
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Viral Tracing von genetisch definierten Neural Circuitry
In diesem Artikel
Zusammenfassung
Verfahren zur Verfolgung synaptisch verbundenen Neuronen beschrieben. Wir verwenden TVA Spezifität eines vorgeschalteten Zelle zu untersuchen, ob eine Zellpopulation von Interesse synaptischen Input von genetisch definierten Zelltypen erhält.
Zusammenfassung
Klassische Methoden zur Untersuchung neuronaler Schaltkreise sind ziemlich geringen Durchsatz. Transsynaptische Viren, insbesondere das Pseudorabiesvirus (PRV) und Tollwutvirus (RABV) und neuerdings vesikuläre Stomatitis-Virus (VSV), eine Schaltung zur Untersuchung, wird immer beliebter. Diese höheren Durchsatz Methoden verwenden Viren, die zwischen den Neuronen entweder in der anterograde oder retrograde Richtung übertragen.
Kürzlich wurde eine modifizierte RABV für monosynaptischen retrograden Tracing entwickelt. (Abbildung 1A). Bei diesem Verfahren wird das Glycoprotein (G)-Gen aus dem viralen Genom gelöscht und wieder zugeführt nur in gezielter Neuronen. Infektion Spezifität wird durch Substituieren eines chimären G, der extrazellulären Domäne des ASLV-A Glykoprotein und die zytoplasmatische Domäne des RABV-G (A / RG) zusammengesetzt ist, für die normale RABV-G 1 gelöst. Dieses chimäre G gezielt infiziert Zellen, die den TVA-Rezeptor-1. Das Gen kodiert TVA kann schon delivered durch verschiedene Verfahren 2-8. Nach RABV-G Infektion einer TVA-exprimierenden Neuronen, kann die RABV zu anderen, synaptisch verbundenen Neuronen in retrograder Richtung von der Natur des eigenen G, die mit dem Rezeptor TVA zusammen geliefert war zu übertragen. Diese Technik kennzeichnet eine relativ große Anzahl von Eingängen (5-10%) 2 auf einem definierten Zelltyp, Bereitstellen eines Probenahme von allen Eingängen auf einen definierten Starter Zelltyp.
Wir haben vor kurzem diese Technik, um VSV als transsynaptische Tracer 9 verwenden modifiziert. VSV hat mehrere Vorteile, einschließlich der Schnelligkeit der Genexpression. Hier haben wir ausführlich eine neue virale Tracing-System mit VSV nützlich für das Sondieren Mikrotechnik mit erhöhter Auflösung. Während die ursprünglich veröffentlichten Strategien Wickersham et al. 4 und Beier et al. 9 Erlaubnis Beschriftung bei allen Neuronen, projizieren eingangs infiziert TVA-exprimierenden-Zellen, hier VSV dazu entworfen wurde, um nur zu übertragen, um TVA-exprimierenden Zellen (Abb. 1B). Das Virus wird zuerst mit RABV-G pseudotypisiert zur Infektion von Neuronen hinter TVA-exprimierenden Neuronen ermöglichen. Nach der Infektion dieses ersten Population von Zellen, das Virus freigesetzt nur infizieren TVA-exprimierenden Zellen. Weil die transsynaptische viralen Ausbreitung auf TVA-exprimierenden Zellen, die Anwesenheit von Abwesenheit von Konnektivität von definierten Zelltypen beschränkt wird dann mit hoher Auflösung untersucht werden. Eine experimentelle Flußdiagramm dieser Versuche ist in Abbildung 2 gezeigt. Hier zeigen wir, ein Modell-Schaltung, dass der Richtungswechsel-Selektivität bei der Maus Netzhaut. Wir untersuchen die Konnektivität starburst Amakrinzellen (SAC) an retinalen Ganglienzellen (RGZ).
Protokoll
Ein. Erstellen Virus aus cDNA: Recovery von VSV aus cDNA mit Vaccinia-T7 System 10
- Einen Tag vor zu experimentieren, aufgeteilt BsrT7 Zellen in 60 mm Schale mit DMEM + 10% FBS. Seed 2E6 Zellen pro Schale. BsrT7 Zellen aus BHK21, oder Baby-Hamster-Nierenzellen, Zellen abgeleitet.
- Warmen PBS mit 1 mM Magnesiumchlorid und 1 mM Calcium und 37 ° C.
- Besorgen Sie sich eine kleine Teilmenge vTF7-3, ein Vaccinia-Virus, das T7-Polymerase und Auftauen auf Raumtemperatur. vTF7-3 ist eine ansteckende Vaccinia-Virus, das T7-Polymerase 11, und sollte nur in Biosafety Level 2 Containment verwendet werden. Dieser Ausdruck wird verwendet, um maximale Werte der gewünschten Transkripte von Plasmiden in Schritt 1,9 transfiziert erzeugen. Vortex-Virus für 30 sec bei max. Beschallen in einer Tischplatte Sonicator für 2 min im Wasserbad bei Raumtemperatur. Vortex wieder für 30 sec zu brechen viralen Klumpen.
- Entfernen Sie das Medium aus den BSRT7 Zellen.
- Fügen 11,7 ul vTF7-3zu 600 ul PBS mit Magnesium und Calcium und fügen Mischung auf die Zellen.
- Bewegen Platten in einem 34 ° C Inkubator mit 5% CO 2. Rock-Platten vorsichtig einmal alle 10 min.
- Nach 45 min vergangen sind, beginnen Transfektion. Fügen Sie 20 ul Lipofectamin 2.000 bis 1 ml DMEM und mischen. Warten Sie 5 min.
- Hinzufügen 5,25 pg pN, 2,3 pg pP, 1,2 ug pl, 1 ug PCAG RABV-G und 6 ug VSV genomischen cDNA zu 500 ul DMEM. pN, pP und pl werden Plasmide die Expression von VSV virale Gene unter der T7-Promotor 10, wobei die Mengen empirisch für optimale Rettung des Virus bestimmt wird.
- Kombinieren Sie beide Schläuche und mischen. Warten 15 min bei Raumtemperatur.
- Entfernen PBS waschen und die Platten einmal mit 1,5 ml DMEM.
- Saugen Spülen Medien. Fügen Transfektion Mix auf Platte. Bewegen Platten in 34 ° C-Inkubator.
- Warten Sie 5 hr, dann entfernen Medien.
- Anderer Medien auf 4 ml DMEM + 2% FBS + 1x Penicillin-Streptomycin + 10 mM HEPES pH 7,4. Die Zugabe von Penicillin-Streptomycin, um eine Kontamination zu verhindern, und HEPES, um den pH des Mediums aufrechtzuerhalten, sind sehr wichtig. Der Inhalt kann FBS bis 2% fallen gelassen werden, da die Zellen in weniger Tage durch das Virus zerstört werden und damit nicht erforderlich 10% FBS.
- Ort Platten in 34 ° C Inkubator für 48-72 Std.
- Collect Medien bei 3 und 6 Tage nach der Transfektion, und sofort Spritzenfilter durch einen 0,20 um Filter. Diese Größe Filter entfernt das Vaccinia-Virus, aber nicht das VSV.
- Da wir RABV-G der Virushülle liefern wollen, und das Genom kodiert nicht die RABV-G-Gen, muss es ständig in trans geliefert werden. Um dies zu tun, stellen Sie eine separate Platte von 293T TVA800 (TVA800)-Zellen. Dies sind Zellen, die konstitutiv das TVA Rezeptor erlauben ENVA-vermittelte Virusinfektion 12. Bei 80% Konfluenz, ändern Medien DMEM nur dann transfizieren die Zellen mit 5 ug Plasmid, das G-Protein (dh PCAG RABV-G) pro Platte. Wir verwenden die PEI Transfektionsreagenz, ein polykationisches Polymer, welches wesentlich günstiger als vergleichbare Lipid-basierte Verfahren ist. Allerdings kann auch Lipofectamin effektiv für diesen Zweck verwendet werden. Die optimale PEI: DNA-Verhältnis muss empirisch ermittelt werden. Tun Sie dies in einem separaten Experiment mit einem fluoreszierenden Reporter, dh PCAG GFP. Wir verwenden 10 ul PEI: 4 ug DNA.
- Übernehmen Sie die Überstand aus Schritt 16 auf den transfizierten Platte TVA800 Zellen.
- Beachten Sie diese Platte am folgenden Tag zum Nachweis der Fluoreszenz des viral ausgedrückt Fluorophors. Virale Fluoreszenz wird zunächst als eine einzelne Zelle beobachtet, und breitet sich allmählich über die Zeit (dh Abbildung 3). Das Virus sollte innerhalb von wenigen Stunden zu verbreiten.
2. Passage und Konzentration des VSV
- Split TVA800 Zellen in Medien mit DMEM + 10% FBS, so dass Sie vier 10-cm Platten bei ~ 80% Konfluenz am nächsten Tag haben. 293T-basierten Zellen gut funktionieren aufgrund ihrer hohen transfectability, und die Verwendung von TVA-exprimierenden Zellen erhöht die Geschwindigkeit, mit der die Zellen in der Schale infiziert. Andere hoch transfizierbar Zelllinien können die Zweck dienen.
- Bei 80% Konfluenz, ändern Medien DMEM nur dann transfizieren die Zellen mit 5 ug Plasmid, welches das G-Protein, dass Sie verwenden möchten (dh PCAG RABV-G) pro Platte. Wir verwenden PEI, aber FUGENE / Lipofectamin Arbeit als gut.
- Warten einen Tag nach der Transfektion für Glykoproteinexpression / Oberflächenwasser Akkumulation. Anderer das Medium (5 ml / Platte) gegen DMEM + 10% FBS, dann mit rVSV (A / RG)-Virus zu infizieren bei einer Multiplizität der Infektion (MOI) von 0,01
- Überprüfen Sie die Menge der Infektion 24 Stunden später. Sie sollten sehen, infizierte Zellen (gekennzeichnet durch GFP-Fluoreszenz), einige mit den umliegenden Flecken von infizierten Zellen (dh Abbildung 3). Normalerweise ist es nur wert Sammeln des Überstandes, wenn Sie> 50% der Zellen infiziert, da sonst die virale Titer sehen obtained von der Konzentration wird suboptimal.
- Wenn> 50%, sammeln die 5 ml Medium / Platte, und ersetzen mit 5 ml frisches Medium. Wenn zu wenige Zellen infiziert sind, für ein anderes 12-24 hr warten und erneut prüfen. Frieren Sie die gesammelten Überstände bei -80 ° C.
- Sammle alle 24 Stunden danach für 3 Tage, für einen Gesamtbetrag von 4 Tagen.
- Insgesamt, wenn Sie mit 4 Platten begonnen haben, sollten Sie 20 ml von Medien aus jeden Tag - so 80 ml insgesamt oder 2 Ultrazentrifuge Rohre im Wert von Überstand. Auftauen der Überstände bei 37 ° C und filtriert über 0,45 um Filter auf Eis. Aliquot 36 ml des Virus in Beckman-Zentrifugenröhrchen.
- Konzentrieren den Überstand (21.000 K in einem SW28 Rotor (= 80.000 xg) für 90 min) bei 4 ° C. Dekantieren des Überstandes in ein Becherglas mit 10% Bleichmittel, und während der Schlauch invertiert ist, verwenden eine Absaugvorrichtung zum soviel Medien von der Seite des Rohrs zu entfernen.
- Während abgedeckt. Schütteln Sie die viralen Röhrchen bei 4 ° C für 1 Stunde. Ein Standard-Schüttler bei about 120 rpm ist ausreichend.
- Gently verreiben Pellet in den restlichen Medien durch vorsichtiges Auf-und Abpipettieren 30 mal. Achten Sie darauf, keine Luftblasen einzuführen. Die übrigen Datenträger sollte etwa 30 ul sein. Aliquot dieses Volumen in mehrere Rohre so mehreren Gefrier-Auftau-Zyklen für einen bestimmten Lager zu verhindern, da dies zu einer Abnahme der Virustiter führen kann. Diese Aliquots werden bei -80 ° C eingefroren werden
- Titer des Virus. Titrierung bei etwa 2 Tage nach der Infektion ist optimal. Erstinfektion kann in einer Angelegenheit von Stunden beobachtet werden, aber Sie bekommen eine Unterrepräsentation der Titer, wenn man nur 1 Tag warten. Um den viralen Titer zu erhalten, führen eine Grenzverdünnung Experiment der konzentrierten Virus an die geeignete Zelllinie (293T-Zellen funktionieren gut) in einer 24-Well-Platte, Verdünnen des Virus 10-fache von 1 bis 1: 1.000.000 beträgt. Die Titer erhalten wir sind typischerweise im Bereich von 10 8 -10 10 Fokus forming units (ffu) / ml.
3. Viruns Injection
- Wir verwenden Mäusen, typischerweise zwischen 6-10 Wochen alt, für diese Experimente. Jedes Alter, nach der Schaltung hergestellt wird ausreichen würde. Wir wählen die Übertragung von retinalen Ganglienzellen (RGCs) bis Starburst Amakrinzellen (SAC) als Beispiel. Dieses Experiment, wie in Abbildung 2 graphisch dargestellt, erfordert einen dreifach transgenen Tieres und nur eine einzige Injektion von Virus. (Alle gezeigten Verfahren wurden von der IACUC an der Harvard Medical School zugelassen und waren in Übereinstimmung mit den Richtlinien des Instituts).
- Zunächst müssen die geeigneten Tieren generiert. Das Ziel ist, dass in der TVA Zelltyp exprimiert daß man wünscht, in zuzuordnen, mittels eines Virus (dh Adeno-assoziierten Virus, AAV) oder durch eine transgene Allel. Die Zelltypspezifität wird üblicherweise durch Cre Ausdruck erhalten. Hier haben wir auch gekreuzt in einem bedingten Ausdruck TVA Allel 7 zu einem Choline Acetyltransferase (ChAT)-Cre-Allel 13, so dass TVA ausgedrückt wirdin allen Zellen mit einem Cre Expression Geschichte (die SAC). Wir überquerten einen bedingten Ausdruck tdTomato Allel (R26TdT) zur Visualisierung der TVA-exprimierenden Zellen.
- Die Koordinaten für die Eindüsestelle müssen identifiziert werden. Diese Koordinaten von Interesse kann in einem Hirnatlas, dh die Franklin und Paxinos Maushirn atlas 14 gefunden werden. Diese Koordinaten müssen relativ zu einer gegebenen Landmarke an der Maus Schädels vermerkt. Wir verwenden Bregma, aber jede Koordinatensystem ist ausreichend.
- Bereiten Sie den stereotaktischen Apparat. Schalten Sie den Mikroinjektor Controller, und bewegen Sie den Kolben und Elektrodenhalter einrastet. Dann zurück-füllen die Injektionsnadel mit Mineralöl, um eine Schnittstelle mit dem Virus bereitzustellen. Setzen Sie die Nadel dann auf den Kolben, um sicherzustellen, dass keine Luftblasen in der Nadel bleiben.
- Auftauen das Virus, und auf Eis mit einem Rückgang der Virustiter zu verhindern. Stellen Sie die Mikroinjektor "Ziehen" und bewegen Sie den Schlauch des Virus, so dass das KontaktI istng die Kapillare an der Mikroinjektorkopfs. Zurückzuziehen die notwendige Menge an rVSV (A / RG) mit RABV-G für das Experiment pseudotypisiert.
- Die Tiere werden einen präoperativen Dosis Buprenorphin (0,05-0,1 mg / kg) gegeben, bevor die Operation beginnt. Für Betäuben das Tier, entweder Isofluran Inhalation oder eine intraperitoneale Injektion einer Ketamin (40-80 mg / kg) und Xylazin (5-10 mg / kg) Gemisch ausreicht. Auswahl des Narkosemittels ist abhängig von dem Benutzer. A toe Prise wird dann durchgeführt, um sicherzustellen, dass die Tiere voll narkotisiert sind, wenn das Tier nicht reagiert, können Sie mit der Prozedur fortzufahren. Achten Sie auf ordnungsgemäße Lizenzen, bevor Sie diese Medikamente erhalten.
- Die Tiere werden auf eine erhitzte Unterlage, die auf einem beweglichen Gestell zur Positionierung der Maus ist gelehnt plaziert. Das Tier bleibt es während der gesamten Dauer des chirurgischen Eingriffs. Bewerben Augensalbe zum Trocknen der Hornhaut zu verhindern, während das Tier betäubt.
- Die Maus Kopf muss dann in der stereotaktischen einer stabilisiert werdenpparatus. Mit den Balken auf dem Ohr stereotaktischen Vorrichtung, um sicherzustellen, daß der Kopf vollständig gesichert ist, so dass der Kopf parallel zum Boden ist und nicht seitlich drehen. Sobald dies abgeschlossen ist, stellen Sie den Biss bar zur Stabilisierung des Kopfes.
- Die Tier das Fell ist in der Region Schnitt auf der Oberseite des Kopfes rasiert. Dieser Bereich wird dann dreimal mit Ethanol gewaschen, gefolgt von drei Mal mit Jod. Ein Einschnitt wird dann in der Haut mit einem Skalpell und enthüllt den Schädel.
- Sobald die Maus und Injektion Setup hergestellt werden, muss Bregma lokalisiert werden. Dies ist der Bereich in der Mitte des Kopfes, dass die sagittalen und zwei Koronarnaht erfüllen. Sobald dieser Bereich liegt, können die Wählscheiben am stereotaktischen Vorrichtung verwendet, um die für das ordnungsgemäße Mikroinjektorkopfs Koordinaten einzustellen. Hier injizieren wir den seitlichen Kniehöcker (LGN), mit Hilfe von Koordinaten A / P -2,46 vom Bregma, L / M 2, D / V 2.75.
- Nachdem die Koordinaten im gewählten, muss der Bohrer zusammengesetzt werden.Setzen Sie den Bohrer in die Bohrmaschine, dann auf den Bohrer drehen. Ein kleines Loch sollte in den Standort wie der Injektionsstelle festgestellt gebohrt werden. Seien Sie sehr sanft mit dem Bohrer, um nicht zu Schäden an der Hirnparenchym. Wenn der Knochen sehr dünn ist, werden die Blutgefäße auf der Dura klar. An dieser Stelle aufmerksam perforieren die Kanten des Kraniotomie mit einem kleinen (30 Gauge) und einer feinen Nadel (# 5), um die Zange verbleibenden Knochen zu entfernen.
- Passen Sie die Kapillare zu einer Position unmittelbar dorsal des Gehirns Oberfläche, wo das Loch gerade gebohrt wurde. Die Nadel sollte scharf genug, um die Dura mater mit Leichtigkeit durchdringen. Anschließend Senken der Nadel auf die gewünschte Tiefe, und beginnen Injektion. Wir injizieren Virus bei einer Rate von 100 nl / min.
- Die Injektion zu diesem Punkt dauert etwa 10 Minuten. Nach der Injektion auf, bleibt die Nadel im Gehirn für etwa sieben Minuten. Dies ist entscheidend, da sie die Diffusion von Virus erlaubt von der Injektionsstelle. Schnelles Entfernen der Nadel bilden die injection Ort führt zu einem reduzierten Wirkungsgrad Infektion am Ort der Injektion, und Infektionen entlang der Nadel Trakt.
- Die Nadel wird sehr langsam aus der Injektionsstelle angehoben, über einen Zeitraum von etwa zwei Minuten. Dies wird wiederum bestimmt die Infektion entlang der Stichkanal zu reduzieren.
- Nach dem Entfernen wird die Haut dann vernäht. Die Tiere werden kontinuierlich bis sie sich erholen von der Narkose überwacht. Buprenorphin (0,05-0,1 mg / kg) wird alle 12 Stunden für 2 Tage verabreicht.
4. Harvesting Tissue / Tissue Vorbereitung
- Zum Ernten das Gewebe von Interesse, hängt die optimale Zeit über die Ziele des Experiments. Im Allgemeinen können VSV transsynaptische gespreizten ersten von weniger als 24 h nach der Injektion beobachtet werden. Allerdings wartet längere Zeiten in mehr Verbreitung führen.
- Um die Netzhaut zu ernten, werden die Tiere eingeschläfert ersten Verwendung von Kohlendioxid Einatmen und Genickbruch durchgeführt. Der Augapfel wird dann entfernt und in ein Rohr con platziertTaining 4% Formaldehyd in PBS für 1 Stunde bei Raumtemperatur.
- Der Augapfel wird dann in eine Petrischale mit PBS gelegt. Die Retina ist dann von anderen Geweben präpariert.
- Für whole mount Analyse, die retinae so geschnitten, dass flach, platziert auf einem Glasträger sind, dann mit zu verlängern gold Eindeckmittel montiert und mit einer Null-Dicke Deckglas. Silikon Abstandshalter zwischen dem Schieber und Deckglas auf Druckkräfte auf die Netzhaut zu minimieren platziert. Für den Abschnitt Analyse wird die Netzhaut in 30% Saccharose in PBS, bis die Netzhaut taucht platziert. Es wird dann in einem Gemisch aus 50% Oktober platziert und 50% 30% Saccharose-Lösung für 3 Stunden. Es wird dann blinken eingefroren und bei -80 ° C bis bereit zu schneiden.
5. Repräsentative Ergebnisse
Das Virus, das aus cDNA gerettet wird, sollte in der Lage sein TVA800 Zellen, aber nicht 293T-Zellen (Figur 3) zu infizieren. Die Versorgung der RABV-G ermöglicht die Infektion von mehreren Zell-types, einschließlich 293T. Da jedoch der A / RG Genoms in das Genom kodiert wird, unter den Zellen verteilt bei einer viel höheren Rate im TVA-exprimierenden Zellen.
Sobald das Virus ist konzentriert, typisch Titer zwischen 10 8 ffu / ml und 10 10 ffu / ml. Diese Titer ausreichend sind für die Verwendung in vivo.
Während LGN Injektion in die Maus, wissen wir nicht unterscheiden dorsalen LGN (DLGN) von ventral LGN (vLGN), da beide in der Regel infiziert. Daher werden mehrere RGC Typen bezeichnet (Abbildung 4), einschließlich derer, die Projekt zum vLGN wie Melanopsin RGCs (Abbildung 4D) und ON-DSGCs (4E), und diejenigen, die Projekt zum DLGN, wie die kleinen arbor RGCs (4C) und ON-OFF-DSGCs (Abbildung 4F). Obwohl mehr als eine Art von RGC übertragen Virus SACs, wie an anderer Stelle berichtet (Beier et al., In review), hier konzentrieren wir uns nurauf den gut untersuchten Verbindungen von ON-OFF-DSGCs um Schutzgebiete.
(-), Wenn wir Mäuse des Genotyps CTVA (+) / ChAT-Cre injizieren, wo kein TVA ausgedrückt werden soll, werden nur markierte RGCs (dh Abbildung 4). Diese sind auf anfängliche Aufnahme des RABV-G pseudotypisierten Virus durch den RGCs und keine Ausbreitung auftritt. Keine RGCs werden aus einer Infektion der LGN rVSV (A / RG) nicht mit Virus-G RABV pseudotypisiert aufgrund einer Unfähigkeit dieser Virionen an der langen transversalen Axone dieser RGCs infiziert. Wenn jedoch Einspritzen CTVA (+) / ChATCre (+) (und R26TdT (+)) Mäuse in der LGN, bedeutet virale Ausbreitung auftreten, nur um rote Blutkörperchen, die das Cre-Reporter (zB Fig. 5) exprimieren. Diese Zellen auch mit-Label mit dem anti-ChAT Antikörper und Schichtung nur in den beiden Plättchen ChAT und bestätigt ihre Identität als SAC (5B ', C'). Die Zahl der viral bezeichnet SACs pro DSGC reichten 1-9.

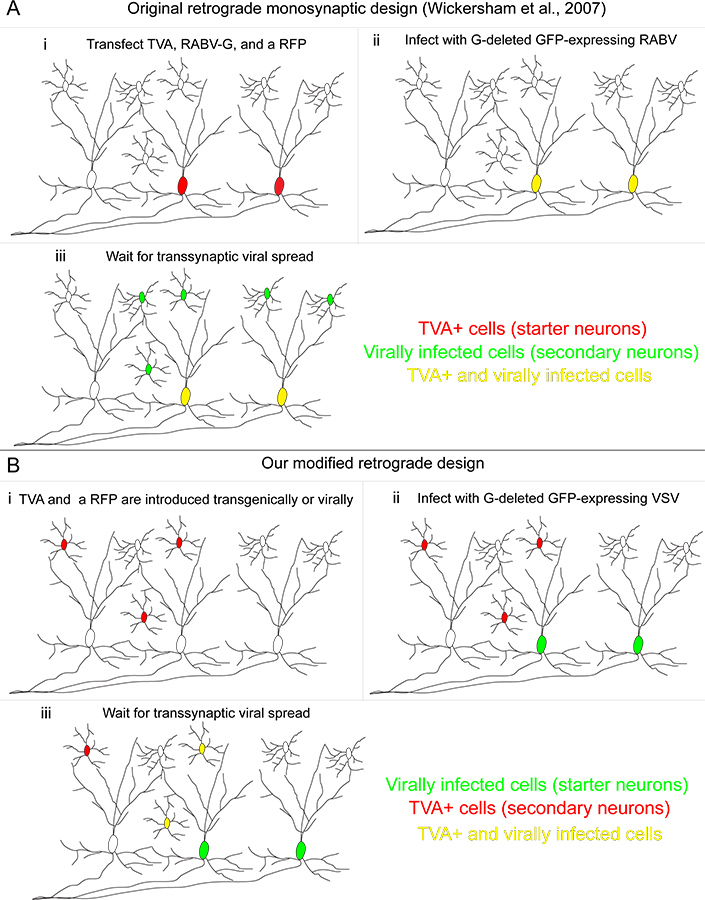
Abbildung 1. Unsere retrograde transsynaptische Tracing-System im Vergleich zu der ursprünglich entwickelten Methode. (A) (i) In der Methode von Wickersham und Kollegen entwickelte "starter Zellen" sind mit drei Genen transfiziert: die TVA-Rezeptor, zur Infektion spezifisch ermöglichen der transfizierten Zellen, die RABV-G, verwendet, um die RABV ergänzen, die selbst hatte das G-Gen deletiert und ein rot fluoreszierendes Protein, um transfizierte Zellen zu identifizieren. (Ii) Ein RABV mit dem A / RG Protein pseudotypisiert infiziert TVA-exprimierenden Zellen, wodurch sie gelb. (Iii) Retrograde transsynaptische Übertragung erfolgt zum vorgeschalteten Neuronen. (B) (i) In unserem Verfahren werden TVA-exprimierenden Zellen genetisch, definiert und mit einem bedingten roten fluoreszierenden markierten protein. (ii) Die "Starter Zellen" sind die von einem VSV Codieren des A / RG Gen im viralen Genom infiziert. Diese Starter-Zellen exprimieren nicht das TVA-Rezeptor. (Iii) Retrograde transsynaptische Übertragung erfolgt TVA-exprimierenden Neuronen nur dann, wenn diese Zellen synaptischen Input liefern auf die Starter-Neuronen. Klicken Sie hier für eine größere Abbildung zu sehen .
{kind=link}

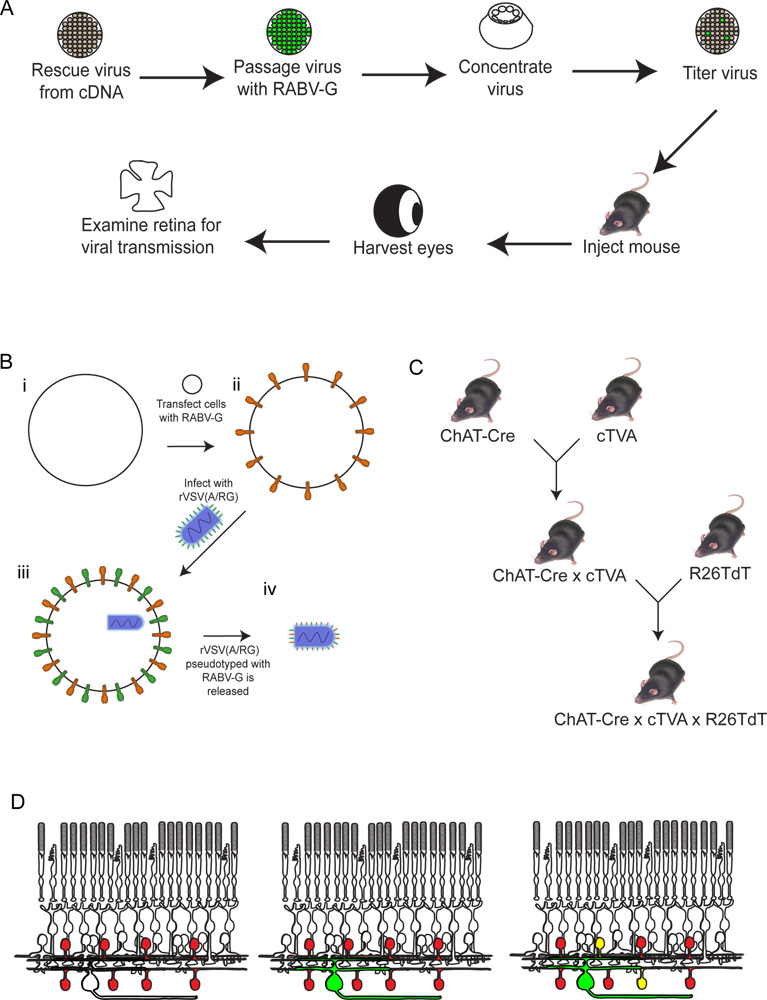
Abbildung 2. Schematische Darstellung des experimentellen Verfahren. (A) Zunächst wird das Virus aus cDNA gerettet. Es wird dann passagiert und mit pseudotypisiert RABV-G und eingeengt und titriert. Das Virus wird dann in das Gehirn injiziert, wo es erlaubt ist, für den gewünschten Zeitraum zu inkubieren. DanachEs wird das Auge geerntet, und die Netzhaut seziert und analysiert. (B) Schematische Darstellung der Pseudotypisierung das Virus mit RABV-G. Dies ist notwendig für die Infektion von retinalen Ganglienzellen von einem Gehirn Injektion. Die RABV-G-Gen zunächst in Zellkultur-Zellen transfiziert Ausdruck der TVA-Rezeptor. Diese Zellen werden dann mit dem rVSV (A / RG) Virus infiziert. Die Virionen freigesetzt wird RABV-G in der Virushülle zu haben, aber nicht haben das Gen für RABV-G in das virale Genom. (C) Eine schematische Darstellung des Maus kreuzt notwendig dreifach transgenen Tieren. Bedingter TVA und tdTomato Allele sind mit einer Cre Fahrer nach Wahl, so dass sowohl TVA und tdTomato nur in Zellen mit einem Cre Expression Geschichte exprimiert werden gekreuzt. In diesem Beispiel verwendeten wir ChAT-Cre. (D) Eine schematische Darstellung des retinalen Infektion um Trace-SAC DSGC Schaltungen. Das Chat-Zellen zu exprimieren TVA und tdTomato gemacht, wie in (C) gezeigt. RGCs aus einem LGN in infiziertfection des Virus hergestellt in (B). Da das Virus exprimiert GFP, werden diese RGCs grün sein. Der Virus wird dann nur breitete sich TVA-exprimierenden ChAT Amakrinzellen, wenn sie an den markierten RGC verbunden sind. Diese Zellen werden sowohl rot und grün, oder gelb. Klicken Sie hier für eine größere Abbildung zu sehen .
{kind=link}

Abbildung 3. Verhalten rVSV (A / RG) Virus auf 293T und TVA800 Zellen. Diese Zellen wurden mit einem niedrigen Titer rVSV (A / RG) mit RABV-G pseudotypisiert infiziert. Das Virus breitet sich zwischen TVA-exprimierenden Zellen in einer Angelegenheit von Stunden, 6 Stunden nach der Infektion (HPI), 1 und 2 Tage nach der Infektion (DPI) gezeigt. Allerdings ist die Verbreitung auf 293T-Zellen, die nicht exprimieren die TVA-Rezeptor, obwohl icht auftritt, passiert viel langsamer. Maßstab = 100 um.

Abbildung 4. Repräsentative Infektionen von RGCs von nicht-exprimierenden Mäusen TVA. Die Tiere wurden in der LGN mit dem rVSV (A / RG) Virus mit RABV-G pseudotypisiert injiziert und Gewebe geerntet 2 dpi. (A) Sparse (RGCs angedeutet durch gelbe Pfeilspitzen) oder (B) dichter Infektionen können erhalten werden, je nach dem Titer der Viren und die Qualität der Injektion. Viele verschiedene Arten von RGCs können basierend auf der Morphologie, einschließlich (C) kleine Laube RGCs, (D) Typ 1 Melanopsin RGCs, (E) ON-DSGCs, und (F) ON-OFF-DSGCs unter anderem identifiziert werden. Maßstabsbalken = 50 um.

Abbildung 5. RVSV (A / RG) Übertragung von ON-OFF-DSGCs um SACs folgt dem erwarteten Muster. (A) ON-OFF-DSGCs (weiß Pfeilspitze mit grünen ausfüllen) übertragen Virus mehrere Schutzgebiete (gelb Pfeilspitzen). (A '). Alle grünen Zellen, die nicht das DSGC Co-Label mit dem Cre-Reporter, der angibt, dass sie TVA-exprimierenden SACs sind. (BC) Die DSGC (weiß Pfeilspitze mit grünen Füllung) und BSG (gelb Pfeilspitzen) in den entsprechenden ChAT Schichten der Netzhaut inneren plexiformen Schicht (IPL) schichten. Die Zellen, die das Virus überträgt umfassen sowohl ON und OFF-Schutzgebiete. Zahlen in Paneele B 'und C' zeigen die IPL Laminae. Die Lage des DSGC Soma ist nur durch die Pfeilspitze angezeigt, aber nicht in Panel C gezeigt, um die BSG markieren. Maßstabsbalken = 50 um.
Diskussion
Verwendung von Viren zu studieren neuronalen Schaltkreise ist eine relativ hohe Durchsatzleistung Verfahren zum Analysieren angeschlossenen Neuronen. Allerdings Erzeugung sowohl VSV und RABV Virionen ist nicht trivial. Obwohl die obige Protokoll zur Rettung Virus aus cDNA aufgeführten vorgesehen ist, ist es immer noch ein geringer Wahrscheinlichkeit Ereignis. Die Niveaus von jedem der N, P, und L Plasmide müssen fein eingestellt werden, und viele Studien und repliziert müssen getan, um virale Rettungs gewährleisten....
Offenlegungen
Keine Interessenskonflikte erklärt.
Danksagungen
Wir möchten Sean Whelan zur Unterstützung bestätigen Sie mit Rettung rekombinanten VSV Varianten und Didem Goz und Ryan Chrenek für technische Unterstützung. Diese Arbeit wurde von HHMI (CLC) unterstützt, und # NS068012-01 (KTB).
Materialien
| Name | Company | Catalog Number | Comments |
| Tissue Culture | |||
| Baby Hamster Kidney (BSRT7) cells | available upon request | ||
| vaccinia (vTF7-3) | available upon request | ||
| pN, pP, pl plasmids | available upon request | ||
| Calcium Chloride | Sigma | C1016 | |
| Magnesium Chloride | Sigma | M8266 | |
| HEK 293T cells | Open Biosystems | HCL4517 | |
| 60 mm TC-Treated Culture Dish | Corning | 430166 | |
| 75 cm2 Rectangular Canted Neck Cell Culture Flask with Vent Cap | Corning | 430641 | |
| Media : DMEM (Dulbecco's Modified Eagle Medium) | Invitrogen | 12491-015 | |
| 1 M HEPES pH 7.4 | Gibo | 15630-080 | |
| FBS: Fetal Bovine Serum | Gibco | 10437-028 | |
| PKS | Invitrogen | 15140-163 | |
| Lipofectamine 2,000 Transfection Reagent | Invitrogen | 11668-019 | |
| Syringe: 5 ml Luer-Lock syringe | Sigma | Z248010-1PAK | |
| Syringe Filters | Nalgene | 190-2520 | |
| PEI: High Potency Linear PEI | Polysciences | 23966 | |
| Viral Centrifugation | |||
| Corning 150 ml Tube Top Vacuum Filter System, 0.45 μm Pore | Corning | 430314 | |
| Thinwall, Ultra-Clear, 38.5 ml, 25 x 89 mm ultracentrifuge tubes | Beckman-Coulter | 344058 | |
| Ultracentrifuge | Beckman-Coulter | optima XL-80K | |
| SW28 Ultracentrifuge rotor | Beckman-Coulter | 342207 | |
| Mouse Injection | |||
| Capillary micropipets | Drummond | 5-000-2005 | |
| Stereotax | Narishige | SR-5M | |
| Micromanipulator | Narishige | SM-15 | |
| Ump injector | World Precision Instruments | Sys-Micro4 | |
| Four channel microcontroller | World Precision Instruments | UMP3 | |
| M.TXB Bench Motor with C.EMX-1 Dial Control, 115 Volt | Foredom | M.TXB-EM | |
| H.10 Handpiece, Quick Change | Foredom | H.10 | |
| Step Drill, 0.5 mm | Foredom | A-58005P | |
| Micr–lectrode holder | World Precision Instruments | MEH2S | |
| Ketamine | Henry Schein | 995-2949 | |
| Xylazine | Henry Schein | 4015809TV | |
| Buprenorphine | Henry Schein | 1118217 | |
| 1 ml syringe | Becton-Dickinson | 309628 | |
| 30 gauge injection needle | Becton-Dickinson | 305106 | |
| Protective Ophthalmic Ointment | Doctors Foster and Smith | 9N-014748 | |
| Ethanol | Sigma | 493511 | |
| Iodine | Sigma | PVP1 | |
| Surgery and Dissection tools | |||
| Scissors | Fine Science Tools | 91402-12 | |
| Standard Forceps | Fine Science Tools | 11000-12 | |
| Fine Forceps | Fine Science Tools | 11255-20 | |
| Vannas spring scissors | Fine Science Tools | 15000-00 | |
| Scalpel handle | Fine Science Tools | 10003-12 | |
| Scalpel blades | Fine Science Tools | 10015-00 | |
| Sutures | Robbins Instruments | 20.SK640 | |
| Dissection and antibody staining | |||
| paraformaldehyde | Sigma | P6148 | |
| Phosphate Buffered Saline | Sigma | P4417 | |
| Triton X-100 | Sigma | T9284 | |
| Donkey Serum | Jackson Immunoresearch | 017-000-121 | |
| Antibodies | |||
| Antibodies | millipore | AB144P | |
| Anti-gfp | Abcam | ab13970 | |
| Donkey anti-chicken Dylight 488 | Jackson immunoresearch | 703-545-155 | |
| Donkey anti-chicken Alexa Fluor 647 | Jackson immunoresearch | 705-605-147 | |
| DAPI | Invitrogen | D1306 | |
| Tissue mounting | |||
| Superfrost plus microscope slides | Fisher | 12-550-100 | |
| Cover glass 22 x 22, 0 thickness | Electron Microscopy Sciences | 72198-10 | |
| Silicone elastomer | Rogers Corp | HT-6220 | |
| Clear nail polish | Electron Microscopy Sciences | 72180 | |
| Prolong Gold antifade reagent | Invitrogen | P36930 | |
| |||
Referenzen
- Wickersham, I. R. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 53, 639-647 (2007).
- Marshel, J. H., Mori, T., Nielsen, K. J., Callaway, E. M. Targeting single neuronal networks for gene expression and cell labeling in vivo. Neuron. 67, 562-574 (2010).
- Wall, N. R., Wickersham, I. R., Cetin, A., De La Parra, M., Callaway, E. M. Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc. Natl. Acad. Sci. U.S.A. 107, 21848-21853 (2010).
- Wickersham, I. R. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 53, 639-647 (2007).
- Yonehara, K. Spatially asymmetric reorganization of inhibition establishes a motion-sensitive circuit. Nature. 469, 407-410 (2011).
- Stepien, A. E., Tripodi, M., Arber, S. Monosynaptic rabies virus reveals premotor network organization and synaptic specificity of cholinergic partition cells. Neuron. 68, 456-472 (2010).
- Beier, K. T., Samson, M. E. S., Matsuda, T., Cepko, C. L. Conditional expression of the TVA receptor allows clonal analysis of descendents from Cre-expressing progenitor cells. Dev. Biol. 353, 309-320 (2011).
- Seidler, B. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc. Natl. Acad. Sci. U.S.A. 105, 10137-10142 (2008).
- Beier, K. T. Anterograde or retrograde transsynaptic labeling of CNS neurons with vesicular stomatitis virus vectors. Proc. Natl. Acad. Sci. U.S.A. 108, 15414-15419 (2011).
- Whelan, S. P., Ball, L. A., Barr, J. N., Wertz, G. T. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. U.S.A. 92, 8388-8392 (1995).
- Fuerst, T. R., Niles, E. G., Studier, F. W., Moss, B. Eukaryotic Transient-Expression System Based on Recombinant Vaccinia Virus That Synthesizes Bacteriophage T7 RNA Polymerase. PNAS. 83, 8122-8126 (1986).
- Young, J. A., Bates, P., Varmus, H. E. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J. Virol. 67, 1811-1816 (1993).
- Madisen, L. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 13, 133-140 (2010).
- Franklin, K., Paxinos, G. . The Mouse Brain in Stereotaxic Coordinates. , (1997).
- van den Pol, A. N. Viral strategies for studying the brain, including a replication-restricted self-amplifying delta-G vesicular stomatis virus that rapidly expresses transgenes in brain and can generate a multicolor golgi-like expression. J. Comp. Neurol. 516, 456-481 (2009).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten