
Method Article
디메틸 설페이트를 사용한 RNA 구조 프로빙 시험관 내 및 세포에서 시퀀싱을 사용한 돌연변이 프로파일링
요약
이 프로토콜은 돌연변이 프로파일링 실험을 위해 디메틸 설페이트로 RNA를 변형하기 위한 지침을 제공합니다. 여기에는 두 가지 대체 라이브러리 준비 방법을 사용한 시험관 내 및 생체 내 프로빙이 포함됩니다.
초록
거의 모든 생물학적 과정에서 RNA 구조의 역할은 특히 지난 10 년 동안 점점 더 분명 해졌습니다. 그러나 RNA 결정학 또는 Cryo-EM과 같은 RNA 구조를 해결하는 기존의 접근 방식은 빠르게 진화하는 분야와 고처리량 솔루션에 대한 요구를 따라가지 못했습니다. 디메틸 설페이트(DMS)를 사용한 시퀀싱을 통한 돌연변이 프로파일링 MaPseq는 염기와 DMS의 반응성에서 RNA 구조를 추론하는 시퀀싱 기반 접근 방식입니다. DMS는 염기가 짝을 이루지 않을 때 왓슨-크릭 얼굴에서 아데노신의 N1 질소와 시토신의 N3을 메틸화합니다. 변형된 RNA를 열안정 II 인트론 역전사효소 (TGIRT-III)로 역전사하면 메틸화된 염기가 cDNA 내로 돌연변이로서 혼입된다. 결과 cDNA를 시퀀싱하고 이를 참조 전사체에 다시 매핑할 때 각 염기에 대한 상대적 돌연변이율은 염기의 "상태"가 쌍 또는 쌍을 이루지 않았음을 나타냅니다. DMS 반응성은 시험관 내 및 세포 내 모두 높은 신호 대 잡음비를 갖지만 이 방법은 처리 절차의 편향에 민감합니다. 이러한 편향을 줄이기 위해 이 논문은 세포 내 DMS 및 체외 전사 RNA를 사용한 RNA 치료 프로토콜을 제공합니다.
서문
RNA가 구조적1,2 및 촉매3 특성을 모두 가지고 있다는 발견 이후, 과다한 생물학적 과정에서 RNA의 중요성과 조절 기능이 점차 밝혀졌습니다. 실제로, 유전자 조절에 대한 RNA 구조의 효과는 점점 더 주목을 받고 있습니다4. 단백질과 마찬가지로 RNA는 각각 뉴클레오티드의 서열, 염기쌍 상호 작용의 2D 매핑 및 이러한 염기쌍 구조의 3D 접힘을 나타내는 1차, 2차 및 3차 구조를 가지고 있습니다. 3차 구조를 결정하는 것이 RNA 의존적 과정의 정확한 메커니즘을 이해하는 데 중요하지만, 2차 구조는 RNA 기능과 관련하여 매우 유익하며 추가 3D폴딩5의 기초가 됩니다.
그러나 RNA 구조를 결정하는 것은 기존의 접근 방식으로는 본질적으로 어려웠습니다. 단백질의 경우 결정학, 핵자기 공명(NMR) 및 극저온 전자 현미경(cryo-EM)을 통해 구조적 모티프의 다양성을 결정할 수 있어서열만으로는 구조 예측이 가능하지만6, 이러한 접근 방식은 RNA에 널리 적용되지 않습니다. 실제로 RNA는 아미노산 대응 물에 비해 훨씬 더 구조적 및 회전 자유도를 갖는 빌딩 블록 (뉴클레오티드)을 가진 유연한 분자입니다. 또한, 염기 쌍을 통한 상호 작용은 아미노산 잔기의 상호 작용보다 더 역동적이고 다재다능합니다. 결과적으로, 고전적인 접근법은 잘 정의되고 고도로 조밀 한 구조를 갖는 비교적 작은 RNA에 대해서만 성공적이었습니다7.
RNA 구조를 결정하는 또 다른 접근법은 차세대 시퀀싱(NGS)과 결합된 화학적 프로빙을 통한 것입니다. 이 전략은 RNA 서열에서 각 염기의 결합 상태 (즉, 2 차 구조)에 대한 정보를 생성합니다. 간단히 말해서, 염기 쌍에 관여하지 않는 RNA 분자의 염기는 작은 화합물에 의해 차등적으로 변형됩니다. 특수 역전사효소(RT)로 이러한 RNA를 역전사하면 변형을 상보적인 디옥시리보 핵산(cDNA)에 돌연변이로 통합합니다. 이러한 cDNA 분자는 중합 효소 연쇄 반응 (PCR)에 의해 증폭되고 시퀀싱됩니다. 결합 또는 비결합으로서의 "상태"에 대한 정보를 얻기 위해, 관심 RNA의 각 염기에서의 돌연변이 빈도가 계산되고 제약 조건8로서 구조 예측 소프트웨어에 입력된다. 최근접 이웃 규칙(9) 및 최소 자유 에너지 계산(10)에 기초하여, 이 소프트웨어는 획득된 실험 데이터(11, 12)에 가장 적합한 구조 모델을 생성한다.
DMS-MaPseq는 매우 특이적인 방식으로 왓슨-크릭 얼굴의 아데노신 및 시토신의 N3 질소를 메틸화하는 DMS를 사용합니다13. 역전사에서 열안정성 그룹 II 인트론 역전사효소(TGIRT-III)를 사용하면 전례 없는 신호 대 잡음비를 갖는 돌연변이 프로파일이 생성되며, 심지어 둘 이상의 대체 형태(14,15)에 의해 생성된 중첩 프로파일의 디콘볼루션이 가능합니다. 또한 DMS는 세포막과 전체 조직을 관통할 수 있어 생리학적 맥락 내에서 프로빙이 가능합니다. 그러나 처리 절차의 변형이 결과에 영향을 미칠 수 있으므로 양질의 데이터를 생성하는 것은 어렵습니다. 따라서 우리는 시험관 내 및 세포 내 DMS-MaPseq 모두에 대한 자세한 프로토콜을 제공하여 편견을 줄이고 신규 이민자가 직면할 수 있는 어려움을 통해 방법을 안내하도록 합니다. 특히 최근의 SARS-CoV2 대유행에 비추어 RNA 바이러스에 대한 고품질 데이터는 유전자 발현을 연구하고 가능한 치료법을 찾는 데 중요한 도구입니다.
프로토콜
참고: 이 프로토콜에 사용된 모든 재료, 소프트웨어, 시약, 기기 및 셀과 관련된 자세한 내용은 재료 표를 참조하십시오.
1. 유전자 특이적 체외 DMS-MaP
- RNA 시험관 내 전사
- 관심 RNA의 서열을 이중 가닥(ds)DNA(예: 기존/게놈 DNA의 DNA 단편, 플라스미드 또는 PCR)로 얻습니다. DNA 서열에 중합효소 촉진제가 포함되어 있으면 3단계로 이동합니다.
- 원하는 DNA 단편의 상류에 RNA 중합효소 프로모터를 부착시키기 위해 중첩 PCR을 수행합니다(T7 중합효소에 대한 정방향 프라이머: 5' TAATACGACTCACTATAGG + 표적 서열 3'의 첫 번째 염기).
- 시험관 내에서 DNA 단편을 RNA로 전사합니다. 항상 RNA를 얼음 위에 두십시오.
- DNase를 사용하여 DNA를 소화합니다.
- 컬럼 기반 접근법 (단계 2.4) 또는 에탄올 침전 (단계 2.5)을 사용하여 RNA를 분리합니다. ~50μg의 수율을 기대하면서 적절한 부피로 용리합니다.
- RNA 무결성을 아가로스 겔 상에서 실행하여 보장하고; 실행하기 전에 70°C에서 2-3분 동안 RNA를 변성시킵니다.
알림: 완충액과 아가로스에는 RNA를 분해하고 RNA 샘플을 오염시킬 수 있는 RNase가 포함될 수 있습니다. 프리 캐스트 아가 로스 겔은 이전에이 실험실에서 사용되었습니다. 결과(특히 RNA의 경우)는 때때로 모호했습니다. 최상의 결과는 아가 로스 또는 PAGE 젤로 얻어졌다. - 해동 후 분해가 보이지 않는 한 RNA를 -80°C에서 몇 달 동안 보관하는 직접 사용.
- 시험관 내 DMS 변형 (105 mM DMS에서)
- 충분한 양의 리폴딩 완충액(0.4 M 나트륨 카코딜레이트, pH 7.2, 6 mMMgCl2 함유)을 준비한다.
참고: 각 반응(최종 부피 100μL)에 대해 89μL의 리폴딩 버퍼를 추가합니다. - 각 반응에 대해 89μL의 리폴딩 버퍼를 지정된 1.5mL 튜브로 옮기고 화학 후드 아래에 놓인 열진탕에서 37°C에서 예열합니다.
알림: DMS는 독성이 높으므로 환원제로 담금질 될 때까지 항상 화학 후드 아래에 보관해야합니다. - 10 μL의 뉴클레아제 무함유 물 (NFH2O)에서 1-10 pmol의 RNA를 용리시키고; PCR 튜브로 옮깁니다.
- 95 ° C의 열 순환기에서 1 분 동안 배양하여 RNA를 변성시킵니다.
- 잘못 접히지 않도록 즉시 얼음 블록 위에 놓으십시오.
- RNA 샘플을 37°C에서 리폴딩 버퍼가 있는 지정된 튜브에 추가하고 잘 혼합한 다음 10-20분 동안 배양하여 RNA를 재포장합니다.
참고: 대부분의 RNA는 밀리초에서 초 단위로 접히지만 예외는 존재합니다16. - RNA 샘플에 100%(10.5M) DMS 1μL를 추가하고 분당 800-1,400회전(rpm)으로 흔들면서 5분 동안 배양합니다.
알림: DMS는 소수성이며 리폴딩 버퍼에 완전히 용해되지 않을 수 있으므로 이 단계에서 흔들림(또는 다른 혼합 수단)이 중요합니다. 반응 시간의 편차는 DMS 반응성의 재현성에 영향을 미칠 수 있습니다. 피펫팅 오류를 최소화하기 위해 DMS의 최종 농도가 1%(105mM)로 유지되는 경우 DMS를 샘플에 추가하기 전에 100% 에탄올에 용해할 수 있습니다. 처리되지 않은 대조군의 경우, DMS는 디메틸 설폭사이드(DMSO) 또는 물로 치환될 수 있다. - 5분의 반응 시간 후, 100% β-메르캅토에탄올(BME) 60μL로 퀀칭하고, 잘 섞고, 즉시 RNA를 얼음 위에 놓는다.
알림: RNA는 BME와의 반응을 소멸시켜 청소한 후 후드에서 안전하게 제거할 수 있습니다. 그러나 BME가 주변에 직접 노출되는 것은 강한 냄새와 자극적 인 특성으로 인해 여전히 피해야합니다. - 아세트산나트륨-에탄올 침전(단계 2.5 참조) 또는 컬럼 기반 접근(단계 2.6 참조)에 의해 RNA를 클린하고, 10 μL의 물에서 용리시킨다.
- 분광 광도계를 사용하여 RNA를 정량합니다.
- 변형된 RNA를 -80°C에 직접 보관한다.
알림: DMS 처리 후 RNA는 덜 안정적이므로 장기 보관은 피해야 합니다.
- 충분한 양의 리폴딩 완충액(0.4 M 나트륨 카코딜레이트, pH 7.2, 6 mMMgCl2 함유)을 준비한다.
- 변형 된 RNA의 유전자 특이 적 RT-PCR
참고: DMS 처리된 단편의 RT-PCR 설정은 그림 1 을 참조하십시오.- 10 μL의 뉴클레아제 없는(NF)H2O에서 변형된 RNA 100ng를 용리하고 PCR 튜브로 옮깁니다.
- 튜브에 4μL의 5x 첫 번째 가닥 완충액(FSB), 1μL의 dNTP 혼합물(각각 10mM), 1μL의 0.1M 디티오트레이톨(DTT)(동결-해동 주기 방지), 1μL의 RNase 억제제, 1μL의 10μM 역방향 프라이머(단일 프라이머 또는 프라이머 풀) 및 1μL의 TGIRT III를 추가합니다.
참고: 프라이머 풀의 경우 각 프라이머의 10μM 1μL를 RT에 직접 추가하지 마십시오. 대신, 프라이머를 먼저 혼합하고 혼합물에서 1μL를 추가합니다(총 프라이머 농도 10μM에서). - 열순환기에서 57°C에서 30분에서 1.5시간(일반적으로 30분이면 500nt 생성물을 만들 수 있음) 동안 배양합니다.
- 1 μL의 4 M NaOH를 첨가하고, 피펫팅에 의해 혼합하고, 95°C에서 3분 동안 배양하여 RNA를 분해한다.
참고: 이 단계는 RNA를 분해하여 cDNA에서 TGIRT를 방출하기 때문에 중요합니다. 건너뛰면 다운스트림 PCR이 영향을 받을 수 있습니다. - 프라이머를 충분히 제거하는 컬럼 기반 접근법(단계 2.6 참조)을 사용하여 세척하고, 10 μL의 NFH2O에서 용리시킨다.
- 수율과 충실도의 균형을 맞추도록 설계된 PCR 키트와 함께 반응 25μL당 1μL의 역전사 산물을 사용하여 cDNA를 PCR 증폭합니다.
알림: 프라이머의 용융 온도는 ~60°C여야 합니다. - PCR 성공을 확인하기 위해 아가로스 겔 또는 프리캐스트 아가로스 겔에서 2μL의 PCR 생성물을 실행합니다.
- 이상적으로는 PCR 후에 하나의 밴드만 표시되어야 합니다. 그렇다면 컬럼 기반 접근 방식을 사용하여 반응을 정리합니다. 대체 밴드가 있는 경우 나머지 PCR 반응을 사용하여 겔에서 올바른 밴드를 절제합니다. 충분히 작은 부피(예: 10 μL)로 용리합니다.
- 분광 광도계를 사용하여 추출 된 단편을 정량화합니다.
- 원하는 시퀀싱 플랫폼에 적합한 접근 방식을 사용하여 시퀀싱을 위해 dsDNA 단편을 인덱싱합니다.

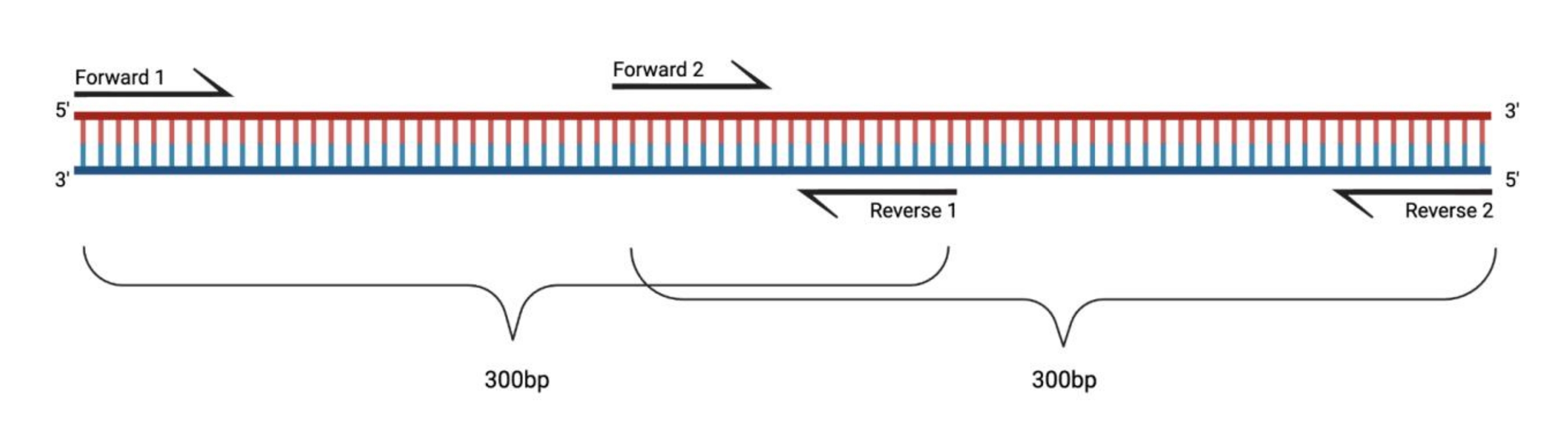
그림 1: 대형 DMS 처리된 단편의 RT-PCR을 위한 실험 설정. 변형된 RNA에 대해 역전사를 수행할 때, 프라이머가 어닐링하는 서열에 대한 변형은 기록되지 않습니다. 따라서, 단편의 길이가 400-500 bp를 초과하는 경우, 여기에 예시된 바와 같이, 프라이머 영역에서 중첩되는 단편이 설계될 필요가 있다. 조각의 길이는 시퀀싱 요구에 따라 다릅니다. 페어 엔드 150 사이클 시퀀싱을 사용할 때 조각은 300bp를 초과하지 않아야 합니다. 약어 : RT-PCR = 역전사 중합 효소 연쇄 반응; DMS = 디메틸 설페이트. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
2. 바이러스 감염 세포를 이용한 전체 게놈 DMS-MaP
참고: 세포에서 DMS 처리는 위에서 설명한 유전자 특이적 증폭 접근법과 조합될 수도 있습니다. 전체 게놈 라이브러리는 단일 유전자에 대한 완전한 커버리지를 달성하기 위해 엄청난 시퀀싱 깊이가 필요합니다. 그러나 바이러스 RNA가 추출 후 리보데플레핑된 RNA의 상당 부분을 구성하는 경우 전체 게놈 시퀀싱이 적절할 것입니다. 또한, 다른 농축 방법은 전체 게놈 라이브러리 생성 방법과 결합 될 수 있습니다.
- DMS 치료
- 원하는 감염 단계까지 바이러스에 감염된 세포를 성장시킵니다.
- 세포 용기를 필요한 생물안전 수준의 바이러스와 DMS와 같은 작용제에서 생성되는 화학 연기를 모두 처리하는 데 적합한 전용 흄 후드로 옮깁니다.
- 2.5% 부피의 DMS를 배양 배지에 첨가하고 파라필름으로 용기(일반적으로 10cm 플레이트)를 밀봉합니다.
참고: DMS를 사용하여 과소 수정 및 과잉 수정하기 쉽습니다. DMS를 세포에 직접 첨가 할 때는 잘 혼합하는 것이 매우 중요합니다. 또는 37°C에서 50mL 원뿔형 튜브에 새 배지를 예열하고 DMS를 직접 세게 흔들어 추가합니다. 사용된 배지를 세포 상에 디캔트하고, DMS 함유 배지에서 천천히 피펫팅한다. - 37 °C 인큐베이터로 5 분 동안 옮깁니다.
알림: 인큐베이터 외부에서 DMS를 처리하는 데 걸리는 시간에 따라 5 분이 과도하게 수정 될 수 있습니다. DMS 추가에서 배양까지의 시간을 ≤1분으로 유지하십시오. 실험을 처음 수행하는 경우 DMS 적정을 수행하고 배양 시간(3분에서 10분 사이)을 변경하여 최적의 수정 속도를 찾고 결과가 농도 창에서 견고한지 확인하는 것이 좋습니다. - DMS 함유 배지를 조심스럽게 피펫팅하고(적절한 화학 폐기물에 넣음) 10mL의 정지 완충액(30% BME가 포함된 PBS[예: 3mL의 BME 및 7mL의 PBS])을 부드럽게 추가합니다.
참고: DMS 및 BME를 추가하면 세포가 강하게 부착되지 않는 경우 플레이트에서 세포를 들어 올릴 수 있습니다. 세포가 리프팅되는 경우 DMS 함유 배지를 제거하는 대신 정지 버퍼를 직접 추가하고 DMS 및 BME로 세포를 50mL 원추형 튜브에 긁어내는 현탁 세포로 처리할 수 있습니다. 세포를 3,000 × g에서 3분 동안 원심분리하여 펠렛화하고; 큰 물방울로 세포 아래에 펠릿이 될 수 있는 잔류 DMS를 제거하십시오. 처음에 DMS 배지를 제거할 수 없는 경우 30% BME에서 추가 세척 단계를 수행하는 것이 좋습니다. - 세포를 긁어 내고 15mL 원추형 튜브로 옮깁니다.
- 펠렛을 3,000 ×g에서 3 분 동안 원 심 분리합니다.
- 상청액을 제거하고 10mL의 PBS로 2x 세척합니다.
- 가능한 한 많은 잔여 PBS를 조심스럽게 제거하십시오.
- 펠렛을 적절한 양의 RNA 분리 시약(예: T75 배양 플라스크의 경우 3mL, 10cm 플레이트의 경우 1mL)에 용해시킵니다.
참고: 시약의 양이 충분하지 않으면 RNA 수율에 영향을 미칠 수 있습니다.
- RNA 추출 및 리보솜 RNA (rRNA) 고갈
- RNA 분리 시약의 균질화된 세포 1mL에 클로로포름 200μL를 넣고 밝은 분홍색이 될 때까지 15-20초 동안 소용돌이한 다음 상 분리가 보일 때까지 최대 3분 동안 배양합니다.
알림: 분홍색 지질 단계는 바닥에 정착해야합니다. 그렇지 않은 경우 소용돌이 시간이 충분하지 않았을 수 있습니다. - 최대 속도(~20,000× g)로 4°C에서 15분 동안 회전합니다.
- 상부 수성 상을 새로운 튜브로 옮깁니다.
- 아세트산나트륨-에탄올 침전(단계 2.5 참조) 또는 컬럼기반 접근(단계 2.6 참조)에 의해 RNA를 클린하고, 충분한 부피의 NFH2O에서 용리시킨다.
- 아가로스 겔에서 RNA 무결성을 확인하십시오. 두 개의 리보솜 서브 유닛에 해당하는 두 개의 밴드를 찾으십시오.
- 바람직한 접근법을 사용하여 rRNA를 고갈시키고, NFH2O의 적절한 부피 (전형적으로 20-50 μL)로 용리시킨다.
참고: 다운스트림 애플리케이션의 경우 총 RNA의 ~500ng가 8μL의 부피로 제안됩니다. 비리보솜 RNA는 일반적으로 전체 RNA의 5%-10%만 구성합니다. - 분광 광도계를 사용하여 정량화합니다.
- RNA 분리 시약의 균질화된 세포 1mL에 클로로포름 200μL를 넣고 밝은 분홍색이 될 때까지 15-20초 동안 소용돌이한 다음 상 분리가 보일 때까지 최대 3분 동안 배양합니다.
- 라이브러리 생성
- 유전자-특이적 RT-PCR 또는 다른 접근법을 사용하여 라이브러리(15)를 생성한다. 프라이밍을 위해 무작위 헥사머를 사용하는 경우 헥사머 어닐링을 허용하기 위해낮은 Tm( 37-42°C)에서 배양 단계를 추가합니다.
참고: 표준 라이브러리 생성 키트는 RT 효소를 TGIRT로 교체하고 RT 온도를 57°C로 변경하여 사용할 수도 있습니다.
- 유전자-특이적 RT-PCR 또는 다른 접근법을 사용하여 라이브러리(15)를 생성한다. 프라이밍을 위해 무작위 헥사머를 사용하는 경우 헥사머 어닐링을 허용하기 위해낮은 Tm( 37-42°C)에서 배양 단계를 추가합니다.
- RNA Clean & Concentrator 컬럼을 사용한 컬럼 기반 RNA 정화
알림: 모든 단계는 실온에서 수행해야 합니다.- NF H2O를 샘플 튜브에 추가하여 50 μL의 부피로 만듭니다.
- 100 μL의 결합 완충액과 150 μL의 100 % 에탄올을 샘플에 첨가한다.
- 혼합하고 스핀 컬럼으로 옮깁니다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- 400μL의 RNA 분취 완충액을 추가합니다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- 700 μL의 RNA 세척 완충액을 추가합니다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- 400 μL의 RNA 세척 완충액을 첨가한다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- (선택 사항) 컬럼을 새 수집 튜브로 옮기고 10,000-16,000×g에서 2 분 동안 회전 합니다.
- 컬럼을 깨끗한 RNAse가 없는 튜브로 옮기고 적절한 양의 NFH2O를 추가합니다.
- 10,000-16,000 × g 에서 1 분 동안 회전시킵니다.
- 산성 페놀 - 클로로포름 RNA 추출.
- 같은 양의 산 페놀 : 클로로포름 : 이소 아밀 알코올을 첨가하십시오.
- 와동을 완전히 하고 14,000× g 에서 5분 동안 원심분리합니다.
- 상 분리가 없으면 2M NaCl 20μL를 추가하고 원심분리를 반복합니다.
- 수성 상을 새로운 튜브로 옮깁니다.
- 500 μL의 이소프로판올과 2 μL의 공동 침전제를 첨가하십시오.
- RT에서 3 분 동안 혼합하고 배양하십시오. 그런 다음 -80 ° C에서 밤새 배양하십시오.
- 4°C에서 30분 동안 최대 속도(~20,000× g)로 원심분리하여 RNA를 펠렛화합니다.
- 펠릿을 얼음처럼 차가운 200% 에탄올 70μL로 세척합니다.
- 최대 속도 (~ 20,000 × g)로 5 분 동안 회전합니다. 플로우스루를 폐기합니다.
- 펠렛을 적절한 양의 NFH2O로 재현탁시킨다.
- 올리고 클린 컬럼 및 농축기 컬럼을 사용한 컬럼 기반 cDNA 클린업
알림: 모든 단계는 실온에서 수행해야 합니다.- NF H2O를 샘플 튜브에 추가하여 50 μL의 부피로 만듭니다.
- 100 μL의 결합 완충액과 400 μL의 100 % 에탄올을 첨가하십시오.
- 혼합하고 스핀 컬럼으로 옮깁니다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- 750 μL의 DNA 세척 완충액을 첨가한다.
- 10,000-16,000 × g 에서 30 초 동안 회전하십시오. 플로우스루를 폐기합니다.
- (선택 사항) 컬럼을 새 수집 튜브로 옮기고 10,000-16,000 × g 에서 2분 동안 회전시킵니다.
- 컬럼을 깨끗한 RNAse가 없는 튜브로 옮기고 적절한 양의 NFH2O를 추가합니다.
- 10,000-16,000 × g 에서 1 분 동안 회전시킵니다.
3. 시퀀싱 데이터 분석
참고: DMS-MaP 시퀀싱 데이터에서 RNA 2차 구조 모델을 생성하려면 결과 .fastq 파일을 여러 단계로 처리해야 합니다. 이러한 단계는 다음을 사용하여 자동으로 수행할 수 있습니다.
- TrimGalore 또는 Cutadapt로 어댑터 시퀀스를 트리밍합니다.
- Bowtie2를 사용하여 읽기를 참조 시퀀스(.fasta 형식)에 매핑합니다.
- 특수 RNA 구조 소프트웨어(예: DREEM14, RNA-Framework17 또는 이와 유사한 소프트웨어)로 판독 횟수를 계산하고 반응성 프로파일을 생성합니다.
- (선택 사항) 판독을 클러스터링하여 DREEM14, DRACO 17, DANCE-MaP18 또는 이와 유사한 것을 사용하여 대체 RNA 형태를 찾습니다.
- RNAStructure12, ViennaRNA 또는 이와 유사한 것을 사용하여 반응성 프로파일을 기반으로 최소 자유 에너지 구조를 예측합니다.
- VARNA (https://varna.lri.fr/) 또는 이와 유사한 것을 사용하여 RNA11 구조를 시각화합니다.
참고: 실용성을 위해 DREEM(www.rnadreem.org) 및 RNA-Framework19 와 같은 소프트웨어는 파이프라인에 1-5단계를 광범위하게 통합하여 분석 프로세스를 간소화합니다. 그러나, 임의의 구조 예측은 주의해서 다루어져야 한다(예를 들어, 데이터(20)와 구조의 일치를 검증함으로써.
결과
유전자 특이적 체외 DMS-MaP
SARS2의 5'UTR을 연구하기 위해 바이러스의 처음 300bp를 3개의 프라이머와 함께 gBlock 서열로 주문했습니다. 여기에는 PCR을 통해 단편("FW" 및 "RV")을 전파하기 위한 두 개의 프라이머와 T7 프로모터("FW-T7")를 부착하는 프라이머가 포함되었습니다. 이들 서열은 표 1에서 확인할 수 있다.
| 이름 | 시퀀스(5'->3') |
| 증권 시세 표시기 | ATTAAAGGTTTTTATACCTTCCCAGGTAAC |
| RV | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
표 1: SARS-CoV2 5'UTR의 DMS-MaP RT-PCR에 대한 프라이머 서열. 여기서, FW-T7 및 RV는 시험관내 전사를 위한 DNA 주형을 생성하기 위해 필요하고, RV는 역전사에 사용되고 FW-RV 프라이머-쌍은 cDNA의 후속 PCR 증폭에 사용된다. 프라이머는 SARS-CoV2 게놈(FW)의 맨 처음과 관심 영역의 바로 하류 서열에 어닐링됩니다. 약어 : DMS-MaP = 디메틸 설페이트를 사용한 시퀀싱을 통한 돌연변이 프로파일 링; RT-PCR = 역전사 중합효소 연쇄반응; SARS-CoV2 = 중증 급성 호흡기 증후군-코로나 바이러스 2; UTR = 비번역 영역; RV = 역방향 프라이머; FW = 순방향 프라이머.
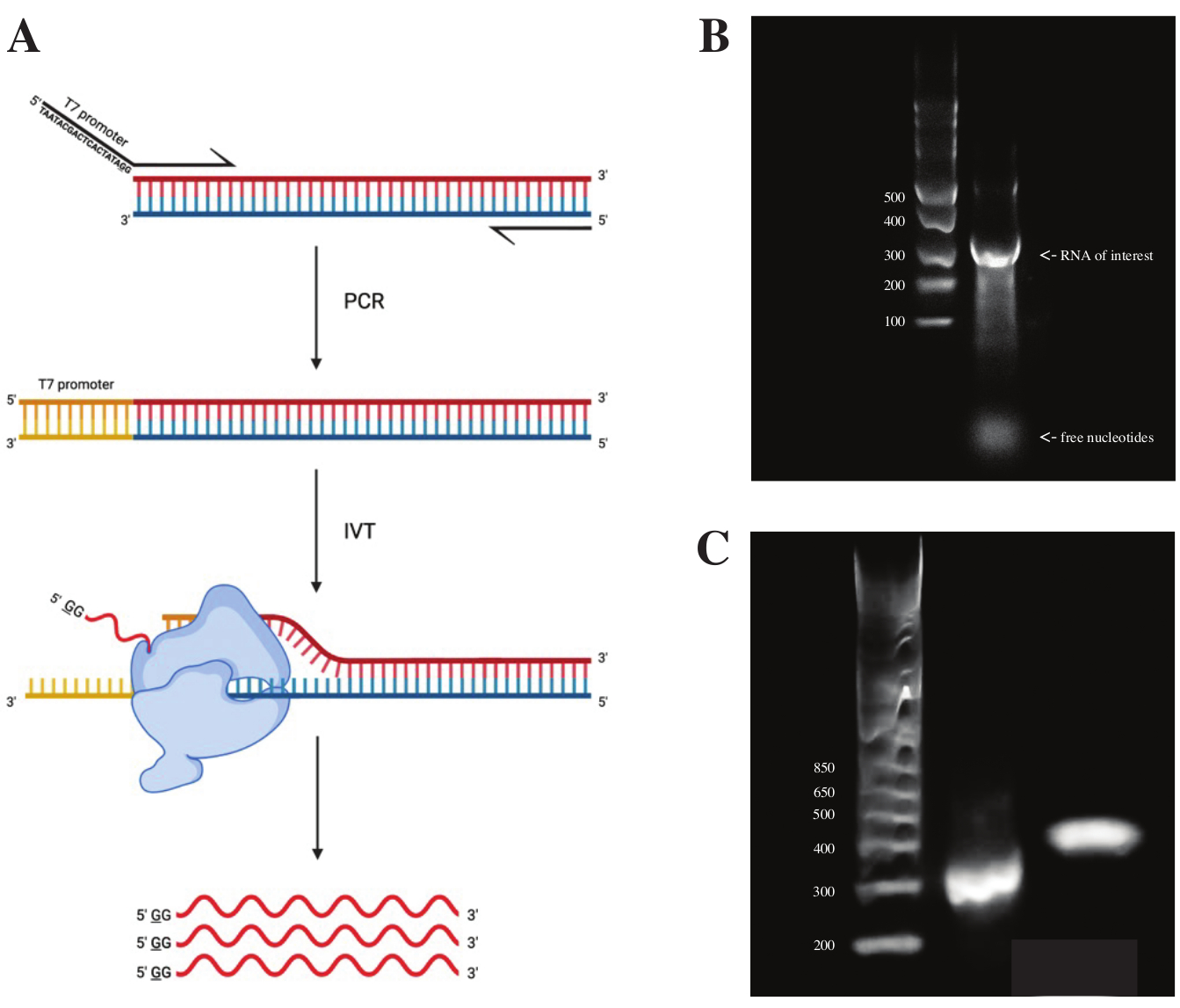
gBlock 단편으로부터 RNA를 생성하기 위해, T7 중합효소 촉진제의 서열을 도 2A에 도시된 반응식에 따라 PCR 프리믹스를 사용하는 중첩 PCR을 사용하여 부착하였다. 길쭉한 단편으로부터, RNA는 T7 전사 키트를 사용하여 생성되었다. DNA 주형은 이후 DNase와 RNA Clean & Concentrator 컬럼을 사용하여 분리된 RNA를 사용하여 분해되었습니다.
시험관내 전사의 품질 관리는 ssRNA 사다리와 함께 1% 아가로스 겔 상에서 RNA 생성물을 실행함으로써 수행되었다. 단지 하나의 밴드가 가시적이었기 때문에, 시험관내 DMS 프로빙 및 RT-PCR을 수행하였다(도 2B 참조).
PCR 반응의 성공을 검증하기 위해, 샘플을 dsDNA 사다리를 사용하여 2% 아가로스 겔 상에서 실행하였다. 인덱싱 후, 밴드는 인덱싱 프라이머의 크기를 고려하여 동일한 젤에서 ~150bp 더 높게 실행되어야 합니다.

그림 2: DNA 주형의 시험관 내 전사. (ᅡ) 아직 고유 RNA 중합효소 프로모터가 없는 DNA 주형을 시험 관내 로 전사하려면 먼저 주형을 중첩 PCR로 부착해야 합니다. 이는 원하는 단편과 겹치는 첫 번째 염기의 상류에 서열 TAATACGACTCACTATAGG (T7 RNA 중합효소의 경우)를 포함하는 순방향 프라이머를 사용하여 수행됩니다. 여기서 밑줄이 그어진 염기는 중합 효소의 전사 시작 부위를 상징합니다. 프로모터가 dsDNA 단편에 부착되면 T7 중합효소에 의해 전사될 수 있습니다. 중요하게도, 중합효소는 언급된 프로모터 서열에 반대되는 가닥을 주형(파란색)으로 사용하여 지시된 프로모터 서열(빨간색)의 바로 하류 서열과 동일한 RNA를 효과적으로 생성합니다. (b) ssRNA 래더 (레인 1) 및 300 nt (레인 2)에서 시험 관 전사 RNA 생성물을 갖는 1 % 아가 로스 겔. (c) GeneRuler 1 kb 플러스 래더 (레인 1), 300 bp (레인 2)에서 실행되는 RT-PCR 후 PCR 산물, 및 470 bp (레인 3)에서 실행되는 라이브러리 준비 후 인덱싱된 단편을 갖는 2% 아가로스 겔. 약어 : RT-PCR = 역전사 중합 효소 연쇄 반응; DMS = 디메틸 설페이트; NT = 뉴클레오티드; dsDNA = 이중 가닥 DNA; ssRNA = 단일 가닥 RNA. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
바이러스 감염 세포를 이용한 생체 내 전체 게놈 DMS-MaP
DMS 처리 전에, HCT-8 세포를 OC43으로 감염시켰다. 감염 후 4일 후에 세포변성 효과(CPE)(cpe)가 관찰되었을 때( 도 3A에서 볼 수 있음), 이들 세포를 처리하고, RNA를 추출하고, 리보데플레팅하였다. 아가 로스 겔에서 총 RNA를 실행할 때 총 RNA 질량의 약 40 %를 구성하는 리보솜의 60S 및 95S 서브 유닛을 차지하는 두 개의 밝은 밴드가 보였습니다 ( 그림 3B 참조). RNA 추출이 실패하거나 분해되었을 때 (예를 들어, 다중 동결-해동 사이클에 의해), RNA 분해 생성물이 겔의 바닥에서 가시화되었다 ( 도 3C, 제2 레인 참조). 또한, rRNA 고갈 후, 두 개의 밝은 밴드가 사라져 차선에 얼룩이 남았습니다 ( 그림 3C, 세 번째 차선 참조). 마지막으로, 라이브러리 준비 후, 샘플은 다양한 크기 분포를 가지며 최종 PAGE 겔에 도말로서 표시되었다. 밴드는 200 뉴클레오티드 (nt)와 500 nt 사이에서 절제되었으며, 이는 이러한 라이브러리를 분석하기 위해 계획된 150 x 150 쌍단 시퀀싱 실행과 일치합니다. 가장 중요한 것은 ~150nt에서 실행되는 어댑터 이량체가 분리되었다는 것입니다( 그림 3D 참조).

그림 3: 바이러스에 감염된 세포가 있는 생체 내 DMS-MaP의 체크포인트. (A) 바이러스에 감염된 HCT-8 세포의 광학 현미경 이미지, 4 일 dpi. 세포 사멸로 인한 부작용을 최소화하면서 전체 RNA에서 바이러스 RNA의 가능한 가장 높은 수율을 얻으려면 이미지에서 볼 수 있듯이 CPE가 시작될 때 또는 그 이전에도 DMS를 추가해야 합니다. (B) 총 RNA 1 μg의 6 개 샘플을 갖는 1 % 아가 로스 겔. 각 레인에서 리보솜 RNA가 전체 RNA의 ~ 95 %를 구성하기 때문에 40S 및 60S 서브 유닛을 차지하는 두 개의 밝은 밴드가 보입니다. 참고: 세포 내 DMS 처리는 일부 RNA 단편화 및 번짐을 유발하지만 두 개의 rRNA 밴드는 여전히 보여야 합니다. 메틸화 마크를 포함하는 정보가 생성되고 세포가 아직 살아있는 동안 DMS 배양 중에 RNA 구조에 대해 보고하기 때문에 경미한 단편화 후 변형이 허용됩니다. (C) GeneRuler 1 kb의 1% 아가로스 겔과 사다리 DNA 마커(레인 1) 총 RNA를 -80°C에서 6개월 동안 미리 보관한 것(레인 2) 및 리보데플레팅된 RNA(레인 3). 여러 번의 동결-해동 주기로 RNA를 장기간 보관하면 RNA가 분해되기 시작하므로 프로빙 실험에 사용해서는 안 됩니다. 또한, 전체 RNA를 리보데플레딩한 후, 리보솜의 40S 및 60S 서브유닛을 차지하는 두 개의 밝은 밴드가 퇴색하고 잔류 RNA의 얼룩이 나타나기 시작합니다. (D) GeneRuler 1 kb 플러스 래더 DNA 마커 (레인 1)의 PAGE 젤 및 전체 게놈 제조 RNA의 라이브러리 샘플. 겔은 시퀀싱 요구에 따라 절제해야합니다. 양쪽에서 150 사이클에 걸친 쌍단 시퀀싱 실행의 경우 겔은 300bp에서 500bp 사이에서 절제되어야 합니다. 어댑터 이량체(170bp에서 실행)는 분리해야 합니다. 약어 : DMS-MaP = 디메틸 설페이트를 사용한 시퀀싱을 통한 돌연변이 프로파일 링; DPI = 감염 후 일수; CPE = 세포 변성 효과. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
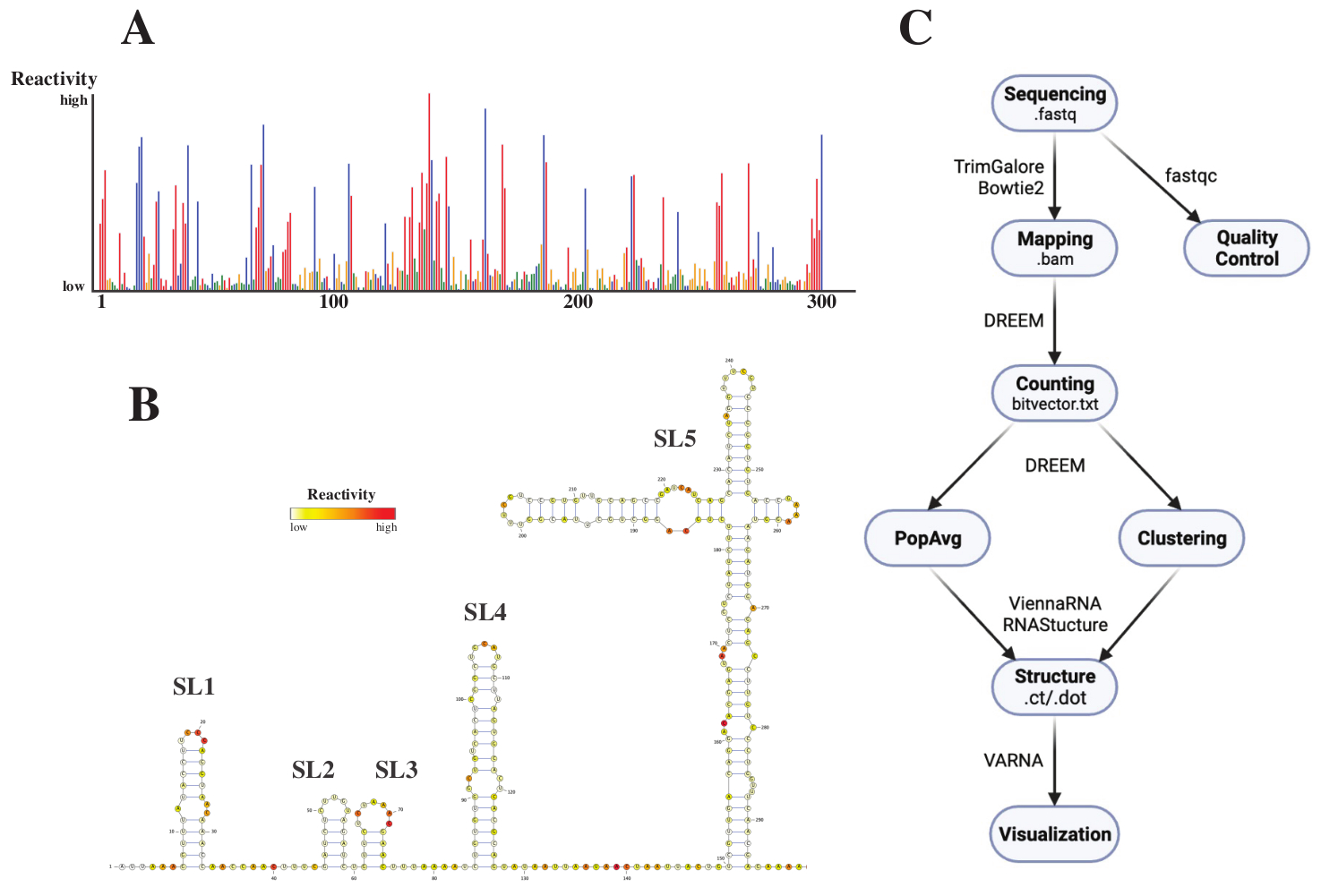
시퀀싱 후 .fastq 파일은 .fasta 참조 파일과 함께 DREEM 웹 서버(http://rnadreem.org/)에 작업을 제출하여 분석되었습니다. 서버에서 생성된 출력에는 fastqc(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) 및 TrimGalore(https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)에서 생성된 품질 관리 파일과 모집단 평균 돌연변이 빈도를 포함하는 기타 출력 파일이 포함됩니다. 대화형 .html(그림 4A 참조) 형식의 돌연변이 빈도와 염기당 원시 재활성이 있는 .csv 파일 및 여러 RNA 구조 예측 소프트웨어에서 읽을 수 있는 struct_constraint.txt 파일 외에도 여기에는 판독 돌연변이에 대한 비트벡터.txt 파일도 포함됩니다. 이들로부터 모집단 평균 구조는 .fasta 및 struct_constraint.txt 파일을 RNAfold 웹 서버에 제출하여 계산되었습니다 (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). 이것은 ViennaRNA 소프트웨어를 사용하여 최소 자유 에너지를 기반으로 구조 예측을 생성하며, 온라인으로 보거나 ct 또는 Vienna 형식으로 다운로드할 수 있습니다. RNA 구조 모델을 생성하기 위해 이러한 다운로드 가능한 파일을 VARNA에 제출했습니다(https://varna.lri.fr/, 그림 4B 참조). 마지막으로, 비트 벡터 .txt 파일은 DREEM (https://codeocean.com/capsule/6175523/tree/v1)의 안정 버전에서 대체 RNA 형태를 검색하는 데 사용할 수 있습니다. DREEM을 사용하여 우수한 구조 모델을 얻으려면 베이스당 10,000회 판독 범위를 달성해야 합니다. 클러스터링의 경우 베이스당 최대 100,000개의 읽기가 필요할 수 있습니다. 전체 작업 흐름에 대한 개요는 그림 4C에서 찾을 수 있습니다.

도 4: SARS-CoV2 5'UTR의 화학적 프로빙 실험으로부터 얻어진 예시적인 데이터. (A) 염기별로 착색된 SARS-CoV2 게놈의 처음 300개 염기의 반응성 프로파일(A: 적색, C: 청색, U: 녹색, G: 황색). 원시 반응성은 절대 돌연변이 빈도를 적용 범위로 나눈 값으로 계산됩니다. 개방 형태를 가진베이스는 높은 반응성 값을 갖는다. 염기 쌍에 관여하는 염기는 반응성 값이 낮습니다. U 및 G는 DMS에 의해 변형되지 않으며 중합 효소 불충실도에서 비롯된 반응성 값이 낮습니다. 예측은 DREEM 웹 서버로 이루어졌습니다. (B) VARNA로 만든 반응성 값으로부터 예측된 SARS-CoV2 5'UTR의 구조 모델. 반응성 값이 높은 염기는 빨간색으로 표시됩니다. 반응성 값이 낮은 염기는 흰색으로 표시됩니다. (C) 시퀀싱에서 얻은 .fastq 파일로 시작하는 DMS-MaP 분석의 워크플로우. fastqc를 사용하여 품질을 관리할 수 있습니다. 어댑터 시퀀스는 TrimGalore를 사용하여 트리밍된 다음 Bowtie2를 사용하여 참조 시퀀스에 다시 매핑됩니다. 가져온 .bam 파일에서 DREEM은 각 읽기의 돌연변이를 계산하여 변형 맵 또는 .bitvector.txt 파일을 만듭니다. 이들은 위치 의존적 방식으로 각 판독의 돌연변이를보고하며,이를 기반으로 모집단 평균 반응성 프로파일을 생성 할 수 있습니다. 대안적으로, 비트벡터는 대체 RNA 형태를 검색하기 위해 DREEM을 사용하여 클러스터링될 수 있다. 마지막으로, 획득된 구조 모델은 소프트웨어(예를 들어, VARNA)를 사용하여 시각화된다. 약어 : DMS-MaP = 디메틸 설페이트를 사용한 시퀀싱을 통한 돌연변이 프로파일 링; SARS-CoV2 = 중증 급성 호흡기 증후군-코로나바이러스 2. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
토론
여기서 프로토콜은 DMS 돌연변이 프로파일링 실험을 사용하여 시험관 내 및 세포에서 RNA를 프로브하는 방법을 설명합니다. 또한 유전자 특이적 데이터를 생성하고 획득한 .fastq 파일을 분석하기 위해 Illumina 시퀀싱을 위한 라이브러리를 준비하는 방법에 대한 지침을 제공합니다. 또한 게놈 전체 라이브러리 접근 방식을 사용할 수 있습니다. 그러나 유전자 특이적 RT-PCR은 최고 품질의 가장 강력한 데이터를 생성합니다. 따라서 샘플을 비교하는 경우 라이브러리 생성으로 인해 약간의 편향이 발생하므로 동일한 시퀀싱 전략으로 준비되었는지 확인하는 것이 중요합니다. 재현성은 항상 반복실험을 사용하여 측정해야 합니다.
몇 가지주의 사항
RNA는 고온과 RNase에 의한 분해에 민감한 불안정한 분자입니다. 따라서 개인 보호 장비 (PPE), RNAse가없는 물질 및 RNAse 억제제의 사용과 같은 특별한 조치가 권장됩니다. 가장 중요한 것은 RNA가 가능할 때마다 얼음 위에 보관되어야한다는 것입니다. 이것은 특히 고온에 훨씬 더 민감한 메틸화 RNA에 적용됩니다.
관심 RNA 구조가 DMS 농도 및 완충액 조건에 민감하지 않은지 확인하는 것이 중요합니다. pH 7-7.5에서 100mM 트리스, 100mM MOPS 및 100mM HEPES와 같은 완충액은 높은 신호를 주지만 반응 동안 pH를 유지하기에 충분하지 않을 수 있다(21). DMS는 물에서 가수분해되어 pH를 낮추기 때문에 변형 반응 중에 중성 pH를 유지하려면 강력한 완충액이 중요합니다. 비신의 첨가는 pH를 약간 염기성21 로 유지하는 데 도움이 되는 것으로 나타났지만 Gs 및 Us에 대한 DMS 변형이 낮으며, 이는 유익할 수 있지만 As 및 Cs보다 훨씬 낮은 신호의 생성으로 인해 별도로 분석되어야 하며 이 프로토콜에서 더 이상 논의되지 않습니다.
유전자 특이적 RT-PCR에서, 변형된 RNA는 DNA로 역전사되고 PCR에 의해 단편으로 증폭된다. RNA의 크기는 이론적으로 무제한일 수 있지만, 이러한 PCR 단편은 역전사 반응 동안 편향을 방지하기 위해 400-500 염기쌍(bp)의 길이를 초과해서는 안 됩니다. 이상적으로, 프래그먼트는 시퀀싱 실행의 범위 내에 있어야 한다(즉, 시퀀싱이 150 x 150 사이클 쌍단 시퀀싱 프로그램을 사용하여 수행되는 경우, 단일 프래그먼트는 300bp를 초과하지 않아야 함). 더 적은 주기로 시퀀싱 프로그램을 사용하는 경우 dsDNase를 사용하여 PCR 산물을 단편화할 수 있습니다. 또한, 프라이머 서열 내의 서열은 어떠한 구조적 정보도 보유하지 않기 때문에, 프로빙된 RNA가 >1 단편을 포함할 때 단편이 겹쳐야 한다. RT 반응은 서로 다른 단편에 대해 여러 개의 RT 프라이머를 포함할 수 있습니다(최대 10개의 서로 다른 RT 프라이머). 서열에 따라 RT 프라이머를 풀링하면 역전사의 효율성이 떨어질 수 있지만 일반적으로 잘 작동합니다. 각 PCR 반응은 별도로 수행되어야합니다.
DMS로 RNA를 프로빙할 때 많은 RNA가 열역학적으로 불안정하고 온도와 같은 환경 요인에 따라 형태가 변하기 때문에 실험 조건이 추가적인 역할을 합니다. 불규칙성을 피하기 위해 실험 조건은 반응 시간과 관련하여 가능한 한 일정하게 유지되어야합니다. 완충 조건은 RNA24의 적절한 접힘을 보장하기 위해 기본 조건(완충 용량 및 1가 이온(Na) 및 2가 이온(Mg)의 존재)이 유지될 때 어느 정도 17,20,22,23까지 교환할 수 있는 것으로 보입니다.
변형 된 RNA의 라이브러리 준비와 관련하여 몇 가지 측면을 고려해야합니다. 첫째, 앞서 언급했듯이 변형된 RNA는 변형되지 않은 RNA보다 안정성이 떨어지므로 최적의 단편 크기 분포를 위해 단편화 시간을 최적화해야 할 수 있습니다. 또한 특정 RNA 라이브러리 준비 키트와 다른 많은 RNAseq 접근법은 역전사 키트에서 무작위 프라이머를 사용합니다. 이것은 특히 유전자의 3'에서 참조의 커버리지를 낮추고 궁극적으로 커버리지 깊이가 불충분해질 수 있습니다. 특정 영역의 적용 범위가 너무 낮으면 구조 예측에서 해당 염기를 제거해야 할 수 있습니다. RT-PCR 및 전체 게놈 RNAseq 키트 외에도 다른 라이브러리 준비 접근법을 사용할 수 있습니다. RNA에 대한 3' 및/또는 5' 어댑터의 결찰을 포함하는 프로토콜은 RNA의 작은 단편을 사용하거나 프라이머 영역에서 프로빙 정보의 손실을 피해야 할 때 유리합니다.
마지막으로, 화학 프로빙 실험의 분석은 항상 신중하게 해석되어야 합니다. 현재, 서열만으로 RNA의 RNA 구조를 높은 정확도로 예측하는 소프트웨어는 없습니다. 화학적 프로빙 제약이 정확도를 크게 향상시키지만 긴 RNA(>500nt)에 대한 좋은 모델을 생성하는 것은 여전히 어렵습니다. 이러한 모델은 다른 접근법 및/또는 돌연변이 유발에 의해 추가로 테스트되어야 합니다. RNA 예측 소프트웨어는 최대 염기쌍 수에 대해 최적화하여RNA 접힘5을 정확하게 나타내지 않을 수 있는 개방 형태에 상당한 벌칙을 줍니다. 따라서, 수득된 구조 모델은 Lan et al.20에 의해 예시된 바와 같이, 기본 화학적 프로빙 데이터(예를 들어, AUROC에 의해) 및 반복체 간(예를 들어, mFMI에 의해)과의 예측 일치를 정량화함으로써 시험되어야 한다.
이상적으로는 얻은 구조 모델에 도전하기 위해 서로 다른 시스템에서 여러 실험을 사용하여 가설을 강화해야합니다. 여기에는 시험관 내 및 세포 내 접근법, 보상 돌연변이, 다양한 세포주 및 종의 사용이 포함될 수 있습니다. 더욱이, 원시 반응성은 RNA 폴딩 앙상블의 "지상 진실" 스냅샷을 기록하기 때문에 구조 예측과 같거나 훨씬 더 유익한 경우가 많습니다. 따라서 원시 반응성은 서로 다른 조건 간의 구조 변화를 비교하는 데 매우 적합하고 유익합니다. 중요한 것은 계산 예측과 함께 화학적 프로빙 제약 조건을 사용하여 계산 된 가장 낮은 자유 에너지 구조는 완전한 구조 모델에 대한 시작 가설로만 사용되어야한다는 것입니다.
공개
저자는 선언 할 이해 상충이 없습니다.
감사의 말
없음
자료
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
참고문헌
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유