתקבל כדי לחזק את ההשערה. אלה יכולים לכלול שימוש בגישות חוץ גופיות ובתוך התא, מוטציות מפצות וקווי תאים ומינים שונים. יתר על כן, תגובות גולמיות הן לעתים קרובות אינפורמטיביות באותה מידה או אפילו יותר מאשר תחזיות מבנה, מכיוון שהן מתעדות את תמונת "האמת הקרקעית" של אנסמבל קיפול הרנ"א. ככזה, תגובתיות גולמית מתאימה מאוד ואינפורמטיבית להשוואת שינויי מבנה בין תנאים שונים. חשוב לציין שמבני האנרגיה החופשית הנמוכים ביותר המחושבים באמצעות אילוצי בדיקה כימית עם חיזוי חישובי צריכים לשמש רק כהשערת מוצא לקראת מודל מבנה שלם.
Disclosures
למחברים אין ניגודי עניינים להצהיר.
Acknowledgements
ללא
Materials
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
References
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved