
Method Article
硫酸ジメチルによるRNA構造のプロービング in vitro および細胞内でのシーケンシングによる突然変異プロファイリング(英語)
要約
このプロトコルは、突然変異プロファイリング実験のために硫酸ジメチルでRNAを修飾するための指示を提供します。これには、2つの代替ライブラリ調製方法を使用したin vitro および in vivo プロービングが含まれます。
要約
事実上あらゆる生物学的プロセスにおけるRNA構造の役割は、特に過去10年間でますます明らかになっています。しかし、RNA結晶構造解析やクライオEMなどのRNA構造を解明するための古典的なアプローチは、急速に進化する分野とハイスループットソリューションの必要性に追いつくことができませんでした。硫酸ジメチル(DMS)を用いたシーケンシングによる変異プロファイリング MaPseqは、塩基とDMSとの反応性からRNA構造を推測するためのシーケンシングベースのアプローチです。DMSは、塩基が不対の場合、ワトソンクリック面でアデノシン中のN1窒素とシトシン中のN3をメチル化します。改変RNAを耐熱性第II群イントロン逆転写酵素(TGIRT-III)で逆転写すると、メチル化塩基がcDNAに変異として組み込まれます。得られたcDNAをシーケンシングし、それを参照転写物にマッピングする場合、各塩基の相対突然変異率は、塩基の「状態」がペアまたは非ペアであることを示します。DMS反応性はin vitro と細胞の両方で高いS/N比を持っていますが、この方法は取り扱い手順のバイアスに敏感です。このバイアスを減らすために、この論文は、細胞内のDMSおよび in vitro 転写RNAを使用したRNA治療のプロトコルを提供します。
概要
RNAが構造1,2と触媒3の両方の特性を持っているという発見以来、RNAの重要性と多数の生物学的プロセスにおけるその調節機能が徐々に明らかにされてきました。実際、遺伝子制御に対するRNA構造の影響はますます注目を集めています4。タンパク質と同様に、RNAは一次、二次、三次構造を持ち、それぞれヌクレオチドの配列、塩基対相互作用の2Dマッピング、およびこれらの塩基対構造の3Dフォールディングを指します。三次構造を決定することは、RNA依存プロセスの背後にある正確なメカニズムを理解するための鍵ですが、二次構造はRNA機能に関しても非常に有益であり、さらなる3Dフォールディングの基礎となります5。
しかし、RNA構造の決定は、従来のアプローチでは本質的に困難でした。タンパク質については、結晶学、核磁気共鳴(NMR)、極低温電子顕微鏡(クライオEM)により構造モチーフの多様性を決定できるようになり、配列のみからの構造予測が可能になりましたが6、これらのアプローチはRNAに広く適用できません。実際、RNAは、対応するアミノ酸と比較してはるかに多くの立体配座および回転自由度を有するビルディングブロック(ヌクレオチド)を有する柔軟な分子である。さらに、塩基対形成による相互作用は、アミノ酸残基の相互作用よりも動的で汎用性が高い。その結果、古典的なアプローチは、明確に定義された非常にコンパクトな構造を持つ比較的小さなRNAに対してのみ成功しています7。
RNA構造を決定するための別のアプローチは、次世代シーケンシング(NGS)と組み合わせた化学的プロービングによるものです。この戦略は、RNA配列中の各塩基の結合状態(すなわち、その二次構造)に関する情報を生成する。簡単に言えば、塩基対形成に関与していないRNA分子中の塩基は、小さな化合物によって差動的に修飾される。これらのRNAを特殊な逆転写酵素(RT)で逆転写すると、その修飾が変異として相補的デオキシリボ核酸(cDNA)に組み込まれます。次に、これらのcDNA分子はポリメラーゼ連鎖反応(PCR)によって増幅され、配列決定されます。結合または非結合としてのそれらの「状態」に関する情報を得るために、目的のRNAの各塩基における突然変異頻度が計算され、制約8として構造予測ソフトウェアに入力される。最近傍ルール9 および最小自由エネルギー計算10に基づいて、このソフトウェアは、得られた実験データ11、12に最も適合する構造モデルを生成する。
DMS-MaPseqはDMSを使用しており、ワトソンクリック面のアデノシン中のN1窒素とシトシン中のN3窒素を非常に特異的な方法でメチル化します13。逆転写に耐熱性第II群イントロン逆転写酵素(TGIRT-III)を使用すると、前例のないS/N比の突然変異プロファイルが作成され、2つ以上の代替立体配座によって生成された重複プロファイルのデコンボリューションも可能になります14,15。さらに、DMSは細胞膜や組織全体を貫通できるため、生理学的状況でのプロービングが可能になります。ただし、処理手順のばらつきが結果に影響を与える可能性があるため、高品質のデータを生成することは困難です。したがって、in vitroおよび細胞内DMS-MaPseqの両方の詳細なプロトコルを提供して、バイアスを減らし、新規参入者が遭遇する可能性のある困難を乗り越えてメソッドに導くようにします。特に最近のSARS-CoV2のパンデミックに照らして、RNAウイルスに関する高品質のデータは、遺伝子発現を研究し、可能な治療法を見つけるための重要なツールです。
プロトコル
注:このプロトコルで使用されるすべての材料、ソフトウェア、試薬、機器、およびセルに関連する詳細については、 材料の表 を参照してください。
1. 遺伝子特異的 インビトロ DMS-MaP
- RNA インビトロ 転写
- 目的のRNAの配列を二本鎖(ds)DNAとして取得します(例:DNAフラグメント、プラスミド、または既存の/ゲノムDNAからのPCRとして)。DNA配列にポリメラーゼプロモーターが含まれている場合は、ステップ3に進みます。
- オーバーラップPCRを実行して、目的のDNA断片の上流にRNAポリメラーゼプロモーターを結合させます(T7ポリメラーゼのフォワードプライマー:5'TAATACGACTCACTATAGG +標的配列3'の最初の塩基)。
- インビトロでDNA断片をRNAに転写する。RNAは常に氷上に置いてください。
- DNaseを使用してDNAを消化します。
- カラムベースのアプローチ(ステップ2.4)またはエタノール沈殿(ステップ2.5)を使用してRNAを単離します。適切な量で溶出し、~50 μgの収量を期待します。
- アガロースゲル上でRNAを走らせてRNAの完全性を確保します。実行する前に、RNAを70°Cで2〜3分間変性させます。
注:バッファーとアガロースには、RNAを分解するRNaseが含まれている可能性があり、RNAサンプルを汚染する可能性があります。プレキャストアガロースゲルは、以前にこのラボで使用されてきました。結果(特にRNAの場合)は時々曖昧でした。最良の結果は、アガロースまたはPAGEゲルで得られました。 - 直接使用 RNAは、解凍後に分解が見られない限り、-80°Cで数ヶ月間保存してください。
- インビトロ DMS の変更 (105 mM の DMS で)
- 十分な量のリフォールディングバッファー(0.4 Mカコジル酸ナトリウム、pH 7.2、6 mM MgCl2を含む)を調製します。
注:各反応(最終容量100 μL)に対して、89 μLのリフォールディングバッファーを追加します。 - 各反応について、89 μLのリフォールディングバッファーを指定された1.5 mLチューブに移し、ケミカルフードの下に置いたサーモシェーカーで37°Cで予温します。
注意: DMSは非常に有毒であり、還元剤で急冷されるまで、常に化学フードの下に保管する必要があります。 - 10 μLのヌクレアーゼフリー水(NF H2O)中の1〜10 pmolのRNAを溶出する。PCRチューブに移します。
- サーモサイクラーで95°Cで1分間インキュベートし、RNAを変性させます。
- 折り間違いを避けるために 、すぐに 氷のブロックの上に置きます。
- RNAサンプルを37°Cのリフォールディングバッファーを含む指定チューブに加え、よく混合し、10〜20分間インキュベートしてRNAをリホールドします。
注:ほとんどのRNAはミリ秒から秒のオーダーで折りたたまれますが、例外は16あります。 - 1 μLの100%(10.5 M)DMSをRNAサンプルに加え、毎分800〜1,400回転(rpm)で振とうしながら5分間インキュベートします。
注:DMSは疎水性であり、リフォールディングバッファーに完全に溶解しない可能性があるため、このステップでの振とう(またはその他の混合手段)は非常に重要です。反応時間の偏差は、DMS反応性の再現性に影響を与える可能性があります。ピペッティングエラーを最小限に抑えるために、DMSは、最終濃度1%(105 mM)のDMSが維持されている場合、サンプルに加える前に100%エタノールに溶解することができます。未処理の対照の場合、DMSはジメチルスルホキシド(DMSO)または水で置換できます。 - 反応時間5分後、60 μLの100%β-メルカプトエタノール(BME)でクエンチし、よく混合し、すぐにRNAを氷上に置きます。
注:RNAは、BMEとの反応をクエンチしてクリーンアップした後、フードから安全に取り外すことができます。ただし、BMEが周囲に直接さらされることは、その強い臭いと刺激性があるため、避ける必要があります。 - 酢酸ナトリウム-エタノール沈殿(ステップ2.5を参照)またはカラムベースのアプローチ(ステップ2.6を参照)によってRNAをクリーンアップし、10 μLの水で溶出します。
- 分光光度計を用いてRNAを定量する。
- 修飾RNAを-80°Cで保存して直接使用する。
注:DMS処理後のRNAの安定性が低下するため、長期保存は避けてください。
- 十分な量のリフォールディングバッファー(0.4 Mカコジル酸ナトリウム、pH 7.2、6 mM MgCl2を含む)を調製します。
- 修飾RNAの遺伝子特異的RT-PCR
注:DMS処理フラグメントのRT-PCRセットアップについては、 図1 を参照してください。- 10 μLのヌクレアーゼフリー(NF)H2O中の100 ngの修飾RNAを溶出し、PCRチューブに移します。
- チューブに、4 μLの5x第一鎖バッファー(FSB)、1 μLのdNTPミックス(各10 mM)、1 μLの0.1 Mジチオスレイトール(DTT)(凍結融解サイクルを避ける)、1 μLのRNase阻害剤、1 μLの10 μMリバースプライマー(単一プライマーまたはプライマーのプール)、および1 μLのTGIRT IIIを追加します。
注:プライマーのプールの場合は、各プライマーの10 μMを1 μL直接RTに追加しないでください。代わりに、最初にプライマーを混合し、混合物から1 μLを追加します(総プライマー濃度10 μM)。 - サーモサイクラーで57°Cで30分から1.5時間(通常、500ntの製品を作るには30分で十分です)インキュベートします。
- 4 M NaOHを1 μL加え、ピペッティングで混合し、95°Cで3分間インキュベートしてRNAを分解します。
注:このステップは、RNAを分解することによってcDNAからTGIRTを放出するため、非常に重要です。スキップすると、ダウンストリーム PCR が影響を受ける可能性があります。 - プライマーを十分に除去するカラムベースのアプローチ(ステップ2.6を参照)を使用してクリーンアップし、10 μLのNF H2Oで溶出します。
- 収量と忠実度のバランスをとるように設計されたPCRキットを使用して、反応25 μLあたり1 μLの逆転写産物を使用してcDNAをPCR増幅します。
注:プライマーの融解温度は~60°Cである必要があります。 - 2 μLのPCR産物をアガロースゲルまたはプレキャストアガロースゲルで実行し、PCRの成功を確認します。
- 理想的には、PCRの後に1つのバンドのみが表示されるはずです。その場合は、列ベースのアプローチを使用して反応をクリーンアップします。代替バンドが存在する場合は、残りのPCR反応を使用して、ゲルから正しいバンドを切り取ります。十分に小さい容量(例えば、10μL)で溶出する。
- 抽出したフラグメントを分光光度計で定量します。
- 目的のシーケンシングプラットフォームに適したアプローチを使用して、シーケンシング用のdsDNAフラグメントにインデックスを付けます。

図1:大きなDMS処理フラグメントのRT-PCRの実験セットアップ。 修飾されたRNAに対して逆転写を行う場合、プライマーがアニールする配列上の修飾は記録されないであろう。したがって、フラグメントの長さが400〜500bpを超える場合、ここで例示するように、プライマー領域内で重複するフラグメントを設計する必要がある。フラグメントの長さは、シーケンシングのニーズによって異なります。ペアエンド150サイクルシーケンシングを使用する場合、フラグメントは300 bpを超えてはなりません。略語:RT-PCR =逆転写ポリメラーゼ連鎖反応;DMS = 硫酸ジメチル。 この図の拡大版を表示するには、ここをクリックしてください。
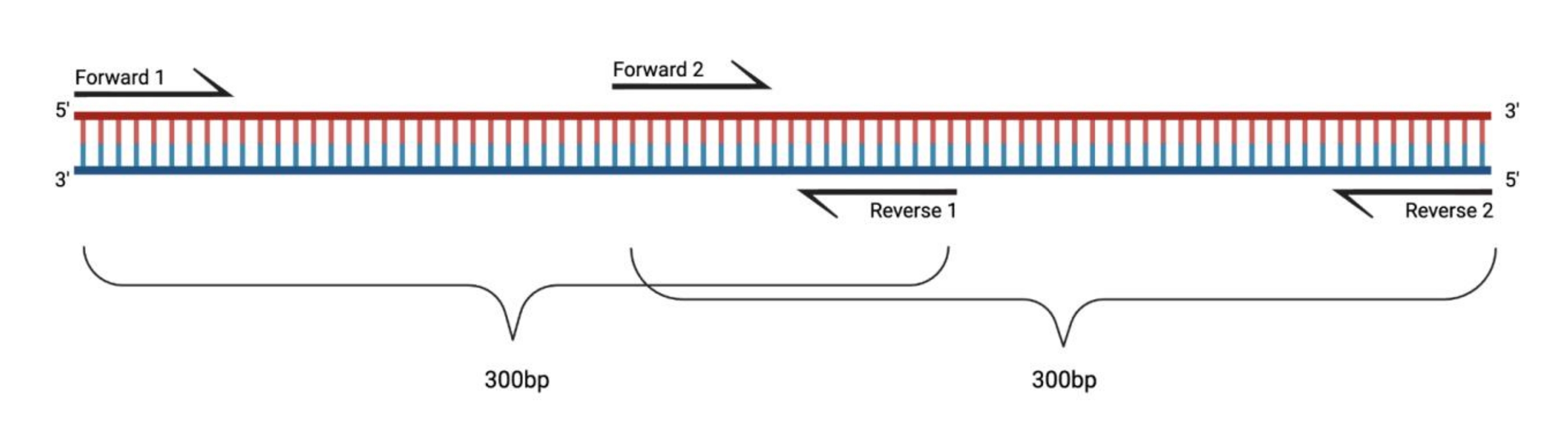{kind=link}
2. ウイルス感染細胞を用いた全ゲノムDMS-MaP
注:細胞では、DMS処理を上記の遺伝子特異的増幅アプローチと組み合わせることもできます。全ゲノムライブラリは、単一の遺伝子を完全にカバーするために膨大なシーケンシングの深さを必要とします。ただし、ウイルスRNAが抽出後にリボデ枯渇RNAのかなりの部分を占める場合は、全ゲノムシーケンスが適切です。さらに、他の濃縮法を全ゲノムライブラリー生成法と組み合わせることができる。
- DMS治療
- ウイルスに感染した細胞を感染の望ましい段階まで増殖させる。
- 必要なバイオセーフティレベルでのウイルスとDMSなどの薬剤によって生成される化学物質の両方の処理に適した専用のヒュームフードにセルコンテナを移します。
- 2.5%容量のDMSを培地に加え、容器(通常は10 cmプレート)をパラフィルムで密封します。
注:DMSを使用すると、簡単に過小変更および過剰変更できます。DMSを細胞に直接添加する場合、よく混合することが非常に重要です。あるいは、新しい培地を50 mLのコニカルチューブで37°Cで予熱し、激しく振とうするDMSを直接加えます。使用済み培地を細胞上でデカントし、DMS含有培地中でゆっくりとピペットでピペットする。 - 37°Cのインキュベーターに5分間移します。
注:インキュベーターの外でDMSを処理するのにかかる時間によっては、5分が過剰変更につながる可能性があります。DMSの追加からインキュベーションまでの時間を≤1分に保ちます。初めて実験を行う場合は、DMS滴定を行い、インキュベーション時間(3分から10分の間)を変えて最適な修飾率を見つけ、濃度のウィンドウ全体で結果が堅牢であることを確認することをお勧めします。 - DMS含有培地を慎重にピペットで取り出し(適切な化学廃棄物に入れ)、10 mLのストップバッファー(30%BMEを含むPBS[例:3 mLのBMEと7 mLのPBS])を加えます。
注意: DMSとBMEを添加すると、細胞が強く接着していない場合、細胞をプレートから持ち上げることができます。細胞が浮き上がっている場合は、DMS含有培地を除去する代わりに浮遊細胞として処理し、ストップバッファーを直接添加し、DMSとBMEで細胞を50 mLコニカルチューブにこすり落とすことができます。3,000 × gで3分間遠心分離して細胞をペレット化します。大きな液滴で細胞の下にペレット化する可能性のある残留DMSを必ず取り除いてください。DMS培地を最初に除去できない場合は、30%BMEで追加の洗浄ステップをお勧めします。 - 細胞をこすり、15 mLのコニカルチューブに移します。
- 3,000 × g で3分間遠心分離してペレット化します。
- 上清を取り除き、10 mLのPBSで2回洗浄します。
- できるだけ多くの残留PBSを慎重に取り除きます。
- ペレットを適量のRNA単離試薬(例えば、T75培養フラスコの場合は3 mL、10 cmプレートの場合は1 mL)に溶解します。
注:試薬の量が不十分な場合、RNA収量に影響を与える可能性があります。
- RNA抽出とリボソームRNA(rRNA)の枯渇
- RNA単離試薬中のホモジナイズした細胞1 mLに、200 μLのクロロホルムを加え、明るいピンク色になるまで15〜20秒間ボルテックスし、相分離が見えるまで最大3分間インキュベートします。
注:ピンク色の脂質相は底に落ち着くはずです。そうでない場合は、ボルテックス時間が不十分であった可能性があります。 - 最高速度(~20,000 × g)で4°Cで15分間回転します。
- 上部水相を新しいチューブに移します。
- 酢酸ナトリウム-エタノール沈殿(ステップ2.5を参照)またはカラムベースのアプローチ(ステップ2.6を参照)によってRNAをクリーンアップし、十分な量のNF H2Oで溶出します。
- アガロースゲルのRNA完全性を確認します。2つのリボソームサブユニットに対応する2つのバンドを探します。
- 好ましいアプローチを用いてrRNAを枯渇させ、適切な量(典型的には20〜50μL)のNF H2Oで溶出する。
注:ダウンストリームアプリケーションでは、8 μLの容量で~500 ngの総RNAが推奨されます。 非リボソームRNAは通常、全RNAの5%〜10%しか占めません。 - 分光光度計を使用して定量します。
- RNA単離試薬中のホモジナイズした細胞1 mLに、200 μLのクロロホルムを加え、明るいピンク色になるまで15〜20秒間ボルテックスし、相分離が見えるまで最大3分間インキュベートします。
- ライブラリ生成
- 遺伝子特異的RT−PCRまたは他のアプローチを使用して、ライブラリーを生成する15。プライミングにランダムな六量体を使用する場合は、六量体アニーリングを可能にするために、低いTm( 37〜42°C)でのインキュベーションステップを追加します。
注:標準ライブラリ生成キットは、RT酵素をTGIRTに置き換え、RT温度を57°Cに変更することによっても使用できます。
- 遺伝子特異的RT−PCRまたは他のアプローチを使用して、ライブラリーを生成する15。プライミングにランダムな六量体を使用する場合は、六量体アニーリングを可能にするために、低いTm( 37〜42°C)でのインキュベーションステップを追加します。
- RNA Clean & Concentrator カラムを使用したカラムベースの RNA クリーンアップ
注意: すべての手順は室温で実行する必要があります。- NF H2Oをサンプルチューブに加えて、50 μLの容量にします。
- 100 μLの結合バッファーと150 μLの100%エタノールをサンプルに加えます。
- 混合してスピンカラムに移します。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- 400 μLのRNA調製バッファーを追加します。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- 700 μLのRNA洗浄バッファーを加えます。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- 400 μLのRNA洗浄バッファーを追加します。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- (オプション)カラムを新しい収集チューブに移し、10,000〜16,000 × g で2分間回転させます。
- カラムを清潔なRNAseフリーチューブに移し、適量のNF H2Oを加える。
- 10,000〜16,000 × g で1分間回転します。
- 酸性フェノール-クロロホルムRNA抽出。
- 等量の酸性フェノール:クロロホルム:イソアミルアルコールを加える。
- 徹底的に渦巻き、14,000 × g で5分間遠心分離します。
- 相分離がない場合は、2 M NaClを20 μL添加し、遠心分離を繰り返します。
- 水相を新しいチューブに移します。
- 500 μLのイソプロパノールと2 μLの共沈剤を追加します。
- 混合し、RTで3分間インキュベートします。その後、-80°Cで一晩インキュベートします。
- 最高速度(~20,000 × g)で4°Cで30分間遠心分離することにより、RNAをペレット化します。
- ペレットを200μLの氷冷70%エタノールで洗浄します。
- 最高速度(~20,000 × g)で5分間回転します。フロースルーを破棄します。
- ペレットを適量のNF H2Oに再懸濁する。
- オリゴクリーンカラムとコンセントレータカラムを使用したカラムベースのcDNAクリーンアップ
注意: すべての手順は室温で実行する必要があります。- NF H2Oをサンプルチューブに加えて、50 μLの容量にします。
- 100 μLの結合バッファーと400 μLの100%エタノールを加えます。
- 混合してスピンカラムに移します。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- 750 μLのDNA洗浄バッファーを追加します。
- 10,000〜16,000 × g で30秒間回転します。フロースルーを破棄します。
- (オプション)カラムを新しい収集チューブに移し、10,000〜16,000 × g で2分間回転させます。
- カラムを清潔なRNAseフリーチューブに移し、適量のNFH2Oを加える。
- 10,000〜16,000 × g で1分間回転します。
3. シーケンシングデータの解析
注:DMS-MaPシーケンシングデータからRNA二次構造モデルを作成するには、結果の.fastqファイルをいくつかの異なるステップで処理する必要があります。これらの手順は、
- アダプターシーケンスをトリムガロアまたはカットアダプトでトリミングします。
- Bowtie2を使用してリードを参照配列(.fasta形式)にマッピングします。
- 特殊なRNA構造ソフトウェア(DREEM14、RNA-Framework17など)でリードをカウントし、反応性プロファイルを作成します。
- (オプション)リードをクラスター化し、DREEM14、DRACO 17、DANCE-MaP18などを使用して代替RNAコンフォメーションを見つけます。
- RNAStructure12、ViennaRNAなどを使用して、反応性プロファイルに基づいて最小自由エネルギー構造を予測します。
- RNA11 構造をVARNA(https://varna.lri.fr/)などを用いて可視化する。
注:実用性のために、DREEM(www.rnadreem.org)やRNA-Framework19 などのソフトウェアは、パイプラインにステップ1〜5を大量に組み込んでおり、分析プロセスを合理化しています。しかしながら、いかなる構造予測も注意して取り扱われるべきである(例えば、構造とデータ20との一致を検証することによって)。
結果
遺伝子特異的イン ビトロ DMS-MaP
SARS2の5'UTRを調べるために、ウイルスの最初の300 bpを3つのプライマーとともにgBlock配列として注文しました。それらには、PCR を介して フラグメントを伝播するための2つのプライマー(「FW」および「RV」)と、T7プロモーター(「FW-T7」)を結合するための1つが含まれていました。これらの配列を 表1に見ることができる。
| 名前 | シーケンス (5'->3') |
| ティッカー | ATTAAAGGTTTATTATTACCTTCCCAGGTAAC |
| ティッカー | GCAAACTGAGTTGGACGTGT |
| FW-T7 | TAATACGACTCACTATAGG ATTAAAGGTTTATACCTTCCCAGGTAAC |
表1:SARS-CoV2 5'UTRのDMS-MaP RT-PCRのプライマー配列。 ここで、FW-T7およびRVは、 インビトロ 転写のためのDNA鋳型を生成するために必要であり、RVは逆転写に使用され、FW-RVプライマーペアは、その後のcDNAのPCR増幅に使用される。プライマーは、SARS-CoV2ゲノム(FW)の冒頭と、関心のある領域のすぐ下流の配列にアニールします。略語:DMS-MaP =硫酸ジメチルを用いたシーケンシングによる変異プロファイリング;RT-PCR = 逆転写ポリメラーゼ連鎖反応;SARS-CoV2 =重症急性呼吸器症候群-コロナウイルス2;UTR = 未翻訳領域;RV =リバースプライマー;FW = フォワードプライマー。
gBlock断片からRNAを生成するために、T7ポリメラーゼプロモーターの配列を、 図2Aに見られるスキームに従ってPCRプレミックスを用いたオーバーラップPCRを用いて結合させた。細長い断片から、T7転写キットを用いてRNAを生成した。続いて、DNAテンプレートをDNaseを用いて消化し、RNAクリーン&コンセントレータカラムを用いて単離したRNAとした。
in vitro転写の品質管理は、RNA産物をssRNAラダーと一緒に1%アガロースゲル上で実行することによって行われました。目に見えるバンドは1つしかなかったため、in vitro DMSプロービングとRT-PCRを実行しました(図2Bを参照)。
PCR反応の成功を検証するために、dsDNAラダーを使用してサンプルを2%アガロースゲル上で実行しました。インデックス作成後、バンドは同じゲル上で~150 bp高く、インデックスプライマーのサイズを考慮します。

図2:DNAテンプレートの インビトロ 転写。 (ア)内因性RNAポリメラーゼプロモーターをまだ有していないDNAテンプレートを in vitro で転写するには、最初にオーバーラップPCRによってテンプレートを添付する必要があります。これは、所望の断片と重なる最初の塩基の上流の配列TAATACGACTCACTATAGG(T7 RNAポリメラーゼの場合)を含むフォワードプライマーを使用することによって行われる。ここでの下線付き基部は、ポリメラーゼの転写開始部位を象徴する。プロモーターがdsDNA断片に結合すると、T7ポリメラーゼによって転写することができます。重要なことに、ポリメラーゼは、言及されたプロモーター配列に対向する鎖を鋳型(青)として使用し、示されたプロモーター配列(赤)のすぐ下流の配列と同一のRNAを効果的に生成する。(B)ssRNAラダー(レーン1)および300ntで in vitro 転写されたRNA産物(レーン2)を有する1%アガロースゲル。(C)GeneRuler 1 kbとラダーを加えた2%アガロースゲル(レーン1)、RT-PCR後のPCR産物を300 bpで実行した後(レーン2)、およびライブラリ調製後のインデックスフラグメントを470 bpで実行(レーン3)。略語:RT-PCR =逆転写ポリメラーゼ連鎖反応;DMS = 硫酸ジメチル;nt =ヌクレオチド;dsDNA = 二本鎖DNA;ssRNA = 一本鎖RNA。 この図の拡大版を表示するには、ここをクリックしてください。
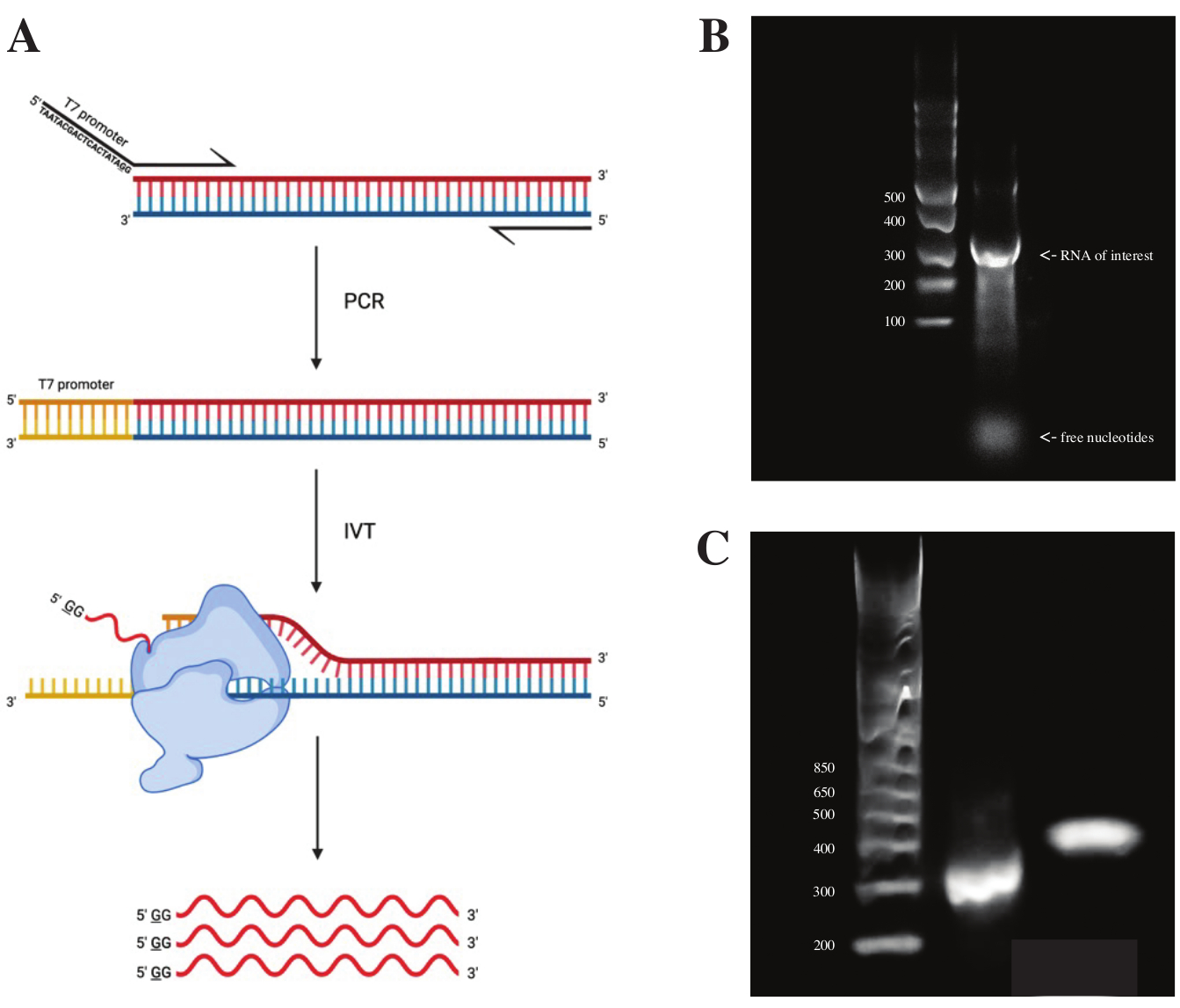{kind=link}
ウイルス感染細胞を用いた全ゲノム インビボ DMS-MaP
DMS処理の前に、HCT-8細胞をOC43に感染させた。感染後4日目に細胞変性効果(CPE)が観察された場合( 図3Aに見られるように)、これらの細胞を処理し、RNAを抽出してリボデロールしました。アガロースゲル上で全RNAを走らせると、リボソームの40Sおよび60Sサブユニットを占める2つの明るいバンドが見え、これらは総RNA質量の約95%を占めます( 図3Bを参照)。RNA抽出が失敗した場合、または分解された場合(例えば、複数回の凍結融解サイクルによって)、RNA分解産物がゲルの底部に見えた( 図3C、2番目のレーンを参照)。さらに、rRNAが枯渇した後、2つの明るいバンドは消失し、レーンに塗抹標本を残しました( 図3C、3番目のレーンを参照)。最後に、ライブラリ調製後、サンプルはさまざまなサイズ分布を持ち、最終的なPAGEゲルに塗抹標本として示されました。バンドは、これらのライブラリを分析するために計画された150 x 150ペアエンドシーケンシングランと一致して、200ヌクレオチド(nt)から500ntの間で切除されました。最も重要なことは、~150 ntで動作するアダプターダイマーが分離されたことです( 図3Dを参照)。

図3:ウイルス感染細胞を用いた 生体内 DMS-MaPのチェックポイント 。 (a)ウイルスに感染したHCT-8細胞の光学顕微鏡像、4日間dpi。細胞死による悪影響を最小限に抑えながら、全RNAから可能な限り最高のウイルスRNAを得るには、画像に見られるように、CPEの開始時またはその前にDMSを追加する必要があります。(B)1 μgの総RNAの6つのサンプルを含む1%アガロースゲル。各レーンには、リボソームRNAが全RNAの~95%を占めるため、40Sおよび60Sサブユニットを占める2つの明るいバンドが見えます。注:細胞内DMS処理では、RNAの断片化と塗抹標本が発生しますが、2つのrRNAバンドはまだ見えるはずです。修飾後の軽度の断片化は、メチル化マークを含む情報が生成され、細胞がまだ生きている間にDMSインキュベーション中にRNA構造について報告するため、許容されます。(C)GeneRuler 1 kbの1%アガロースゲルとラダーDNAマーカー(レーン1)を-80°Cで6ヶ月間保存した全RNAとリボデポリマーRNA(レーン3)。数回の凍結融解サイクルでRNAを長期間保存すると、RNAは分解を開始するため、プロービング実験には使用しないでください。さらに、全RNAをリボデロールした後、リボソームの40Sおよび60Sサブユニットを占める2つの明るいバンドがフェードし、残留RNAの塗抹標本が現れ始める。(D)GeneRuler 1 kbとラダーDNAマーカー(レーン1)のPAGEゲルと、全ゲノムで調製されたRNAのライブラリサンプル。ゲルは、シーケンシングのニーズに基づいて切除する必要があります。両側から150サイクルに及ぶペアエンドシーケンシングランの場合、ゲルは300 bpから500 bpの間で切除する必要があります。アダプターダイマー(170 bpで実行)を分離する必要があります。略語:DMS-MaP =硫酸ジメチルを用いたシーケンシングによる変異プロファイリング;DPI =感染後の日数。CPE =細胞変性効果。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
シーケンス処理後、.fastq ファイルは、.fasta 参照ファイルと共に DREEM ウェブサーバー (http://rnadreem.org/) にジョブをサブミットすることによって分析されました。サーバーによって生成される出力には、fastqc(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)およびTrimGalore(https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)によって生成された品質管理ファイル、および母集団平均突然変異頻度を含むその他の出力ファイルが含まれます。インタラクティブ.html(図4Aを参照)形式の突然変異頻度を示す図と、塩基あたりの生の再活性化を含む.csvファイルと、いくつかのRNA構造予測ソフトウェアで読み取り可能なstruct_constraint.txtファイルとは別に、これには、バイリード突然変異について報告するbitvector.txtファイルも含まれています。これらから、.fastaファイルとstruct_constraint.txtファイルをRNAfoldウェブサーバーに提出することによって、母集団平均構造を計算しました(http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)。これは、ViennaRNAソフトウェアを使用して、最小自由エネルギーに基づいて構造予測を生成し、オンラインで表示したり、ctまたはVienna形式でダウンロードしたりできます。RNA構造モデルを生成するために、これらのダウンロード可能なファイルをVARNAに提出しました(https://varna.lri.fr/、図4Bを参照)。最後に、ビットベクトル.txtファイルをDRIEM(https://codeocean.com/capsule/6175523/tree/v1)の安定バージョンで使用して、代替RNAコンフォメーションを検索できます。DREEMを使用して優れた構造モデルを取得するには、ベースあたり10,000リードのカバレッジを達成する必要があります。クラスタリングの場合、ベースあたり最大 100,000 回の読み取りが必要になる場合があります。ワークフロー全体の概要を図 4C に示します。

図4:SARS-CoV2 5'UTRの化学的プロービング実験から得られた例示的なデータ。 (A)塩基で着色されたSARS-CoV2ゲノムの最初の300塩基の反応性プロファイル(A:赤、C:青、U:緑、G:黄)。生の反応性は、絶対突然変異頻度をカバレッジで割ったものとして計算されます。開環立体配座を有する塩基は高い反応性値を有する。塩基対形成を行う塩基は反応性値が低い。uとGはDMSによって修飾されておらず、ポリメラーゼの不忠実性に起因する反応性値が低い。予測はDREEMウェブサーバーで行われました。(B)VARNAで得られた反応性値から予測されたSARS-CoV2 5'UTRの構造モデル。反応性値の高い塩基は赤色で表示されます。反応性の低い塩基は白色に着色されます。(C)シーケンシングから得られた.fastqファイルから始まるDMS-MaP解析のワークフロー。これらはfastqcを使用して品質管理できます。アダプター配列は TrimGalore を使用してトリミングされ、Bowtie2 を使用して参照配列にマップされます。取得した .bam ファイルから、DREEM は各読み取りのミューテーションをカウントし、ミューテーション マップまたは .bitvector.txt ファイルを作成します。これらは、位置依存的な方法で各リードの突然変異を報告し、それに基づいて母集団平均反応性プロファイルを作成することができます。あるいは、DREEMを使用してビットベクターをクラスタリングし、代替RNAコンフォメーションを検索することもできます。最後に、得られた構造モデルをソフトウェア(VARNAなど)を使用して視覚化します。略語:DMS-MaP =硫酸ジメチルを用いたシーケンシングによる変異プロファイリング;SARS-CoV2 =重症急性呼吸器症候群-コロナウイルス2。 この図の拡大版を表示するには、ここをクリックしてください。
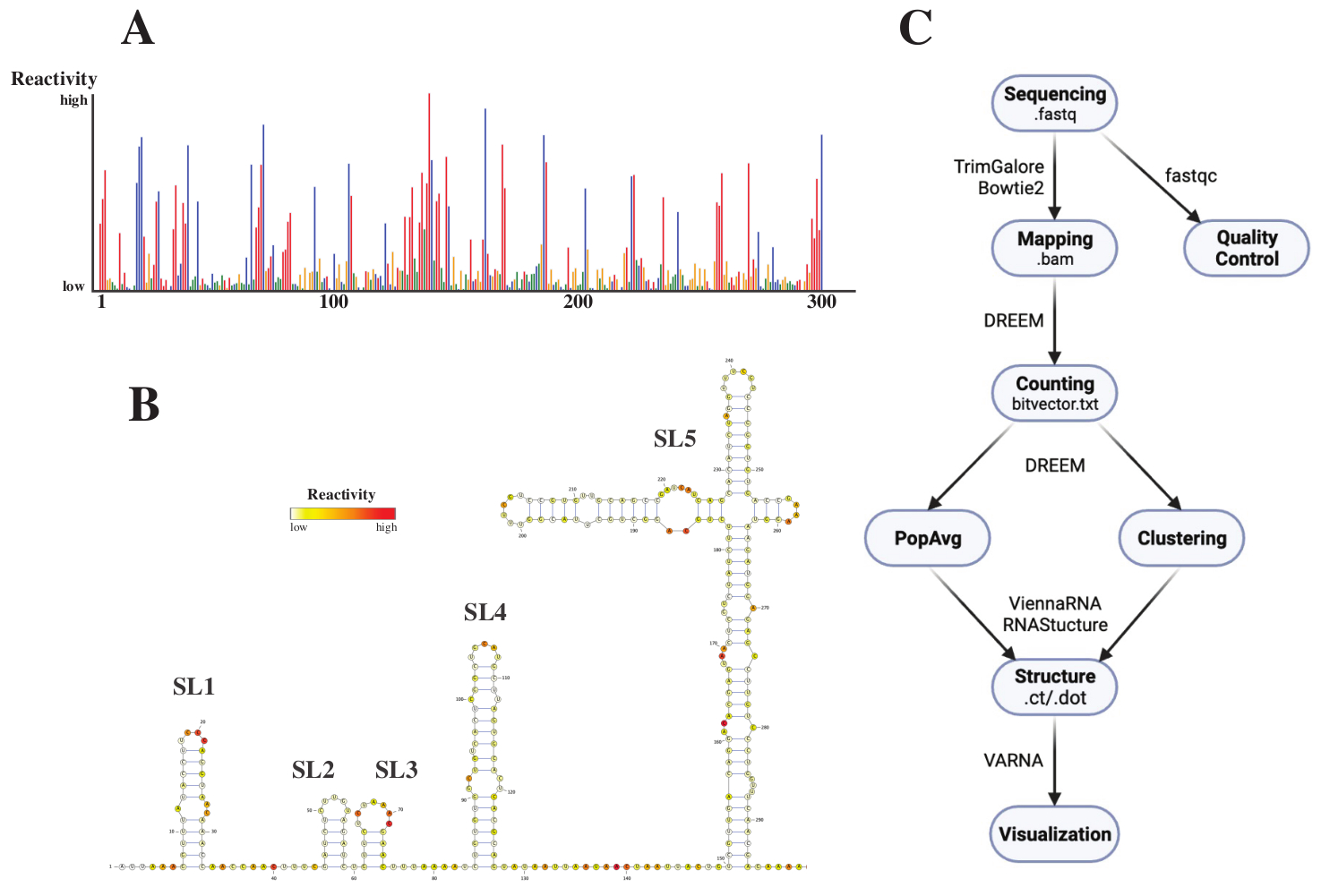{kind=link}
ディスカッション
ここでのプロトコルでは、DMS突然変異プロファイリング実験を使用して 、in vitro および細胞内のRNAをプローブする方法について説明します。さらに、Illuminaシーケンシング用のライブラリを準備して、遺伝子特異的データを生成し、取得した.fastqファイルを分析するための手順を説明します。さらに、ゲノムワイドライブラリーアプローチを使用することができる。ただし、遺伝子特異的RT-PCRは、最高品質で最も堅牢なデータを生成します。したがって、サンプル間で比較する場合は、ライブラリの生成によってバイアスが発生するため、サンプルが同じシーケンシング戦略で調製されていることを確認することが重要です。再現性は、常に反復を使用して測定する必要があります。
いくつかの注意事項
RNAは不安定な分子であり、高温とRNaseの両方による分解に敏感です。したがって、個人用保護具(PPE)、RNAseフリー材料、およびRNAse阻害剤の使用などの特別な対策が推奨されます。最も重要なことは、RNAは可能な限り氷上に保管する必要があるということです。これは特に、高温に対してさらに敏感なメチル化RNAに当てはまります。
目的のRNA構造がDMS濃度および緩衝条件に敏感でないことを確認することが重要です。pH 7〜7.5の100mMトリス、100mM MOPS、および100mM HEPESなどの緩衝液は高いシグナルを与えるが、反応中にpHを維持するには十分ではない可能性がある21。DMSは水中で加水分解してpHを低下させるため、修飾反応中に中性pHを維持するためには強力な緩衝液が重要です。ビシンの添加は、pHをわずかに塩基性21 として維持するのに役立つことが示されていますが、GsおよびUsのDMS修飾が低くなり、有益である可能性がありますが、AsおよびCsよりもはるかに低いシグナルが生成されるため、個別に分析する必要があり、このプロトコルではこれ以上説明しません。
遺伝子特異的RT-PCRでは、修飾されたRNAはDNAに逆転写され、PCRによって断片に増幅されます。RNAのサイズは理論的には無制限ですが、逆転写反応中のバイアスを防ぐために、これらのPCRフラグメントは400〜500塩基対(bp)の長さを超えてはなりません。理想的には、フラグメントはシーケンシング実行の範囲内にある必要があります(すなわち、シーケンシングが150 x 150サイクルのペアエンドシーケンシングプログラムを使用して行われる場合、単一のフラグメントは300 bpを超えてはなりません)。サイクル数が少ないシーケンシングプログラムを使用する場合、dsDNaseを使用してPCR産物を断片化できます。さらに、プライマー配列内の配列は構造情報を保持しないため、プローブされたRNAが>1フラグメントを含む場合、フラグメントは重なり合う必要があります。RT反応には、異なるフラグメント(最大10種類のRTプライマー)に対する複数のRTプライマーを含めることができます。配列によっては、RTプライマーをプールすると逆転写の効率が低下する可能性がありますが、通常はうまく機能します。各PCR反応は別々に実施する必要があります。
DMSでRNAをプロービングする場合、多くのRNAは熱力学的に不安定であり、温度などの環境要因に基づいて立体構造を変化させるため、実験条件がさらに役割を果たします。不規則性を避けるために、実験条件は、反応時間に関しても、可能な限り一定に保つ必要があります。バッファー条件は、RNA24の適切な折り畳みを確実にするために、緩衝能と一価イオン(Na)および二価イオン(Mg)の存在という基本条件が維持されている場合、ある程度交換可能であるようです17、20、22、23。
改変RNAのライブラリー調製に関しては、いくつかの側面を考慮する必要があります。まず、前述のように、修飾されたRNAは修飾されていないRNAよりも安定性が低いため、最適なフラグメントサイズ分布のために断片化時間の最適化が必要になる場合があります。さらに、特定のRNAライブラリ調製キット、および他の多くのRNAseqアプローチでは、逆転写キットでランダムプライマーを使用します。これにより、特に遺伝子の3'で参照のカバレッジが低下し、最終的にはカバレッジの深さが不十分になる可能性があります。特定の領域のカバレッジが低すぎる場合は、構造予測からそれらのベースを削除する必要がある場合があります。RT-PCRおよび全ゲノムRNAseqキットとは別に、他のライブラリ調製アプローチを使用できます。RNAへの3'および/または5'アダプターのライゲーションを含むプロトコルは、RNAの小さな断片を使用する場合、またはプライマー領域でのプロービング情報の損失を回避する必要がある場合に有利です。
最後に、化学プロービング実験の分析は常に慎重に解釈する必要があります。現在、配列単体から任意のRNAのRNA構造を高精度に予測するソフトウェアはありません。化学的プロービングの制約により精度が大幅に向上しますが、長いRNA(>500 nt)の優れたモデルを生成することは依然として困難です。これらのモデルは、他のアプローチおよび/または突然変異誘発によってさらにテストする必要があります。RNA予測ソフトウェアは、塩基対の最大数を最適化するため、RNAフォールディングを正確に表さない可能性のあるオープンコンフォメーションに著しくペナルティを課します5。したがって、得られた構造モデルは、Lanら20に例示されているように、基礎となる化学プロービングデータ(例えば、AUROCによる)および反復間(例えば、mFMIによる)との予測一致を定量化することによってテストされるべきである。
理想的には、得られた構造モデルに挑戦するための異なるシステムでのいくつかの実験を使用して、仮説を強化する必要があります。これらには、 in vitro および細胞内アプローチ、代償的突然変異、および異なる細胞株および種の使用が含まれます。さらに、生の反応性は、RNAフォールディングアンサンブルの「グラウンドトゥルース」スナップショットを記録するため、構造予測と同等かそれ以上に有益であることがよくあります。そのため、生の反応性は、異なる条件間の構造変化を比較するのに非常に適しており、有益です。重要なことに、計算予測を伴う化学的プロービング制約を使用して計算された最も低い自由エネルギー構造は、完全な構造モデルに向けた出発仮説としてのみ使用する必要があります。
開示事項
著者は宣言する利益相反はありません。
謝辞
何一つ
資料
| Name | Company | Catalog Number | Comments |
| 1 Kb Plus DNA Ladder | 10787018 | Thermo | |
| 2-mercaptoethanol | M6250-250ML | Sigma | |
| Acid-Phenol:Chloroform, pH 4.5 | AM9720 | Thermo | |
| Advantage PCR | 639206 | Takara | |
| CloneAmp HiFi PCR Premix | 639298 | Takara | |
| DMS | D186309 | Sigma | |
| dNTPs 10 mM each | U151B | Promega | |
| E-Gel EX Agarose Gels, 2% | G402022 | Thermo | precast agarose gels |
| Ethanol (200 proof) | E7023-4X4L | Sigma | |
| Falcon tubes, 15 mL, 50 mL | |||
| GlycoBlue | co-precipitant | ||
| HCT-8 cells | ATCC #CCL-244 | ||
| Invitrogen MgCl2 (1 M) | AM9530G | fisherscientific | |
| Isopropanol | 278475 | Sigma | |
| Megascript T7 transcription | AM1334 | Thermo | |
| NanoDrop spectrophotometer | |||
| Novex TBE Gels, 8%, 10 well | EC6215BOX | Thermo | |
| OC43 | ATCC #VR-1558 | ||
| RiboRuler Low Range RNA Ladder | SM1831 | Thermo | |
| RNAse H | M0297L | NEB | |
| Sodium Cacodylate, 0.4 M, pH 7.2 | 102090-964 | VWR | |
| Sodium hydroxide solution | S8263-150ML | Sigma | |
| SuperScript II Reverse Transcriptase for FSB and DTT | 18064014 | Thermo | |
| TGIRT-III Enzyme | TGIRT50 | Ingex | |
| The Oligo Clean & Concentrator | D4060 | Genesee | |
| The RNA Clean & Concentrator kits are RNA clean up kits | R1016 | Genesee | |
| TRIzol Reagents | 15596018 | Thermo | RNA isolation reagent |
| Water, (For RNA Work) (DEPC-Treated, DNASE, RNASE free/Mol. Biol.) | BP561-1 | fisherscientific | |
| xGen Broad-range RNA Library Prep 16rxn | 10009865 | IDT | |
| Zymo RNA clean and concentrator columns |
参考文献
- Kim, S. H., et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 185 (4149), 435-440 (1974).
- Robertus, J. D., et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 250 (467), 546-551 (1974).
- Zaug, A. J., Cech, T. R. In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell. 19 (2), 331-338 (1980).
- Zhao, Y., et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 44, D203-D208 (2016).
- Vandivier, L. E., Anderson, S. J., Foley, S. W., Gregory, B. D. The conservation and function of RNA secondary structure in plants. Annual Review of Plant Biology. 67, 463 (2016).
- Jumper, J., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596 (7873), 583-589 (2021).
- Das, R. RNA structure: A renaissance begins. Nature Methods. 18 (5), 439-439 (2021).
- Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A., Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature Protocols. 10 (11), 1643-1669 (2015).
- Mathews, D. H., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 101 (19), 7287-7292 (2004).
- Zuker, M., Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research. 9 (1), 133-148 (1981).
- Lorenz, R., et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology. 6, (2011).
- Reuter, J. S., Mathews, D. H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, (2010).
- Wells, S. E., Hughes, J. M. X., Igel, A. H., Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods in Enzymology. , 479-493 (2000).
- Tomezsko, P. J., et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. 582 (7812), (2020).
- Zubradt, M., et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nature Methods. 14 (1), (2017).
- Woodson, S. A. Compact intermediates in RNA folding. Annual Reviews in Biophysics. 39, (2010).
- Morandi, E., et al. Genome-scale deconvolution of RNA structure ensembles. Nature Methods. 18 (3), 249-252 (2021).
- Olson, S. W., et al. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Molecular Cell. 82 (9), 1708-1723 (2022).
- Incarnato, D., Morandi, E., Simon, L. M., Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Research. 46 (16), (2018).
- Lan, T. C. T., et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nature Communications. 13 (1), 1128 (2022).
- Homan, P. J., et al. Single-molecule correlated chemical probing of RNA. Proceedings of the National Academy of Sciences of the United States of America. 111 (38), 13858-13863 (2014).
- Yang, S. L., et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nature Communications. 12 (1), 5113 (2021).
- Manfredonia, I., et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Research. 48 (22), 12436-12452 (2020).
- Fischer, N. M., Polěto, M. D., Steuer, J., vander Spoel, D. Influence of Na+ and Mg2+ ions on RNA structures studied with molecular dynamics simulations. Nucleic Acids Research. 46 (10), 4872-4882 (2018).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved