
Method Article
Criblage de la structure de l’ARN avec le profilage mutationnel du sulfate de diméthyle avec séquençage in vitro et dans les cellules
Dans cet article
Résumé
Le protocole fournit des instructions pour modifier l’ARN avec du sulfate de diméthyle pour les expériences de profilage mutationnel. Il comprend le sondage in vitro et in vivo avec deux méthodes alternatives de préparation de bibliothèque.
Résumé
Le rôle de la structure de l’ARN dans pratiquement tous les processus biologiques est devenu de plus en plus évident, en particulier au cours de la dernière décennie. Cependant, les approches classiques pour résoudre la structure de l’ARN, telles que la cristallographie de l’ARN ou la cryo-EM, n’ont pas réussi à suivre l’évolution rapide du domaine et le besoin de solutions à haut débit. Profilage mutationnel avec séquençage à l’aide de sulfate de diméthyle (DMS) MaPseq est une approche basée sur le séquençage pour déduire la structure de l’ARN à partir de la réactivité d’une base avec le DMS. Le DMS méthyle l’azote N1 dans les adénosines et le N3 dans les cytosines au niveau de leur face Watson-Crick lorsque la base n’est pas appariée. La transcription inverse de l’ARN modifié avec la transcriptase inverse d’intron du groupe II thermostable (TGIRT-III) conduit à l’incorporation des bases méthylées sous forme de mutations dans l’ADNc. Lors du séquençage de l’ADNc résultant et de sa mise en correspondance avec une transcription de référence, les taux de mutation relatifs pour chaque base indiquent le « statut » de la base en tant que paire ou non appariée. Même si les réactivités DMS ont un rapport signal sur bruit élevé à la fois in vitro et dans les cellules, cette méthode est sensible aux biais dans les procédures de manipulation. Pour réduire ce biais, cet article fournit un protocole pour le traitement de l’ARN avec DMS dans les cellules et avec de l’ARN transcrit in vitro .
Introduction
Depuis la découverte que l’ARN a à la fois des propriétés structurelles1,2 et catalytiques3, l’importance de l’ARN et sa fonction de régulation dans une pléthore de processus biologiques ont été progressivement découvertes. En effet, l’effet de la structure de l’ARN sur la régulation des gènes a attiré une attention croissante4. Comme les protéines, l’ARN a des structures primaires, secondaires et tertiaires, se référant respectivement à la séquence des nucléotides, à la cartographie 2D des interactions d’appariement de bases et au repliement 3D de ces structures paires de bases. Bien que la détermination de la structure tertiaire soit essentielle pour comprendre les mécanismes exacts derrière les processus dépendants de l’ARN, la structure secondaire est également très informative en ce qui concerne la fonction de l’ARN et constitue la base d’un repliement 3Dultérieur 5.
Cependant, la détermination de la structure de l’ARN a été intrinsèquement difficile avec les approches conventionnelles. Alors que pour les protéines, la cristallographie, la résonance magnétique nucléaire (RMN) et la microscopie électronique cryogénique (cryo-EM) ont permis de déterminer la diversité des motifs structuraux, permettant de prédire la structure à partir de la séquence seule6, ces approches ne sont pas largement applicables aux ARN. En effet, les ARN sont des molécules flexibles avec des blocs de construction (nucléotides) qui ont beaucoup plus de liberté conformationnelle et rotationnelle par rapport à leurs homologues d’acides aminés. De plus, les interactions par appariement de bases sont plus dynamiques et polyvalentes que celles des résidus d’acides aminés. En conséquence, les approches classiques n’ont été couronnées de succès que pour des ARN relativement petits avec des structures bien définies et très compactes7.
Une autre approche pour déterminer la structure de l’ARN consiste à utiliser un sondage chimique combiné au séquençage de nouvelle génération (NGS). Cette stratégie génère des informations sur l’état de liaison de chaque base dans une séquence d’ARN (c’est-à-dire sa structure secondaire). En bref, les bases d’une molécule d’ARN qui ne s’engagent pas dans l’appariement de bases sont modifiées différentiellement par de petits composés chimiques. La transcription inverse de ces ARN avec des transcriptases inverses spécialisées (RT) incorpore les modifications dans l’acide désoxyribonucléique complémentaire (ADNc) sous forme de mutations. Ces molécules d’ADNc sont ensuite amplifiées par la réaction en chaîne de la polymérase (PCR) et séquencées. Pour obtenir des informations sur leur « statut » lié ou non lié, les fréquences de mutation à chaque base d’un ARN d’intérêt sont calculées et saisies dans un logiciel de prédiction de structure sous forme de contraintes8. Basé sur les règlesdu plus proche voisin 9 et les calculs d’énergie libre minimale 10, ce logiciel génère des modèles de structure qui correspondent le mieux aux données expérimentales obtenues11,12.
Le DMS-MaPseq utilise le DMS, qui méthyle l’azote N1 dans les adénosines et l’azote N3 dans les cytosines au niveau de leur face Watson-Crick d’une manière très spécifique13. L’utilisation de la transcriptase inverse d’intron thermostable du groupe II (TGIRT-III) en transcription inverse crée des profils mutationnels avec des rapports signal sur bruit sans précédent, permettant même la déconvolution de profils chevauchants générés par deux ou plusieurs conformations alternatives14,15. De plus, le DMS peut pénétrer dans les membranes cellulaires et les tissus entiers, ce qui rend possible le sondage dans des contextes physiologiques. Cependant, la génération de données de bonne qualité est difficile, car les variations dans la procédure de traitement peuvent avoir un impact sur les résultats. Par conséquent, nous fournissons un protocole détaillé pour le DMS-MaPseq in vitro et dans la cellule afin de réduire les biais et de guider les nouveaux arrivants vers la méthode à travers les difficultés qu’ils peuvent rencontrer. Surtout à la lumière de la récente pandémie de SRAS-CoV2, des données de haute qualité sur les virus à ARN sont un outil important pour étudier l’expression des gènes et trouver des traitements possibles.
Protocole
REMARQUE : Consultez le tableau des matériaux pour plus de détails sur tous les matériaux, logiciels, réactifs, instruments et cellules utilisés dans ce protocole.
1. DMS-MaP in vitro spécifique au gène
- Transcription de l’ARN in vitro
- Obtenir la séquence de l’ARN d’intérêt sous forme d’ADN double brin (ds) (p. ex., sous forme de fragments d’ADN, de plasmides ou de PCR à partir d’ADN préexistant/génomique). Si la séquence d’ADN contient un promoteur de polymérase, passez à l’étape 3.
- Effectuer une PCR de chevauchement pour attacher un promoteur d’ARN polymérase en amont du fragment d’ADN désiré (amorce directe pour la polymérase T7 : TAATACGACTCACTATAGG 5' + premières bases de la séquence cible 3').
- Transcrire in vitro le fragment d’ADN en ARN. Gardez toujours l’ARN sur la glace.
- Digérer l’ADN à l’aide d’une DNase.
- Isoler l’ARN en utilisant une approche basée sur la colonne (étape 2.4) ou par précipitation à l’éthanol (étape 2.5). Éluer dans un volume approprié, en espérant un rendement de ~50 μg.
- Assurer l’intégrité de l’ARN en l’exécutant sur un gel d’agarose; dénaturer l’ARN pendant 2-3 min à 70 °C avant de l’exécuter.
REMARQUE: Le tampon et l’agarose peuvent contenir des RNases qui dégradent l’ARN et peuvent contaminer l’échantillon d’ARN. Des gels d’agarose préfabriqués ont déjà été utilisés dans ce laboratoire; les résultats (en particulier avec l’ARN) ont parfois été ambigus. Les meilleurs résultats ont été obtenus avec des gels d’agarose ou de PAGE. - Utiliser directement ou stocker l’ARN à −80 °C pendant plusieurs mois, sauf si une dégradation est visible après décongélation.
- In vitro Modification DMS (à 105 mM DMS)
- Préparer une quantité suffisante de tampon de repliement (cacodylate de sodium 0,4 M, pH 7,2, contenant 6 mM de MgCl2).
REMARQUE : Pour chaque réaction (volume final de 100 μL), ajouter 89 μL de tampon repliable. - Pour chaque réaction, transférer 89 μL de tampon repliable dans un tube désigné de 1,5 mL et préchauffer à 37 °C dans un thermoshaker placé sous une hotte chimique.
REMARQUE: Le DMS est hautement toxique et doit toujours être conservé sous une hotte chimique jusqu’à ce qu’il soit éteint par un agent réducteur. - Éluer 1-10 pmol d’ARN dans 10 μL d’eau sans nucléase (NFH2O); transférer dans un tube PCR.
- Incuber dans un thermocycleur à 95 °C pendant 1min pour dénaturer l’ARN.
- Placer immédiatement sur un bloc de glace pour éviter les mauvais pliages.
- Ajouter l’échantillon d’ARN dans le tube désigné avec un tampon de repliement à 37 °C, bien mélanger et incuber pendant 10-20 minutes pour remodeler l’ARN.
REMARQUE: La plupart des ARN se replient de l’ordre de millisecondes à secondes, bien qu’il existedes exceptions 16. - Ajouter 1 μL de DMS à 100 % (10,5 M) à l’échantillon d’ARN et incuber pendant 5 minutes tout en agitant à 800-1 400 rotations par minute (rpm).
REMARQUE: L’agitation (ou d’autres moyens de mélange) à cette étape est cruciale car le DMS est hydrophobe et peut ne pas se dissoudre complètement dans le tampon de repliement. Des écarts dans les temps de réaction peuvent affecter la reproductibilité des réactivités DMS. Pour minimiser les erreurs de pipetage, le DMS peut être dissous dans de l’éthanol à 100 % avant de l’ajouter à l’échantillon si une concentration finale de 1 % (105 mM) de DMS est maintenue. Pour un contrôle non traité, le DMS peut être remplacé par du diméthylsulfoxyde (DMSO) ou de l’eau. - Après 5 min de temps de réaction, tremper avec 60 μL de β-mercaptoéthanol (BME) à 100 %, bien mélanger et placer immédiatement l’ARN sur la glace.
REMARQUE: L’ARN peut être retiré en toute sécurité de la hotte après avoir éteint la réaction avec BME pour le nettoyer. Cependant, l’exposition directe de BME à l’environnement doit toujours être évitée en raison de sa forte odeur et de ses propriétés irritantes. - Nettoyer l’ARN par précipitation acétate-éthanol de sodium (voir étape 2.5) ou par une approche en colonne (voir étape 2.6), et éluer dans 10 μL d’eau.
- Quantifier l’ARN à l’aide d’un spectrophotomètre.
- Utilisation directe ou stockage de l’ARN modifié à −80 °C.
REMARQUE: Le stockage à long terme doit être évité, car l’ARN est moins stable après le traitement DMS.
- Préparer une quantité suffisante de tampon de repliement (cacodylate de sodium 0,4 M, pH 7,2, contenant 6 mM de MgCl2).
- RT-PCR spécifique du gène de l’ARN modifié
REMARQUE : Voir la figure 1 pour la configuration RT-PCR des fragments traités par DMS.- Éluer 100 ng d’ARN modifié dans 10 μL deH2Osans nucléase (NF). Transfert dans un tube PCR.
- Dans le tube, ajouter 4 μL de tampon premier brin (FSB) 5x, 1 μL de mélange dNTP (10 mM chacun), 1 μL de dithiothréitol 0,1 M (DTT) (éviter les cycles de congélation-décongélation), 1 μL d’inhibiteur de RNase, 1 μL d’amorce inverse 10 μM (amorce simple ou un pool d’amorces) et 1 μL de TGIRT III.
REMARQUE : Pour un ensemble d’amorces, n’ajoutez pas 1 μL de 10 μM de chaque amorce directement au TA; au lieu de cela, mélangez d’abord les amorces et ajoutez 1 μL du mélange (à une concentration totale de 10 μM d’amorce). - Incuber à 57 °C pendant 30 min à 1,5 h (généralement, 30 min suffisent pour fabriquer un produit de 500 nt) dans un thermocycleur.
- Ajouter 1 μL de NaOH 4 M, mélanger par pipetage et incuber à 95 °C pendant 3 min pour dégrader l’ARN.
REMARQUE: Cette étape est cruciale car elle libère TGIRT de l’ADNc en dégradant l’ARN. Si elle est ignorée, la PCR en aval peut être affectée. - Nettoyez en utilisant une approche basée sur des colonnes (voir étape 2.6) qui élimine suffisamment les amorces, et éluez 10 μL de NFH2O.
- La PCR amplifie l’ADNc en utilisant 1 μL de produit de transcription inverse par 25 μL de réaction avec un kit PCR conçu pour équilibrer le rendement et la fidélité.
REMARQUE: Les amorces doivent avoir une température de fusion de ~60 °C. - Exécutez 2 μL du produit PCR sur un gel d’agarose ou un gel d’agarose préfabriqué pour vérifier le succès de la PCR.
- Idéalement, une seule bande devrait apparaître après la PCR. Si c’est le cas, nettoyez la réaction à l’aide d’une approche basée sur les colonnes. Si d’autres bandes sont présentes, utilisez la réaction PCR restante pour exciser la bande correcte du gel. Éluer dans un volume suffisamment petit (p. ex. 10 μL).
- Quantifier les fragments extraits à l’aide d’un spectrophotomètre.
- Indexez les fragments dsDNA pour le séquençage en utilisant une approche adaptée à la plate-forme de séquençage souhaitée.

Figure 1 : Configuration expérimentale pour la RT-PCR de gros fragments traités par DMS. Lors de la transcription inverse sur un ARN modifié, les modifications sur la séquence à laquelle les recuits d’amorce ne seront pas enregistrées. Ainsi, lorsque les fragments dépassent 400-500 pb de longueur, des fragments se chevauchant dans les régions d’amorce doivent être conçus, comme illustré ici. La longueur des fragments dépend des besoins de séquençage. Lors de l’utilisation d’un séquençage à 150 cycles appariés, les fragments ne doivent pas dépasser 300 pb. Abréviations : RT-PCR = réaction en chaîne de la polymérase à transcription inverse; DMS = sulfate de diméthyle. Veuillez cliquer ici pour voir une version agrandie de cette figure.
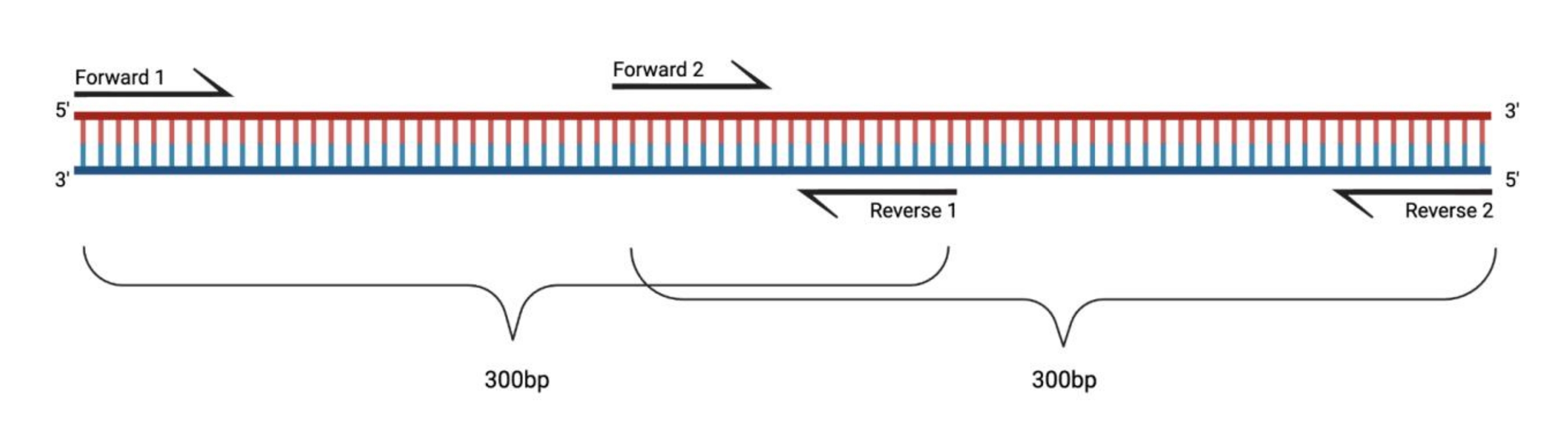{kind=link}
2. DMS-MaP du génome entier utilisant des cellules infectées par le virus
REMARQUE: Dans les cellules, le traitement DMS peut également être combiné avec l’approche d’amplification spécifique du gène décrite ci-dessus. La bibliothèque du génome entier nécessite une énorme profondeur de séquençage pour obtenir une couverture complète sur un seul gène. Cependant, si les ARN viraux constituent une fraction significative de l’ARN ribodéplé après extraction, le séquençage du génome entier serait approprié. En outre, d’autres méthodes d’enrichissement peuvent être combinées avec la méthode de génération de bibliothèque de génomes entiers.
- Traitement DMS
- Cultiver des cellules infectées par le virus jusqu’au stade d’infection souhaité.
- Transférer le contenant de la cellule dans une hotte dédiée qui convient à la manipulation des virus au niveau de biosécurité requis et des vapeurs chimiques générées par des agents tels que le DMS.
- Ajouter un volume de DMS de 2,5 % au milieu de culture et sceller le récipient (généralement une plaque de 10 cm) avec du parafilm.
REMARQUE: Il est facile de sous-modifier et de sur-modifier avec DMS. Lors de l’ajout de DMS directement aux cellules, il est très important de bien mélanger. Sinon, préchauffer le nouveau milieu dans un tube conique de 50 ml à 37 °C et ajouter le DMS directement en agitant vigoureusement. Décanter le milieu usé sur les cellules et pipeter lentement dans le milieu contenant du DMS. - Transférer dans un incubateur à 37 °C pendant 5 min.
REMARQUE: Selon le temps nécessaire pour manipuler le DMS à l’extérieur de l’incubateur, il est possible que 5 minutes entraînent une modification excessive. Gardez le temps entre l’ajout du DMS et l’incubation à ≤1 min. Si vous effectuez l’expérience pour la première fois, il est recommandé de faire un titrage DMS et de varier le temps d’incubation (entre 3 min et 10 min) pour trouver le taux de modification optimal et s’assurer que les résultats sont robustes sur une fenêtre de concentrations. - Extraire soigneusement le milieu contenant du DMS (dans les déchets chimiques appropriés) et ajouter doucement 10 mL de tampon stop (PBS contenant 30 % de BME [p. ex. 3 mL de BME et 7 mL de PBS]).
REMARQUE: L’ajout de DMS et de BME peut soulever les cellules de la plaque si les cellules ne sont pas fortement adhérentes. Si les cellules se soulèvent, elles peuvent être traitées comme des cellules en suspension - au lieu d’enlever le milieu contenant du DMS, ajoutez directement le tampon d’arrêt et grattez les cellules avec DMS et BME dans un tube conique de 50 mL. Enduire les cellules par centrifugation pendant 3 min à 3 000 × g ; assurez-vous de vous débarrasser de tout DMS résiduel, qui peut granuler sous les cellules en grosses gouttelettes. Une étape de lavage supplémentaire dans 30% de BME est recommandée si le milieu DMS ne peut pas être retiré initialement. - Grattez les cellules et transférez-les dans un tube conique de 15 mL.
- Granulé par centrifugation à 3 000 × g pendant 3 min.
- Retirer le surnageant et laver 2x avec 10 ml de PBS.
- Retirez soigneusement autant de PBS résiduel que possible.
- Dissoudre la pastille dans une quantité appropriée du réactif d’isolement de l’ARN (p. ex. 3 mL pour une fiole de culture T75, 1 mL pour une plaque de 10 cm).
REMARQUE: Des quantités insuffisantes de réactif peuvent avoir un impact sur le rendement en ARN.
- Extraction d’ARN et épuisement de l’ARN ribosomique (ARNr)
- À 1 mL de cellules homogénéisées dans le réactif d’isolement de l’ARN, ajouter 200 μL de chloroforme, vortex pendant 15 à 20 s jusqu’à ce qu’il soit rose vif, puis incuber jusqu’à 3 minutes jusqu’à ce que la séparation de phase soit visible.
REMARQUE: La phase lipidique rose devrait se déposer au fond. Si ce n’est pas le cas, le temps de vortex était probablement insuffisant. - Rotation à vitesse maximale (~ 20 000 × g) pendant 15 min à 4 °C.
- Transférer la phase aqueuse supérieure dans un nouveau tube.
- Nettoyer l’ARN par précipitation acétate-éthanol de sodium (voir étape 2.5) ou par une approche en colonne (voir étape 2.6), et éluer dans un volume suffisant de NFH2O.
- Vérifiez l’intégrité de l’ARN sur un gel d’agarose. Recherchez deux bandes correspondant aux deux sous-unités ribosomiques.
- Épuiser les ARNr en utilisant l’approche préférée et éluer dans un volume adéquat (typiquement 20-50 μL) de NF H2O.
REMARQUE: Pour les applications en aval, ~500 ng d’ARN total est suggéré dans un volume de 8 μL. Les ARN non ribosomales ne représentent généralement que 5% à 10% de l’ARN total. - Quantifier à l’aide d’un spectrophotomètre.
- À 1 mL de cellules homogénéisées dans le réactif d’isolement de l’ARN, ajouter 200 μL de chloroforme, vortex pendant 15 à 20 s jusqu’à ce qu’il soit rose vif, puis incuber jusqu’à 3 minutes jusqu’à ce que la séparation de phase soit visible.
- Génération de bibliothèques
- Utiliser la RT-PCR spécifique aux gènes ou d’autres approches pour générer des bibliothèques15. Si vous utilisez des hexamères aléatoires pour l’amorçage, ajouter une étape d’incubation à faible Tm (37-42 °C) pour permettre le recuit de l’hexamère.
REMARQUE: Les kits de génération de bibliothèque standard peuvent également être utilisés en remplaçant l’enzyme RT par TGIRT et en modifiant la température RT à 57 ° C.
- Utiliser la RT-PCR spécifique aux gènes ou d’autres approches pour générer des bibliothèques15. Si vous utilisez des hexamères aléatoires pour l’amorçage, ajouter une étape d’incubation à faible Tm (37-42 °C) pour permettre le recuit de l’hexamère.
- Nettoyage de l’ARN basé sur les colonnes à l’aide des colonnes RNA Clean & Concentrator
REMARQUE: Toutes les étapes doivent être effectuées à température ambiante.- Ajouter NFH2Oau tube à échantillon pour l’amener à un volume de 50 μL.
- Ajouter 100 μL de tampon de liaison et 150 μL d’éthanol à 100 % à l’échantillon.
- Mélangez et transférez dans une colonne de rotation.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- Ajouter 400 μL de tampon de préparation de l’ARN.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- Ajouter 700 μL de tampon de lavage d’ARN.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- Ajouter 400 μL de tampon de lavage d’ARN.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- (Facultatif) Transférer la colonne dans un nouveau tube de collecte et faire tourner à 10 000-16 000 × g pendant 2 min.
- Transférer la colonne dans un tube propre sans RNAse et ajouter une quantité appropriée de NFH2O.
- Faire tourner à 10 000-16 000 × g pendant 1 min.
- Extraction acide de l’ARN phénol-chloroforme.
- Ajouter un volume égal d’acide phénol:chloroforme:alcool isoamylique.
- Vortex à fond, et centrifuger à 14 000 × g pendant 5 min.
- S’il n’y a pas de séparation de phase, ajouter 20 μL de NaCl 2 M et répéter la centrifugation.
- Transférer la phase aqueuse dans un nouveau tube.
- Ajouter 500 μL d’isopropanol et 2 μL de coprécipitant.
- Mélanger et incuber à TA pendant 3 min; puis incuber à −80 °C pendant la nuit.
- Enduire l’ARN par centrifugation à vitesse maximale (~ 20 000 × g) pendant 30 min à 4 °C.
- Lavez la pastille avec 200 μL d’éthanol glacé à 70 %.
- Rotation à vitesse maximale (~ 20 000 × g) pendant 5 min; Ignorez le flux.
- Remettez la pastille en suspension dans la quantité appropriée de NFH2O.
- Nettoyage de l’ADNc en colonne à l’aide des colonnes Oligo Clean et Concentrator
REMARQUE: Toutes les étapes doivent être effectuées à température ambiante.- Ajouter NFH2Oau tube à échantillon pour l’amener à un volume de 50 μL.
- Ajouter 100 μL de tampon de liaison et 400 μL d’éthanol à 100 %.
- Mélangez et transférez dans une colonne de rotation.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- Ajouter 750 μL de tampon de lavage d’ADN.
- Essorer à 10 000-16 000 × g pendant 30 s; Ignorez le flux.
- (Facultatif) Transférer la colonne dans un nouveau tube de collecte et faire tourner à 10 000-16 000 × g pendant 2 min.
- Transférer la colonne dans un tube propre sans RNAse et ajouter une quantité appropriée de NFH2O.
- Faire tourner à 10 000-16 000 × g pendant 1 min.
3. Analyse des données de séquençage
Remarque : Pour créer des modèles de structure secondaire ARN à partir des données de séquençage DMS-MaP, les fichiers .fastq résultants doivent être traités par plusieurs étapes différentes. Ces étapes peuvent être effectuées automatiquement à l’aide de l’icône
- Découpez les séquences de l’adaptateur avec TrimGalore ou Cutadapt.
- Mappez les lectures aux séquences de référence (format .fasta) à l’aide de Bowtie2.
- Comptez les lectures avec un logiciel spécialisé de structure d’ARN (par exemple, DREEM14, RNA-Framework17 ou similaire) et créez des profils de réactivité.
- (Facultatif) Regroupez les lectures pour trouver d’autres conformations d’ARN en utilisant DREEM14, DRACO 17, DANCE-MaP18 ou similaire.
- Prédire la structure minimale d’énergie libre en fonction des profils de réactivité à l’aide de RNAStructure12, ViennaRNA ou similaire.
- Visualisez la structure de l’ARN11 en utilisant VARNA (https://varna.lri.fr/) ou similaire.
REMARQUE: Pour des raisons pratiques, des logiciels tels que DREEM (www.rnadreem.org) et RNA-Framework19 intègrent largement les étapes 1 à 5 dans leurs pipelines, ce qui rationalise le processus d’analyse. Cependant, toute prédiction de structure doit être manipulée avec précaution (par exemple, en vérifiant l’accord de la structure avec les données20.
Résultats
DMS-MaP in vitro spécifique au gène
Pour étudier le 5'UTR du SRAS2, les 300 premiers pb du virus ont été ordonnés sous forme de séquence gBlock, aux côtés de trois amorces. Ceux-ci comprenaient deux amorces pour propager le fragment (« FW » & « RV ») par PCR, ainsi qu’une pour fixer le promoteur T7 (« FW-T7 »). Ces séquences sont présentées dans le tableau 1.
| Nom | Séquence (5'->3') |
| FW | ATTAAAGGTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
Tableau 1 : Séquence d’amorce pour la DMS-MaP RT-PCR du SARS-CoV2 5'UTR. Ici, FW-T7 et RV sont nécessaires pour générer un modèle d’ADN pour la transcription in vitro , le RV est utilisé dans la transcription inverse et la paire d’amorces FW-RV est utilisée dans l’amplification PCR ultérieure de l’ADNc. Les amorces recuisent jusqu’au tout début du génome du SARS-CoV2 (FW) et la séquence juste en aval de la région d’intérêt. Abréviations : DMS-MaP = Profilage mutationnel avec séquençage à l’aide de sulfate de diméthyle; RT-PCR = réaction en chaîne de la polymérase à transcription inverse; SRAS-CoV2 = syndrome respiratoire aigu sévère-coronavirus 2; UTR = région non traduite; RV = amorce inversée; FW = amorce avant.
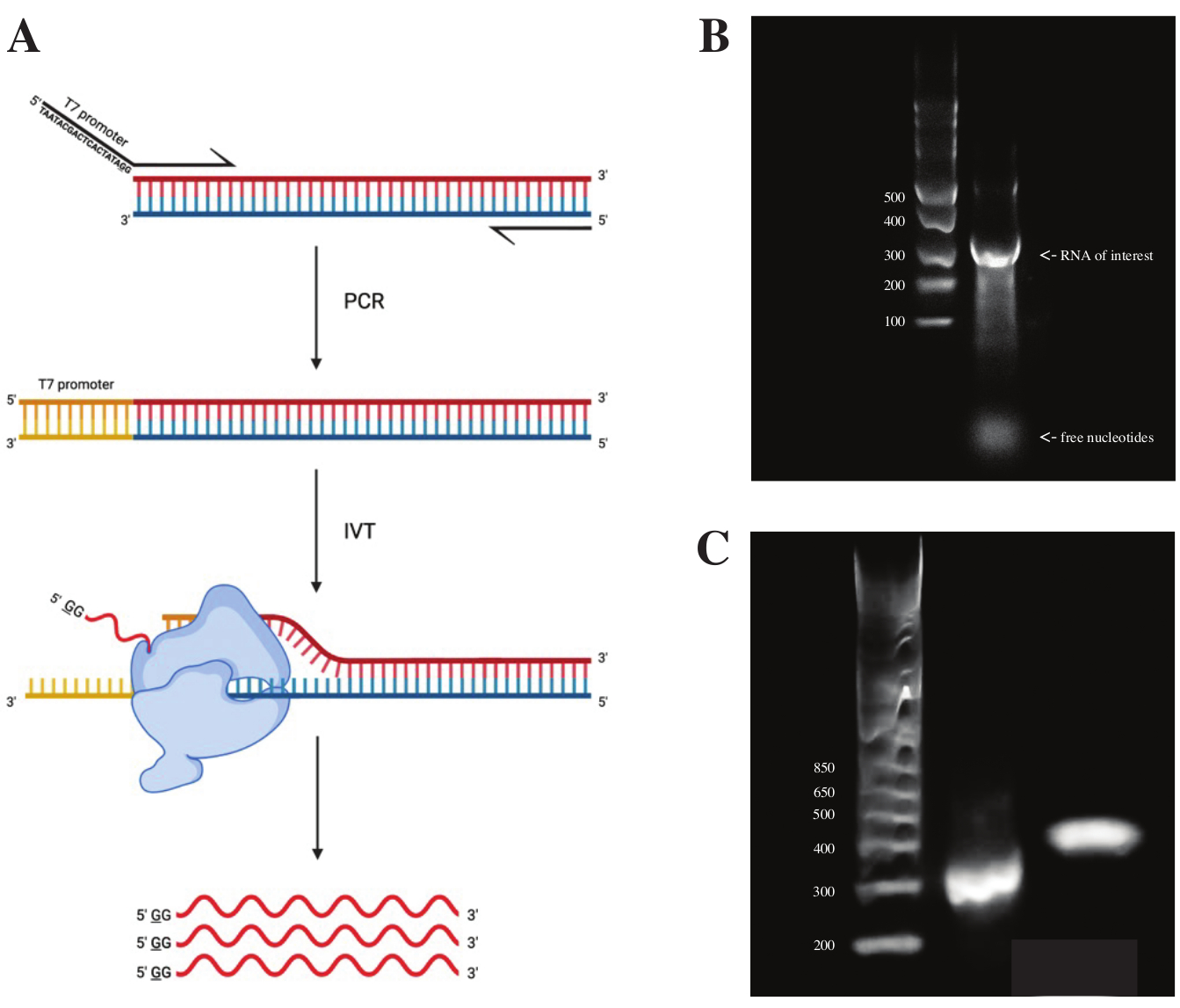
Pour générer de l’ARN à partir du fragment gBlock, la séquence du promoteur de la polymérase T7 a été fixée à l’aide de la PCR à chevauchement en utilisant le prémélange PCR selon le schéma illustré à la figure 2A. À partir du fragment allongé, l’ARN a été généré à l’aide du kit de transcription T7. Le modèle d’ADN a ensuite été digéré à l’aide de la DNase et l’ARN isolé à l’aide de colonnes RNA Clean & Concentrator.
Le contrôle de la qualité de la transcription in vitro a été effectué en exécutant le produit d’ARN sur un gel d’agarose à 1% le long d’une échelle d’ARNs. Comme il n’y avait qu’une seule bande visible, un sondage in vitro DMS et RT-PCR ont été effectués (voir Figure 2B).
Pour vérifier le succès de la réaction PCR, l’échantillon a été exécuté sur un gel d’agarose à 2% à l’aide d’une échelle d’ADNds. Après l’indexation, la bande doit fonctionner ~ 150 pb plus haut sur le même gel, en tenant compte de la taille des amorces d’indexation.

Figure 2 : Transcription in vitro du modèle d’ADN. (A) Pour transcrire in vitro un modèle d’ADN qui n’a pas encore de promoteur intrinsèque de l’ARN polymérase, le modèle doit d’abord être attaché par PCR de chevauchement. Cela se fait en utilisant une amorce directe, qui comprend la séquence TAATACGACTCACTATAGG (dans le cas de l’ARN polymérase T7) en amont des premières bases chevauchant le fragment désiré. La base soulignée symbolise ici le site de début de la transcription de la polymérase. Une fois que le promoteur s’est attaché au fragment d’ADNds, il peut être transcrit par la polymérase T7. Il est important de noter que la polymérase utilise le brin opposé à la séquence promotrice mentionnée comme matrice (bleu), créant ainsi un ARN identique à la séquence immédiatement en aval de la séquence promotrice indiquée (rouge). (B) Un gel d’agarose à 1% avec une échelle d’ARNss (voie 1) et le produit d’ARN transcrit in vitro à 300 nt (voie 2). (C) Un gel d’agarose à 2 % avec GeneRuler 1 kb plus Ladder (voie 1), le produit PCR après RT-PCR fonctionnant à 300 pb (voie 2) et le fragment indexé après préparation de la bibliothèque fonctionnant à 470 pb (voie 3). Abréviations : RT-PCR = réaction en chaîne de la polymérase à transcription inverse; DMS = sulfate de diméthyle; nt = nucléotides; dsDNA = ADN double brin; ssRNA = ARN simple brin. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Génome entier in vivo DMS-MaP utilisant des cellules infectées par le virus
Avant le traitement DMS, les cellules HCT-8 étaient infectées par OC43. Lorsqu’un effet cytopathique (ECP) a été observé 4 jours après l’infection (dpi) (comme le montre la figure 3A), ces cellules ont été traitées, et l’ARN a été extrait et ribodé. Lors de l’exécution de l’ARN total sur un gel d’agarose, deux bandes brillantes étaient visibles, représentant les sous-unités 40S et 60S du ribosome, qui représentent environ 95% de la masse totale d’ARN (voir Figure 3B). Lorsque l’extraction de l’ARN a échoué ou a été dégradée (p. ex. par plusieurs cycles de gel-dégel), les produits de dégradation de l’ARN étaient visibles au fond du gel (voir la figure 3C, deuxième voie). De plus, après l’épuisement de l’ARNr, les deux bandes brillantes ont disparu, laissant un frottis dans la voie (voir Figure 3C, troisième voie). Enfin, après la préparation de la bibliothèque, les échantillons avaient des distributions de taille variables et ont été montrés sous forme de frottis sur le gel PAGE final. La bande a été excisée entre 200 nucléotides (nt) et 500 nt, en accord avec le séquençage d’extrémités appariées 150 x 150 prévu pour analyser ces bibliothèques. Plus important encore, les dimères de l’adaptateur fonctionnant à ~150 nt ont été séparés (voir Figure 3D).

Figure 3 : Points de contrôle du DMS-MaP in vivo avec des cellules infectées par le virus. (A) Image de microscopie optique de cellules HCT-8 infectées par le virus, 4 jours dpi. Pour obtenir le rendement le plus élevé possible d’ARN viral à partir de l’ARN total tout en minimisant les effets indésirables dus à la mort cellulaire, le DMS doit être ajouté au début de l’EPC ou même avant, comme on le voit sur l’image. (B) Un gel d’agarose à 1 % avec six échantillons de 1 μg d’ARN total. Dans chaque voie, deux bandes brillantes, représentant les sous-unités 40S et 60S, sont visibles, car l’ARN ribosomique représente ~95% de l’ARN total. Remarque: Le traitement DMS dans la cellule provoque une fragmentation et un frottis de l’ARN, mais les deux bandes d’ARNr doivent toujours être visibles. Une légère fragmentation post-modification est tolérée car l’information contenant la marque de méthylation est générée et rend compte de la structure de l’ARN pendant l’incubation du DMS alors que les cellules sont encore en vie. (C) Un gel d’agarose à 1% de GeneRuler 1 kb plus un marqueur d’ADN à échelle (voie 1), de l’ARN total précédemment stocké à -80 °C pendant 6 mois (voie 2) et de l’ARN ribodéplé (voie 3). Lors du stockage prolongé de l’ARN avec plusieurs cycles de gel-dégel, l’ARN commence à se dégrader et ne devrait peut-être pas être utilisé pour sonder des expériences. De plus, après avoir ribodé l’ARN total, les deux bandes brillantes, représentant les sous-unités 40S et 60S du ribosome, s’estompent et un frottis des ARN résiduels commence à apparaître. (D) Un gel PAGE de GeneRuler 1 kb plus un marqueur ADN à échelle (voie 1) et un échantillon de bibliothèque d’ARN préparé par le génome entier. Le gel doit être excisé en fonction des besoins de séquençage. Pour un séquençage d’extrémité appariée couvrant 150 cycles des deux côtés, le gel doit être excisé entre 300 pb et 500 pb. Les dimères d’adaptateur (fonctionnant à 170 bp) doivent être séparés. Abréviations : DMS-MaP = Profilage mutationnel avec séquençage à l’aide de sulfate de diméthyle; dpi = jours après l’infection; CPE = effet cytopathique. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
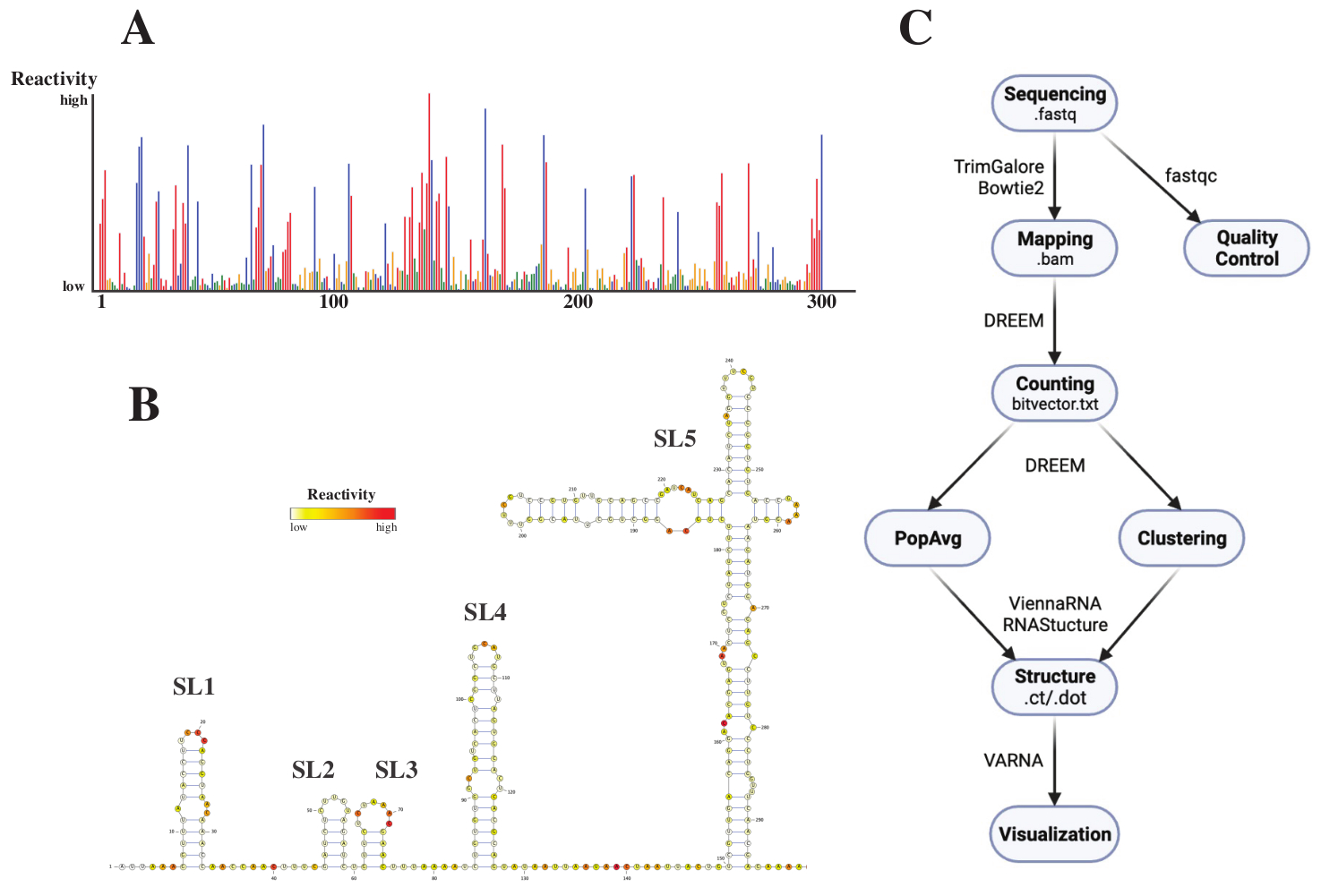
Après le séquençage, les fichiers .fastq ont été analysés en soumettant une tâche au serveur Web de DREEM (http://rnadreem.org/), ainsi qu’un fichier de référence .fasta. La sortie générée par le serveur comprend des fichiers de contrôle qualité générés par fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) et TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), ainsi que d’autres fichiers de sortie contenant les fréquences moyennes de mutation de la population. Outre le diagramme montrant les fréquences de mutation avec un format .html interactif (voir Figure 4A) et un fichier .csv avec les réactivites brutes par base et un fichier struct_constraint.txt, lisible par plusieurs logiciels de prédiction de structure d’ARN, cela comprend également un fichier bitvector.txt rapportant les mutations en lecture directe. À partir de ceux-ci, les structures moyennes de la population ont été calculées en soumettant les fichiers .fasta et struct_constraint.txt au serveur Web RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Celui-ci utilise le logiciel ViennaRNA pour générer des prédictions de structure basées sur l’énergie libre minimale, qui peuvent être visualisées en ligne ou téléchargées au format ct ou Vienna. Pour générer des modèles de structure d’ARN, ces fichiers téléchargeables ont été soumis à VARNA (https://varna.lri.fr/, voir Figure 4B). Enfin, les fichiers bitvector.txt peuvent être utilisés par la version stable de DREEM (https://codeocean.com/capsule/6175523/tree/v1) pour rechercher des conformations d’ARN alternatives. Pour obtenir de bons modèles de structure à l’aide de DREEM, une couverture de 10 000 lectures par base doit être atteinte; Pour le clustering, jusqu’à 100 000 lectures par base peuvent être nécessaires. Une vue d’ensemble de l’ensemble du flux de travail peut être trouvée à la figure 4C.

Figure 4 : Données exemplaires obtenues à partir d’expériences de sondage chimique du SARS-CoV2 5'UTR. (A) Profil de réactivité des 300 premières bases du génome du SARS-CoV2 colorées par base (A : rouge, C : bleu, U : vert, G : jaune). Les réactivités brutes sont calculées en divisant la fréquence absolue de mutation par la couverture. Les bases à conformation ouverte ont des valeurs de réactivité élevées; Les bases engagées dans l’appariement de bases ont de faibles valeurs de réactivité. u et G ne sont pas modifiés par DMS et ont de faibles valeurs de réactivité, provenant de l’infidélité de la polymérase. Les prédictions ont été faites avec le serveur web de la DREEM. (B) Modèle de structure du SARS-CoV2 5'UTR prédit à partir des valeurs de réactivité faites avec VARNA. Les bases avec des valeurs de réactivité élevées sont colorées en rouge; Les bases à faible valeur de réactivité sont colorées en blanc. (C) Workflow de l’analyse DMS-MaP en commençant par les fichiers .fastq obtenus à partir du séquençage. Ceux-ci peuvent être contrôlés en qualité à l’aide de fastqc; les séquences de l’adaptateur sont découpées à l’aide de TrimGalore puis mappées à une séquence de référence à l’aide de Bowtie2. À partir des fichiers .bam obtenus, DREEM compte les mutations dans chaque lecture, créant ainsi une carte de mutations ou un fichier .bitvector.txt. Ceux-ci rapportent les mutations de chaque lecture d’une manière dépendante de la position, sur la base de laquelle les profils de réactivité moyenne de la population peuvent être créés. Alternativement, les vecteurs binaires peuvent être regroupés en utilisant DREEM pour rechercher des conformations d’ARN alternatives. Enfin, les modèles de structure obtenus sont visualisés à l’aide d’un logiciel (par exemple, VARNA). Abréviations : DMS-MaP = Profilage mutationnel avec séquençage à l’aide de sulfate de diméthyle; SARS-CoV2 = syndrome respiratoire aigu sévère-coronavirus 2. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
Le protocole décrit ici comment sonder l’ARN in vitro et dans les cellules à l’aide d’expériences de profilage mutationnel DMS. En outre, il donne des instructions sur la façon de préparer les bibliothèques pour le séquençage Illumina afin de générer des données spécifiques aux gènes et d’analyser les fichiers .fastq obtenus. De plus, des approches de bibliothèque à l’échelle du génome peuvent être utilisées. Cependant, la RT-PCR spécifique aux gènes produit des données de la plus haute qualité et les plus robustes. Par conséquent, si vous comparez des échantillons, il est important de s’assurer qu’ils sont préparés avec des stratégies de séquençage identiques, car la génération de bibliothèque provoque un certain biais. La reproductibilité doit toujours être mesurée à l’aide de réplications.
Plusieurs précautions
L’ARN est une molécule instable qui est sensible à la dégradation à la fois par des températures élevées et par les RNases. Par conséquent, des mesures spéciales – l’utilisation d’équipement de protection individuelle (EPI), de matériel exempt d’ARNase et d’inhibiteurs de l’ARNase – sont recommandées. Plus important encore, l’ARN devrait être gardé sur la glace dans la mesure du possible. Cela s’applique particulièrement à l’ARN méthylé, qui est encore plus sensible aux températures élevées.
Il est important de confirmer que la structure d’intérêt de l’ARN n’est pas sensible à la concentration de DMS et aux conditions tampons. Des tampons tels que 100 mM Tris, 100 mM MOPS et 100 mM HEPES à pH 7-7,5 donnent un signal élevé mais peuvent ne pas être suffisants pour maintenir le pH pendant la réaction21. Comme le DMS s’hydrolyse dans l’eau, ce qui diminue le pH, un tampon puissant est essentiel pour maintenir un pH neutre pendant la réaction de modification. Il a été démontré que l’ajout de bicine aide à maintenir le pH légèrement basique21 , mais entraîne une faible modification du DMS sur Gs et Us, ce qui pourrait être informatif mais devrait être analysé séparément en raison de la production d’un signal beaucoup plus faible que As et Cs et n’est pas discuté plus en détail dans ce protocole.
Dans la RT-PCR spécifique au gène, l’ARN modifié est transcrit à l’envers dans l’ADN et amplifié en fragments par PCR. Bien que la taille de l’ARN puisse théoriquement être illimitée, ces fragments de PCR ne doivent pas dépasser une longueur de 400 à 500 paires de bases (pb) pour éviter les biais lors de la réaction de transcription inverse. Idéalement, les fragments devraient entrer dans le cadre de l’exécution de séquençage (c.-à-d. si le séquençage est effectué à l’aide d’un programme de séquençage d’extrémité appariée de 150 x 150 cycles, un seul fragment ne devrait pas dépasser 300 pb). Lors de l’utilisation de programmes de séquençage avec moins de cycles, les produits de PCR peuvent être fragmentés à l’aide d’une dsDNase. De plus, comme les séquences dans les séquences d’amorce ne contiennent aucune information structurelle, les fragments doivent se chevaucher lorsque l’ARN sondé comprend >1 fragment. Les réactions RT peuvent contenir plusieurs amorces RT pour différents fragments (jusqu’à 10 amorces RT différentes). Selon les séquences, la mise en commun des amorces RT peut rendre la transcription inverse moins efficace, mais fonctionne généralement bien. Chaque réaction PCR doit être conduite séparément.
Lors du sondage de l’ARN avec DMS, les conditions expérimentales jouent un rôle supplémentaire, car de nombreux ARN sont thermodynamiquement instables et modifient leur conformation en fonction de facteurs environnementaux tels que la température. Pour éviter les irrégularités, les conditions expérimentales doivent être maintenues aussi constantes que possible, y compris en ce qui concerne les temps de réaction. Les conditions tampons semblent être échangeables dans une certaine mesure 17,20,22,23 lorsque les conditions de base sont maintenues — la capacité tampon et la présence d’ions monovalents (Na) et divalents (Mg) — pour assurer un repliement correct de l’ARN 24.
En ce qui concerne la préparation en bibliothèque des ARN modifiés, plusieurs aspects doivent être pris en considération. Tout d’abord, comme mentionné précédemment, les ARN modifiés sont moins stables que leurs homologues non modifiés, ce qui signifie qu’ils pourraient nécessiter l’optimisation des temps de fragmentation pour une distribution optimale de la taille des fragments. De plus, certains kits de préparation de bibliothèque d’ARN, ainsi que de nombreuses autres approches RNAseq, utilisent des amorces aléatoires dans le kit de transcription inverse. Cela pourrait conduire à une couverture plus faible de la référence, en particulier dans les 3' d’un gène, et, finalement, à une profondeur de couverture insuffisante. Si la couverture d’une certaine région est trop faible, il peut être nécessaire de supprimer ces bases de la prévision de la structure. Outre la RT-PCR et les kits RNAseq du génome entier, d’autres approches de préparation de bibliothèque peuvent être utilisées. Les protocoles qui incluent la ligature d’adaptateurs 3' et/ou 5' à l’ARN sont avantageux lors de l’utilisation de petits fragments d’ARN ou lorsque la perte d’informations de sondage dans les régions d’amorce doit être évitée.
Enfin, l’analyse des expériences de sondage chimique doit toujours être interprétée avec prudence. Actuellement, il n’existe aucun logiciel qui prédit la structure de l’ARN d’un ARN à partir de la séquence seule avec une grande précision. Bien que les contraintes de sondage chimique améliorent considérablement la précision, générer de bons modèles pour les ARN longs (>500 nt) est encore difficile. Ces modèles devraient être testés davantage par d’autres approches et/ou la mutagénèse. Le logiciel de prédiction de l’ARN optimise le nombre maximum de paires de bases, pénalisant ainsi considérablement les conformations ouvertes, qui peuvent ne pas représenter avec précision le repliement de l’ARN5. Ainsi, le modèle de structure obtenu devrait être testé en quantifiant l’accord de prédiction avec les données de sondage chimique sous-jacentes (par exemple, par AUROC) et entre les répétitions (par exemple, par mFMI), comme l’illustrent Lan et al.20.
Idéalement, plusieurs expériences dans différents systèmes pour remettre en question le modèle de structure obtenu devraient être utilisées pour renforcer son hypothèse. Celles-ci peuvent inclure l’utilisation d’approches in vitro et intracellulaires, de mutations compensatoires et de différentes lignées cellulaires et espèces. De plus, les réactivités brutes sont souvent tout aussi informatives, voire plus, que les prédictions de structure, car elles enregistrent l’instantané de « vérité de terrain » de l’ensemble de repliement de l’ARN. En tant que telles, les réactivités brutes sont très appropriées et informatives pour comparer les changements de structure entre différentes conditions. Il est important de noter que les structures d’énergie libre les plus basses calculées à l’aide de contraintes de sondage chimique avec prédiction informatique ne devraient être utilisées que comme hypothèse de départ vers un modèle de structure complet.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à déclarer.
Remerciements
Aucun
matériels
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
Références
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.